

Abstract
The muscle-specific receptor tyrosine kinase (MuSK) is part of a receptor complex, activated by neural agrin, that orchestrates the differentiation of the neuromuscular junction (NMJ). To gain insight into the function of the MuSK complex, we have developed a proteomic approach to identify new MuSK partners. MS analysis of MuSK crosslink products from postsynaptic membranes of the Torpedo electrocytes identified the adaptor protein 14-3-3 γ. The 14-3-3 γ protein was found localized at the adult rat NMJ. Cotransfection experiments in COS-7 cells showed that MuSK codistributed with the 14-3-3 γ protein at the plasma membrane. Furthermore, 14-3-3 γ was copurified by affinity chromatography with MuSK from transfected COS-7 cells and myotubes. The 14-3-3 γ protein did not colocalize with agrin-elicited acetylcholine receptor (AChR) aggregates in cultured myotubes, suggesting that it is not involved in AChR clustering. Expression of 14-3-3 γ specifically repressed the transcription of several synaptic reporter genes in cultured myotubes. This repression was potentiated by MuSK expression. Moreover, the expression of 14-3-3 γ in muscle fibers in vivo caused both the repression of synaptic genes transcription and morphological perturbations of the NMJ. Our data extend the notion that, apart from its well documented role in AChR clustering, the MuSK complex might also be involved in the regulation of synaptic gene expression at the NMJ.
Keywords: neuromuscular junction, synaptogenesis
An important step in differentiation of the neuromuscular junction (NMJ) is the accumulation of synaptic proteins, including acetylcholine receptors (AChRs) at postsynaptic sites (reviewed in refs. 1–3). Clustering of AChRs requires the muscle-specific kinase (MuSK), a tyrosine kinase receptor specifically expressed in skeletal muscle cells (4, 5). The steps that follow MuSK activation by neural agrin and lead to clustering of synaptic components are still incompletely understood. In the last few years, several MuSK partners have been identified, including Dishevelled (Dvl1), a signaling molecule important for planar cell polarity in Drosophila that interacts both with the cytoplasmic region of MuSK and the downstream kinase PAK1 (6), the Ablesson (Abl) kinases required for agrin-stimulated enhancement of MuSK tyrosine phosphorylation, and AChR clustering by activation of Rac/Cdc42 pathway (7). Geranylgeranyltransferase (8), Src family kinases (9), the scaffolding molecules MAGI-1c, a membrane-associated guanylate kinase (10), and AChR (11) are also associated with MuSK.
A common view is that agrin promotes AChR clustering at the NMJ without major effects on transcriptional regulation. However, several studies supported the notion that agrin or constitutively active MuSK can stimulate the transcription of synaptic genes through the downstream expression and aggregation of ErbB tyrosine kinase receptors activated by the nerve-derived factor, neuregulin-1 (NRG; reviewed in ref. 3). Moreover, activation of MuSK by agrin elicits AChR expression in vivo in the absence of a nerve terminal and, thus, of neural NRG (12). Recently, Lacazette et al. (13) have shown that agrin-induced synaptic gene expression is controlled in part by a secondary NRG/ErbB pathway organized by agrin/MuSK and in part by a shunt path in which MuSK signals to the muscle nuclei more directly by Rac, independent of NRG/ErbB.
In this new context, it is important to pursue the identification of potential new effectors of MuSK that may account for its pleiotropic effects. In this work, chemical crosslinking experiments in AChR-rich membrane of Torpedo electrocytes followed by MALDI-TOF MS analysis of the MuSK crosslink products allowed us to identify the adaptor protein 14-3-3 γ as a candidate for MuSK signaling at the NMJ. The 14-3-3 proteins constitute a family of conserved regulatory proteins involved in such cellular processes as cell division, signaling, and apoptosis (reviewed in refs. 14–16). Forced expression of 14-3-3 γ in myotubes in vitro and in muscle fibers in vivo induced both the specific repression of synaptic genes transcription and morphological perturbations of the NMJ. The present data thus extend the notion that the MuSK complex is involved in the regulation of synaptic gene expression at the NMJ.
Materials and Methods
Antibodies. Anti-MuSK antibody 2847 (17) was a gift from S. Burden (Skirball Institute, New York University Medical School, New York). Polyclonal antibodies cyt-MuSK have been previously characterized (18). Anti-HA and anti-14-3-3 γ were purchased from Santa Cruz Biotechnology. Anti-myc and anti-GFP antibodies were from Invitrogen and Roche Molecular Biochemicals, respectively. Goat antibody directed against the extracellular domain of MuSK (EC-MuSK) was purchased from R & D Systems.
