

Abstract
Overproduction of nitric oxide (NO) by inducible nitric oxide synthase (iNOS) has been implicated in the pathogenesis of many disorders. iNOS is notably distinguished from constitutive NOSs by its production of large amounts of NO for a prolonged period; hence, it was termed the high-output NOS. Understanding how cells regulate iNOS is a prerequisite for strategies aimed at modulating NO synthesis. iNOS is thought to be regulated primarily at the transcriptional level in response to cytokines and inflammatory mediators. In this study, we report a posttranslational regulatory mechanism for control of iNOS expression through a rapid cellular rate of turnover. Unexpectedly, iNOS cellular half-life was found to be relatively short. In primary bronchial epithelial cells, iNOS half-life was 1.6 ± 0.3 h. A similar half-life was found for iNOS in several cell lines. This fast rate of turnover is in sharp contrast to that reported for the constitutive NOS isoforms. iNOS half-life was not affected by intracellular depletion of tetrahydrobiopterin, a critical cofactor required for iNOS activity. Further, iNOS monomers and dimers had a similar half-life. Importantly, we discovered a previously unrecognized cotranslational down-regulation mechanism by which the newly discovered pyrimidineimidazole-based allosteric dimerization inhibitors of iNOS lead to reduced iNOS expression. This study provides insights into the cellular posttranslational mechanisms of iNOS and has important implications for design of selective iNOS inhibitors and their use in therapeutic strategies.
Keywords: degradation, proteasome, half-life
Nitric oxide (NO) is an important signaling and cytotoxic molecule that is synthesized from l-arginine by isoforms of nitric oxide synthase (NOS) (1–3). As a signaling molecule, NO is produced by two constitutive calcium (Ca2+)-dependent isoforms, neuronal NOS and endothelial NOS (or NOSI and NOSIII, respectively). Ca2+-activated calmodulin binds to and transiently activates constitutive NOS dimers (3). Because of the transient nature of elevated Ca2+ levels, these isoforms produce small amounts of NO for short periods of time. As an agent of inflammation and cell-mediated immunity, NO is produced by a Ca2+-independent cytokine-inducible NOS (iNOS or NOSII) that is widely expressed in diverse cell types under transcriptional regulation by inflammatory mediators (2, 4). Calmodulin is tightly bound to iNOS even at basal Ca2+ levels; therefore, iNOS is notably distinguished from the constitutive isoforms by its prolonged production of a relatively large amount of NO (5). iNOS has been implicated in the pathogenesis of many diseases including Alzheimer's disease, tuberculosis, asthma, transplant rejection, stroke, glaucoma, inflammatory bowel disease, arthritis, and septic shock (6, 7). Such wide implication has produced a corresponding intense interest in understanding the regulation of NO synthesis by iNOS with the goal of developing therapeutic strategies aimed at selective modulation of iNOS activity (8). Understanding how cells regulate iNOS is a prerequisite for strategies aimed at modulating NO synthesis.
The activity of an enzyme can be controlled through the regulation of its synthesis, catalytic activity, or degradation. Although much is known about factors affecting the synthesis and catalytic activity of iNOS, little is known about mechanisms regulating its degradation. Recently, the ubiquitin–proteasome pathway has been identified as the major pathway responsible for iNOS degradation (9, 10). However, the cellular regulatory mechanisms governing the targeting of iNOS for degradation have yet to be studied.
Here, we report the presence of a posttranslational level of regulation for nitric oxide synthesis by iNOS that is accomplished by maintaining a fast cellular turnover of iNOS. This fast rate of turnover is in sharp contrast to the half-life reported for the constitutive isoforms, endothelial NOS (28 h) (11) and the neuronal NOS (20 h) (12). We propose that these regulatory mechanisms allow cells to maintain a tighter control over NO synthesis. Furthermore, we show that the newly discovered allosteric selective dimerization inhibitors of iNOS have an additional regulatory effect on iNOS protein production by cotranslational down-regulation of iNOS expression.
Materials and Methods
Pulse–Chase Analysis. Studies of iNOS protein stability in cultured cells were conducted in confluent monolayers that were starved for methionine and cysteine in serum-free media for 0.5 h at 37°C, then pulsed for 1 h, in methionine and cysteine-free DMEM, with 0.25 mCi/ml (1 Ci = 37 GBq) l-[35S]methionine/cysteine mix (Amersham Pharmacia). Cells were then vigorously washed twice with PBS, incubated in DMEM supplemented with 10% FBS, 2 mM glutamine, and 300 mg/liter nonradioactive methionine and cysteine, and chased at specific time intervals. In experiments involving proteasomal inhibition, 10 μM MG132 was added 2 h before the start of the pulse–chase and maintained in culture media throughout the procedure. At the end of each chase period, cells were lysed and subjected to immunoprecipitation of iNOS. The immunoprecipitated proteins were analyzed by SDS/PAGE (3–8%), and gels were dried and analyzed by PhosphorImager (Molecular Dynamics) or x-ray film (Kodak MR) densitometry.
In Vitro Transcription–Translation of iNOS. [35S]methionine-labeled iNOS was expressed in vitro by using rabbit reticulocyte system (TnT Quick Coupled Transcription/Translation System, Promega). Each reaction (50 μl) contained 1 μg of human iNOS cDNA and 2 μl of [35S]methionine (10 mCi/ml, Amersham Pharmacia). Reactions were performed at 30°C for 90 min.
Cell culture, transfection, iNOS induction, cell lysis, iNOS-activity assays, Western analysis, and immunoprecipitation were performed as described in refs. 13–15 and in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Results and Discussion
Determination of Human iNOS Half-Life in Human Embryonic Kidney (HEK) 293 Cells. HEK 293 cells are human embryonic kidney epithelial cells that do not contain any of the NOS genes, and they have been previously used for the characterization of human iNOS (13–15). We used a HEK 293 cell line that stably expresses human iNOS (9, 10). Pulse–chase analysis revealed that human iNOS in these cells has a half-life of ≈2.0 ± 0.1 h (Fig. 1). The relatively short half-life of iNOS was a rather surprising finding because iNOS is known for its prolonged production of large amounts of NO. By demonstrating the accumulation of iNOS after proteasomal inhibition, we have previously shown that human iNOS is primarily degraded by the proteasome pathway (9). In this study, to directly evaluate the effect of proteasomal inhibition on iNOS half-life, we analyzed iNOS half-life in HEK 293 cells expressing iNOS in the presence or absence of the proteasome inhibitor MG132 (10 μM). With proteasomal inhibition, the half-life of iNOS was markedly prolonged to 6.5 ± 1.2 h (Fig. 1). These results confirm that iNOS is primarily degraded through the proteasome pathway.
Fig. 1.

Determination of human iNOS half-life. HEK 293 cells, stably expressing human iNOS, were pulsed with [35S]methionine/cysteine for 1 h and chased with unlabeled media at various time points in the presence or absence of 10 μM proteasome inhibitor MG132. iNOS was immunoprecipitated with anti-iNOS antibody. To verify the stringency of immunoprecipitation, in some samples the immunoprecipitating antibody was omitted (lane 0*) or HEK 293 cells that do not express iNOS were used (lane 0**). Eluted proteins were separated by SDS/PAGE, and 35S-labeled iNOS was detected by PhosphorImager. (Lower) iNOS half-life was calculated, for cells cultured in the absence (filled circles) or the presence of MG132 (open squares), as the time needed for decay of 50% of 35S-labeled iNOS. The half-life of human iNOS in HEK 293 cells was 2.0 ± 0.1 h and was prolonged to 6.5 ± 1.2 h in cells treated with the proteasomal inhibitor MG132 (average ± SD, n = 3).
Determination of Half-Life of Cytokine-Induced Human iNOS. iNOS is known to be up-regulated in vivo in response to cytokines and inflammatory mediators. Therefore, we sought to examine the half-life of iNOS after cytokine induction, thus mimicking the in vivo state and eliminating concerns over the consequences of exogenous expression of iNOS in HEK 293 cells. We used A549 and RT4 cell lines. A594 is a human alveolar type II epithelium-like lung carcinoma cell line, and RT4 is a human urinary bladder transitional cell papilloma cell line. A549 and RT4 cells were stimulated by a cytokine mixture of IFN-γ, IL-1β, TNF-α, and IL-6 (4, 9). Eighteen hours later, cytokine-containing media were removed and a pulse–chase analysis was performed. Human iNOS had a half-life of 1.6 ± 0.4h(n = 3) in A549 cells and 1.6 ± 0.1 h (n = 3) in RT4 cells (Fig. 2 A and B). These results are consistent with data obtained with HEK 293 cells and extend findings to cells that express iNOS as a result of a more “natural” induction. Thus, they indicate that the observed half-life of iNOS is not peculiar to HEK 293 and is not simply the consequence of overexpression.
Fig. 2.

Determination of half-life of cytokine-induced human iNOS. A549 cells (A), RT4 cells (B), or primary bronchial epithelial cells (C) were stimulated for 18–24 h by a cytokine mixture to induce iNOS. Pulse–chase analysis and iNOS immunoprecipitation was performed as in Fig. 1. In some samples the immunoprecipitating antibody was omitted (lane 0*) or unstimulated cells were used (lane 0**). Immunoprecipitated proteins were separated by SDS/PAGE, and bands, representing 35S-labeled iNOS, were quantitated to calculate half-life of iNOS. The half-life of iNOS was 1.6 ± 0.4 h in A549 cells, 1.6 ± 0.1 h in RT4 cells, and 1.6 ± 0.3hin primary bronchial cells (average ± SD, n = 3).
Determination of Human iNOS Half-Life in Primary Bronchial Epithelial Cells. In human lungs, airway bronchial epithelial cells are a major source of iNOS (4, 7, 16). In airway inflammation of asthma, iNOS expression is markedly increased in these cells (7). Therefore, we sought to determine the half-life of iNOS in primary bronchial epithelial cells cultured at the air–liquid interface system (10). These cells clearly represent a closer model for the in vivo state in the lung airways. More importantly, when cells are cultured in the air–liquid interface they can achieve a state of differentiation that can be obtained neither in transformed cell lines nor in primary cells cultured submerged in tissue culture medium (17). In primary cells, stimulated by a cytokine mixture as above, human iNOS had a half-life of 1.6 ± 0.3 h (n = 3) (Fig. 2C), confirming the results obtained with cell lines and further indicating that the observed half-life of iNOS is not restricted to transformed cell lines.
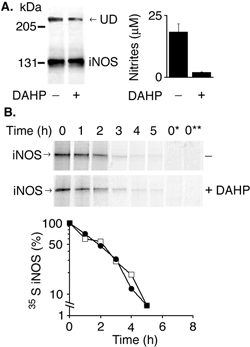
Tetrahydrobiopterin (H4B) Depletion in Cultured Cells Has no Effect on iNOS Half-Life. H4B is an important cofactor that is required for iNOS activity. Its exact role in NO synthesis, however, is not entirely clear. A role for H4B in stabilizing iNOS dimers has been debated (18–20). We aimed to determine whether reducing the intracellular levels of H4B would affect iNOS half-life. Intracellular H4B synthesis can be inhibited by 2,4-diamino-6-hydroxypyrimidine (DAHP), which is a selective inhibitor of GTP cyclohydrolase I, the rate-limiting enzyme for de novo H4B synthesis (21). Incubation of HEK 293 cells stably expressing iNOS with 10 mM DAHP for 18 h resulted in virtual elimination of iNOS activity but did not have detectable effect on iNOS half-life (Fig. 8, which is published as supporting information on the PNAS web site). Similar results were obtained by using the murine macrophage cell line RAW264.7, stimulated by LPS and IFN-γ, to produce iNOS (data not shown). The virtual elimination of iNOS activity by DAHP treatment of cells indicates that H4B levels were critically reduced (21). Furthermore, iNOS activity was restored by incubating cells with sepiapterin, an agent that circumvents the inhibition of H4B synthesis by DAHP (data not shown), confirming that iNOS inhibition was due to a lack of H4B. These results suggest that H4B does not play a major role in stabilizing iNOS protein. They also indicate that inactive iNOS is not preferentially degraded compared with active iNOS.
Effect of iNOS Dimerization Inhibitors on Cellular iNOS Activity and Expression. The use of selective inhibitors of iNOS has been advocated as a novel therapeutic approach for several inflammatory diseases, e.g., asthma, rheumatoid arthritis, multiple sclerosis, and glaucoma (6–8). The catalytic activity of iNOS can be inhibited by using l-arginine analogues (3). A major difficulty in the use of these compounds is their lack of specificity for the iNOS isoform. The heme ligands imidazole and imidazole-containing antifungal compounds such as clotrimazole and miconazole are known inhibitors of heme-containing enzymes including iNOS (22). These compounds have been shown to inhibit iNOS dimerization by binding to its heme ligand. However, they have low affinity to NOS and lack specificity for the iNOS isoform (22–24). Recently, screening of an encoded combinatorial chemical library, designed based on a structurally related series of compounds of a pyrimidineimidazole core, resulted in the discovery of a class of allosteric high-affinity potent and selective iNOS inhibitors. The prototype compound of this class, BBS-1 (compound 2 in ref. 23), acts by binding to the iNOS monomer. Crystallographic studies showed that BBS-1 coordinates directly to the heme at the sixth axial position and disrupts helices 7a and 8 in the active site (23, 24). Thus, in occupying the iNOS active site, BBS-1 appears to allosterically prevent appropriate protein–protein interactions between monomers and prevents dimer formation. The inhibitor does not, however, bind to and has no effect on the catalytic activity of the already formed iNOS dimer, because it cannot gain access to the heme site in the dimer (23, 24).
In the context of therapeutic inhibition of iNOS, the finding that iNOS has a much faster rate of turnover compared with the constitutive isoforms, endothelial NOS (28 h) (11) and the neuronal NOS (20 h) (12), has important implications for the design of selective inhibitors for iNOS. Inhibitors that would bind to and inactivate newly synthesized NOSs, e.g., imidazole-like compounds (23), would be inherently more iNOS-selective because they would theoretically inactivate 50% of iNOS in a given NOS synthesis biological system in only 2 h, provided that the inhibitor would bind to and inactivate 100% of newly synthesized iNOS. To test this prediction, we incubated HEK 293 cells with the iNOS allosteric dimerization inhibitor, BBS-1 (1 μM), for various time points and analyzed cell lysates for iNOS activity and by Western analysis (Fig. 3). In the presence of the inhibitor, iNOS activity gradually decreased over time. After 2 h, iNOS activity decreased to 61% of its original value. The calculated time needed for BBS-1 to induce a 50% reduction of iNOS activity was 2.3 ± 0.1 h, which is in reasonable agreement with iNOS half-life data of 2.0 ± 0.1 h in these cells. As expected, Western analysis of the same samples showed gradual disappearance of iNOS undisruptable dimers (UD). iNOS UD have been shown to correlate with the ability of iNOS to form dimers (20). Unexpectedly, however, there was a gradual reduction in the amount of iNOS protein detected in the presence of the inhibitor over time. To explore this observation further, we incubated HEK 293 cells stably expressing iNOS at higher concentrations of BBS-1. Under these conditions, we observed a more pronounced reduction in the expression of iNOS protein (Fig. 4A). Similar results were observed when we performed the same experiments using the murine macrophage cell line RAW264.7 in which iNOS was induced by LPS and IFN-γ (Fig. 4B) (9). In this cell line, we took advantage of the inducible nature of iNOS by adding BBS-1 immediately before iNOS induction. Thus, BBS-1 should potentially bind to almost all newly synthesized iNOS monomers. This resulted in a higher degree of iNOS inactivation, compared with that achieved in HEK 293 cells. More importantly, in both cell lines, there was marked decrease in iNOS protein expression. In a recent independent study, and consistent with our findings, BBS-1 was noted to reduce iNOS protein expression in cardiac allograft in vivo in a rat model of acute cardiac allograft rejection (25).
Fig. 3.

Time course of the effect of iNOS dimerization inhibitor, BBS-1. HEK 293 cells stably expressing human iNOS were incubated for the indicated time periods in the presence of 1 μM BBS-1. A representative Western blot of cells lysates (50 μg per lane), using an anti-iNOS antibody, is shown (Top). UD, presence of UD of iNOS. Densitometry analysis of bands representing iNOS on the Western blot is shown (Middle). iNOS activity was evaluated in cell lysates by the conversion of [3H]arginine to [3H]citrulline (Bottom) (means ± SD, n = 3).
Fig. 4.

Effect of iNOS dimerization inhibitor BBS-1 on iNOS expression. HEK 293 cells stably expressing iNOS (A) or RAW264.7 cells stimulated to produce iNOS by incubation in media containing LPS (100 ng/ml) and IFN-γ (10 units/ml) (B) were incubated for 18 h in the presence of BBS-1 (0–100 μM). A representative Western blot of cells lysates (50 μg/lane), using an anti-iNOS antibody, is shown (Upper). UD, presence of UD of iNOS. iNOS activity was evaluated by measuring nitrite accumulation in culture media (Lower) (means ± SD, n = 2).
It has been previously shown that BBS-1 class of inhibitors does not affect iNOS mRNA in cell culture system (23). Furthermore, in each of the above two cell lines, iNOS is transcribed from a different promoter, endogenous iNOS promoter in RAW264.7 vs. exogenous CMV promoter in iNOS cDNA plasmid expressed in HEK 293 cells. Therefore, the reduction in iNOS expression induced by BBS-1 could not be accounted for by reduction in iNOS mRNA transcription and thus must be due to a posttranscriptional effect.
BBS-1 binding to iNOS monomer is previously assumed to lead to the formation of a “dead-end” inhibitor–monomer complex (23, 24). Therefore, it was logical to speculate that the inhibitor–monomer complex may be rapidly degraded compared with the active dimeric iNOS. To investigate this possibility, we evaluated the effect of BBS-1 on iNOS half-life in HEK 293 cells stably expressing iNOS. Unexpectedly, the half-life of active dimeric iNOS and that of inhibitor inactivated monomeric iNOS were almost identical (Fig. 5). Similar results were obtained when using RAW264.7 cells stimulated by LPS and IFN-γ to produce iNOS in the presence or absence of BBS-1 (data not shown). These results indicate that the iNOS monomers inactivated by BBS-1 are not preferentially targeted for degradation compared with active dimeric iNOS.
Fig. 5.

Effect of iNOS dimerization inhibitor, BBS-1, on iNOS half-life. HEK 293 cells stably expressing iNOS were incubated for 18 h in the presence or absence of 1 μM BBS-1. (A) Western blotting of cells lysates, using anti-iNOS antibody (Left). UD, presence of UD of iNOS. iNOS activity was evaluated by measuring nitrite accumulation in culture media (n = 3) (Right). (B) Pulse–chase analysis was performed in the presence or absence of BBS-1. iNOS was immunoprecipitated with anti-iNOS antibody. Eluted proteins were separated by SDS/PAGE, and bands, representing 35S-labeled iNOS were quantitated to calculate iNOS half-life (Lower). Filled circles and open squares denote cells incubated in the absence or presence of BBS-1, respectively.
iNOS Dimers and Monomers Are Degraded at Similar Rates. iNOS, similar to other NOSs, is only active as a homodimer (3). Unlike the constitutive isoforms, iNOS dimers, because of bound calmodulin, are always active even at low calcium levels (5). However, the subsequent fate of iNOS dimers is not clear. It is not known whether dimers are primarily targeted for degradation or whether their dissociation into inactive monomers is required before their degradation. This is an important question because termination of dimerization either by degradation or by dissociation seems to be the major cellular mechanism of inactivating iNOS. If dimer dissociation is required for iNOS degradation, a purely monomeric iNOS should have a shorter half-life than a mostly dimeric iNOS. The above results obtained with BBS-1 bound monomers implied that iNOS monomers, in general, are not preferentially targeted for degradation compared with dimers. However, we wanted to verify this observation by generating iNOS monomers independent of the use of BBS-1 and thus eliminating the bound inhibitor as a potential confounding factor. We generated a human iNOS mutant with alanine replacing glycine at position 456 (iNOS G456A). A similar mutation of the cognate residue in murine iNOS was previously shown to prevent iNOS dimerization, producing only monomeric iNOS (26). Both wild-type iNOS and iNOS G456A were transiently transfected in HEK 293T cells. Wild-type iNOS expressed in HEK 293T cells is mostly dimeric by gel permeation chromatography (20). In contrast, iNOS G456A was inactive and could not form dimers (Fig. 6A). However, on pulse–chase analysis both wild-type iNOS and iNOS G456A had almost identical cellular half-life (Fig. 6B). These results indicate that iNOS monomers are not preferentially targeted for degradation, consistent with data obtained on BBS-1-bound iNOS monomers.
Fig. 6.

Evaluation of half-life of the monomeric human iNOS mutant G456A. HEK 293T cells were transfected with plasmids encoding cDNA of wild-type human iNOS or G456A iNOS mutant. (A) Twenty hours after transfection, cells were lysed and evaluated by Western blotting, using anti-iNOS antibody (Left). iNOS activity was evaluated by measuring nitrite accumulation in culture media (Right). UD, presence of UD of iNOS. (B) Pulse–chase analysis was performed. iNOS was immunoprecipitated with anti-iNOS antibody. Eluted proteins were separated by SDS/PAGE, and bands, representing 35S-labeled iNOS, were quantitated to calculate iNOS half-life (Lower). Filled circles and open squares denote wild-type iNOS and iNOS G456A mutant, respectively.
Cotranslational Down-Regulation of iNOS by Allosteric Dimerization Inhibitors. In eukaryotic cells, a large number of proteins are multidomain proteins and, therefore, protein folding and assembly occur cotranslationally (27). For iNOS, both heme insertion and calmodulin binding are thought to occur during or promptly after protein translation (3, 5). We therefore hypothesized that the reduction in iNOS protein expression induced by BBS-1 binding may occur cotranslationally. In our pulse–chase analysis above, cells were radiolabeled for 1 h. This period was necessary to have enough amounts of iNOS synthesized to be reliably detected during later points of chase time. During the chase, iNOS expression at various time points was compared with its expression at the 0-h time point, which is at the end of the 1-h labeling period. Therefore, any changes in iNOS production that would occur during or promptly after translation would not be reflected in its measured half-life. To reconcile the findings of the reduction of iNOS protein by BBS-1 and the absence of a significant change in iNOS half-life, we postulated that reduction in iNOS protein expression induced by BBS-1 binding must occur cotranslationally, i.e., during or promptly after translation. This effect could be due to either reduction in translated iNOS or a cotranslation-associated degradation that degrades the BBS-1-bound iNOS monomer preferentially.
To investigate this hypothesis, we labeled HEK 293 cells stably expressing iNOS with [35S]methionine/cysteine for 1 h, in the presence of BBS-1 or its vehicle only, and then immunoprecipitated 35S-labeled iNOS from cell lysates and evaluated the products on SDS/PAGE. This procedure allowed us to estimate the amount of iNOS that was synthesized exclusively during the 1-h labeling period as evidenced by its 35S label. Furthermore, direct analysis of 35S-labeled iNOS by SDS/PAGE by using this method is inherently more reliable for quantitation purposes than Western analysis in which significant signal amplification is used. The 35S-labeled iNOS was markedly reduced in cells treated with BBS-1 compared with vehicle only (Fig. 7A). These results confirmed our hypothesis that the reduction in iNOS expression induced by BBS-1 is an early event that occurs during or promptly after translation. To confirm this observation in an another independent model of iNOS translation, we used an in vitro coupled transcription/translation system in rabbit reticulocytes lysates to synthesize a 35S-labeled iNOS for 90 min. Consistent with data obtained in cultured cells, in vitro translated iNOS was markedly reduced in the presence of BBS-1 compared with vehicle only. The reduction of iNOS product in this system was still detectable at the lowest tested concentration of BBS-1 (50 nM) (Fig. 7B). To examine the effect of potential cotranslational proteasomal degradation of BBS-1-bound iNOS, we tested whether the specific proteasome inhibitor lactacystin could prevent the cotranslational reduction in iNOS induced by BBS-1. Lactacystin could only partially rescue iNOS treated with BBS-1 (Fig. 7C). Similar findings were obtained by using a different proteasome inhibitor, MG132 (data not shown). These data suggest that only a minor part of the iNOS reduction by BBS-1 is due to cotranslational degradation by the 26S proteasome. The major cause of reduction, however, seems to be due to reduced translation. We hypothesize that the reduction in translation may be due to a slow rate of protein assembly, cofactors binding, and/or protein folding caused by the disorder in the active site associated with BBS-1 binding.
Fig. 7.

Effect of BBS-1 on iNOS translation in cultured cells and in vitro.(A) HEK 293 cells, stably expressing iNOS, were labeled by [35S]methionine/cysteine mix for 1 h in the presence or absence of 10 μM BBS-1. iNOS was immunoprecipitated from cell lysates with anti-iNOS antibody. Eluted proteins were separated by SDS/PAGE, and bands, representing 35S-labeled iNOS, were detected by PhosphorImager. (B) Rabbit reticulocyte-based in vitro coupled transcription/translation system and iNOS cDNA plasmid were used to express human iNOS in the presence of 0–100 nM BBS-1. Reactions were performed at 30°C for 90 min. Translation products were separated by SDS/PAGE, and bands, representing 35S-labeled iNOS, were detected by PhosphorImager. (C) In vitro translation of iNOS was done in the presence of BBS-1(10 μM), in the presence or absence of the proteasome inhibitor lactacystin (10 μM). (D) HEK 293 cells, stably expressing iNOS, were incubated for 8 h in the presence or absence of BBS-1 (10 μM) or the translation inhibitor emetine (20 μM). Western blotting of cell lysates (50 μg per lane) was performed by using an anti-iNOS antibody.
To further confirm this finding, we examined the effect of the translation inhibitor emetine on iNOS expression in HEK 293 cells stably expressing iNOS and treated with BBS-1 or vehicle only (Fig. 7D). Cells were incubated in the presence of reagents for 8 h. As expected, iNOS expression was markedly diminished in cells treated with emetine because of general inhibition of protein translation. Interestingly, when translation was inhibited by emetine, there was no significant difference in iNOS expression in cells treated with or without BBS-1. These data suggest that the BBS-1 effect on iNOS expression is largely due to a reduction in iNOS translation.
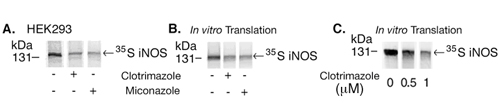
BBS-1 is related to the antifungal imidazoles clotrimazole and miconazole, previously shown to inhibit iNOS dimerization presumably because of steric hindrance from the aromatic side chains (22). BBS-1, however, has higher affinity for iNOS monomers over clotrimazole and miconazole, presumably because of specific interactions with residues in iNOS (23, 24). We reasoned that, because of similarities in structure and effects on iNOS activity, clotrimazole and miconazole would induce cotranslational down-regulation of iNOS similar to that caused by BBS-1. In a similar experiment to that performed with BBS-1, both clotrimazole and miconazole reduced iNOS translation in HEK 293 cells and in an iNOS in vitro expression system (Fig. 9, which is published as supporting information on the PNAS web site). These results suggest common mechanisms among imidazole-related compounds in the down-regulation of iNOS translation.
In summary, the results of this study are important in understanding the regulation of iNOS turnover and in designing therapeutic strategies aimed at iNOS regulation. The constitutive isoforms are tightly controlled by the level of intracellular Ca2+, whereas iNOS is Ca2+-independent. Therefore, it seems plausible that cells may exert their control on iNOS by ensuring its rapid degradation. Thus, we propose that cells developed a dual strategy for controlling iNOS expression. First, lack of dependence on intracellular levels of Ca2+ ensures that once iNOS is induced, it is continuously actively producing large amounts of NO, essential for host defense. Second, cells simultaneously degrade iNOS rapidly, thus ensuring that once the need for NO is abated, its production is rapidly terminated. This strategy may be beneficial to injury avoidance in host cells. Thus, rapid proteasomal degradation of iNOS could act as a safety valve.
Furthermore, our study reveals a previously unrecognized mechanism for the allosteric pyrimidineimidazole-based iNOS dimerization inhibitors that involve cotranslational down-regulation of iNOS. This issue is critical for understanding the clinical utility of these inhibitors. In this regard, an inhibitor that inactivates iNOS and reduces its expression would be clearly superior to one that only inhibits iNOS activity. By reducing iNOS expression, subsequent use of reduced dose of the inhibitor will be allowed and thus minimize side effects including potential unintended effects on other NOS isoforms.
Supplementary Material
Acknowledgments
We thank J. Parkinson (Berlex) for the BBS-1 inhibitor, R. Sifers for useful discussions, M. Moczygemba for review of the manuscript, and A. Musial and T. Mazumdar for assistance with some of the preliminary experiments. This work was supported by the American Lung Association; the American Heart Association; the National Heart, Lung, and Blood Institute; and the National Institute of Allergy and Infectious Diseases.
Author contributions: P.J.K. and N.T.E. designed research; P.J.K., J.-S.K., and N.T.E. performed research; P.J.K. and N.T.E. analyzed data; and P.J.K. and N.T.E. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: H4B, tetrahydrobiopterin; HEK, human embryonic kidney; NOS, NO synthase; iNOS, inducible NOS; UD, undisruptable dimers.
References
- 1.Ignarro, L. J., Buga, G. M., Wood, K. S., Byrns, R. E. & Chaudhuri, G. (1987) Proc. Natl. Acad. Sci. USA 84, 9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie, Q.-w., Cho, H. J., Calaycay, J., Mumford, R. A., Swiderek, K. M., Lee, T. D., Ding, A., Troso, T. & Nathan, C. (1992) Science 256, 225–228. [DOI] [PubMed] [Google Scholar]
- 3.Stuehr, D. J. (1999) Biochim. Biophys. Acta 1411, 217–230. [DOI] [PubMed] [Google Scholar]
- 4.Eissa, N. T., Strauss, A. J., Haggerty, C. M., Choo, E. K., Chu, S. C. & Moss, J. (1996) J. Biol. Chem. 271, 27184–27187. [DOI] [PubMed] [Google Scholar]
- 5.Cho, H. J., Xie, Q.-w., Calaycay, J., Mumford, R. A., Swiderek, K. M., Lee, T. D. & Nathan, C. (1992) J. Exp. Med. 176, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nathan, C. (1997) J. Clin. Invest. 100, 2417–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo, F. H., Comhair, S. A. A., Zheng, S., Dweik, R. A., Eissa, N. T., Thomassen, M. J., Calhoun, W. & Erzurum, S. C. (2000) J. Immunol. 164, 5970–5980. [DOI] [PubMed] [Google Scholar]
- 8.Hobbs, A. J., Higgs, A. & Moncada, S. (1999) Annu. Rev. Pharamacol. Toxicol. 39, 191–220. [DOI] [PubMed] [Google Scholar]
- 9.Musial, A. & Eissa, N. T. (2001) J. Biol. Chem. 276, 24268–24273. [DOI] [PubMed] [Google Scholar]
- 10.Kolodziejski, P. J., Musial, A., Koo, J. S. & Eissa, N. T. (2002) Proc. Natl. Acad. Sci. USA 99, 12315–12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fairchild, T. A., Fulton, D., Fontana, J. T., Gratton, J. P., McCabe, T. J. & Sessa, W. C. A. (2001) J. Biol. Chem. 276, 26674–26679. [DOI] [PubMed] [Google Scholar]
- 12.Noguchi, S., Jianmongkol, S., Bender, A. T., Kamada, Y., Demady, D. R. & Osawa, Y. (2000) J. Biol. Chem. 275, 2376–2380. [DOI] [PubMed] [Google Scholar]
- 13.Eissa, N. T., Yuan, J., Haggerty, C. M., Choo, E. K. & Moss, J. (1998) Proc. Natl. Acad. Sci. USA 95, 7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eissa, N.T., Haggerty, C. M., Palmer, C. D., Patton, W. & Moss, J. (2001) Am. J. Respir. Cell Mol. Biol. 24, 616–620. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh, D. K., Rashid, M. B., Crane, B., Taskar, V., Mast, M., Misukonis, M., Weinberg, J. B. & Eissa, N. T. (2001) Proc. Natl. Acad. Sci. USA 98, 10392–10397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu, S. C., Wu, H.-P., Banks, T. C., Eissa, N. T. & Moss, J. (1995) J. Biol. Chem. 270, 10625–10630. [DOI] [PubMed] [Google Scholar]
- 17.Koo, J. S., Jetten, A. M., Belloni, P., Yoon J. H., Kim, Y. D. & Nettesheim, P. (1999) Biochem. J. 338, 351–357. [PMC free article] [PubMed] [Google Scholar]
- 18.Panda, K., Rosenfeld, R. J., Ghosh, S., Meade, A. L., Getzoff, E. D. & Stuehr, D. J. (2002) J. Biol. Chem. 277, 31020–31030. [DOI] [PubMed] [Google Scholar]
- 19.Hurshman, A. R., Krebs, C., Edmondson, D. E. & Marletta, M. A. (2003) Biochemistry 42, 13287–13303. [DOI] [PubMed] [Google Scholar]
- 20.Kolodziejski, P. J., Rashid, M. B. & Eissa, N. T. (2003) Proc. Natl. Acad. Sci. USA 100, 14263–14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross, S. S. & Levi, R. (1992) J. Biol. Chem. 267, 25722–25729. [PubMed] [Google Scholar]
- 22.Sennequier, N., Wolan, D. & Stuehr, D. J. (1999) J. Biol. Chem. 274, 930–938. [DOI] [PubMed] [Google Scholar]
- 23.McMillan, K., Adler, M., Auld, D. S., Baldwin, J. J., Blasko, E., Browne, L. J., Chelsky, D., Davey, D., Dolle, R. E., Eagen, K. A., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 1506–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blasko, E., Glaser, C. B., Devlin, J. J., Xia, W., Feldman, R. I., Polokoff, M. A., Phillips, G. B., Whitlow, M., Auld, D. S., McMillan, K., et al. (2002) J. Biol. Chem. 277, 295–302. [DOI] [PubMed] [Google Scholar]
- 25.Szabolcs, M. J., Sun, J., Ma, N., Albala, A., Sciacca, R. R., Philips, G. B., Parkinson, J., Edwards, N. & Cannon, P. J. (2002) Circulation 106, 2392–2396. [DOI] [PubMed] [Google Scholar]
- 26.Xie, Q.-w., Leung, M., Fuortes, M., Sassa, S. & Nathan, C. (1996) Proc. Natl. Acad. Sci. USA 93, 4891–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis, R. J. & Hartl, F. U. (1999) Curr. Opin. Struct. Biol. 9, 102–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}