Cross-Linking Experiments and MALDI-TOF MS. Purification of AChR-rich membranes from Torpedo electric tissue, crosslinking, and MALDI-TOF MS analysis of MuSK cross-linked products were carried out as described by Strochlic et al. (10) For more information, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Vectors and Transfection Assays in COS-7 Cells. The construct 14-3-3 γ-GFP contains the sequence from the rat 14-3-3 γ (19) fused to the C-terminal domain of GFP in pEGFP-C1 (Clontech) to preserve the 14-3-3 γ C-terminal fragment important for ligand binding (20). MuSK, MuSK-HA, and MuSK-myc constructs were gifts from W. Hoch (University of Houston, Houston). COS-7 cells (African green monkey kidney) cultures and transfection experiments were carried out as described by using FuGENE 6 (21). The cDNA plasmid concentrations were 1 μg/ml for all MuSK constructs and 0.5 μg/ml for 14-3-3 γ. Immunoprecipitation and immunofluorescence analysis were carried out 48 h later.
Immunoprecipitation of the MuSK/14-3-3 γ Complex. C2C12 mouse muscle cells [American Type Culture Collection (ATCC) Catalog No. CRL-1772] or COS-7 cells transfected with 14-3-3 γ-GFP and MuSK-HA were harvested in lysis buffer (50 mM Tris/150 mM NaCl/2 mM EDTA/2 mM orthovanadate/1% Triton/0.3 mM PMSF, pH 7.5) for 30 min at 4°C. Cell lysates (500 μl) were incubated with Cyt-MuSK or the extracellular domain of MuSK. Proteins A or G (Amersham Pharmacia Biotech) were added to the lysate and incubated for 3 h at 4°C. Immunoprecipitates were taken up in SDS sample buffer, resolved by SDS/PAGE, and analyzed by Western blotting. In some experiments, Torpedo agrin (10 ng/ml) was added to C2C12 cultures for 30 min before cell lysis.
Vectors and Transfection Assays in C2C12 Cells. The cytomegalovirus (CMV)-14-3-3 γ construct contains the sequence from the rat 14-3-3 γ cloned into the pCMV plasmid (Clontech). Constructs ε-AChR or δ-AChR and muscle creatine kinase (MCK) contain the promoter sequences from the rat ε- and δ-subunits of AChR and the MCK genes fused to the luciferase (22). The utrophin construct containing the promoter A from the rat utrophin gene fused to β-gal was a gift from B. J. Jasmin (University of Ottawa, Ottawa). C2C12 cells (80% confluence) were transiently transfected with Lipofectamine (GIBCO/BRL). cDNA plasmid concentrations were 1 μg/ml for all constructs except for CMV-14-3-3 γ (typically 0.5 μg/ml) and for MuSK (1.5 μg/ml). A pSV-β-gal control vector (Promega) was used to normalize for transfection efficiency. After transfection, myoblasts were switched to differentiation media (5% horse serum, l-glutamine) on day 1 and stimulated with or without neuregulin-1 (3 nM; Upstate Biotechnology, Lake Placid, NY) on day 2. Myotubes were harvested 3 days later and assayed for luciferase and β-gal activities according to the manufacturer's instructions (Promega).
Immunofluorescence Analyses. Indirect immunoflourescence experiments were carried out as described in Strochlic et al. (10) and Cartaud et al. (21). See Supporting Materials and Methods for details.
Statistical Analyses. Reported means were compared by analysis of variance (see Figs. 4 and 7). The equal variance test is significant with P < 0.05. Pairwise multiple comparisons were made with the Student–Newman–Keuls method (t test).
Fig. 4.

Repression of synaptic genes expression by 14-3-3 γ in C2C12 myotubes. Luciferase for AChR subunits (Upper Left and Upper Right) and muscle creatine kinase (MCK) (Lower Right) or β-gal (for utrophin) (Lower Left) genes reporter constructs placed under the control of various promoters (see Materials and Methods) were transiently transfected into C2C12 cells. All genes tested but MCK showed a transcriptional activation by NRG. The expression of the 14-3-3 γ protein (0.5 μg/ml plasmid) induced a strong inhibition of the transcription of all tested such synaptic genes as ε-AChR and δ-AChR subunits and utrophin in the presence or absence of NRG. MCK reporter gene showed no sensitivity to 14-3-3 γ. The relative luciferase or β-gal activities after normalization for transfection efficiency were reported as arbitrary units. All data were expressed as the mean ± SEM of four to six independent experiments.
In Vivo Electroporation of Muscles. Five micrograms of total plasmid DNA (pcDNA3 or pCMV/14-3-3 γ) and 2 μg of pECFPNuc (Clontech) in 30 μl of 0.9% NaCl were injected in the tibialis anterior (TA) muscles of anesthetized 6-week-old male OF1 mice. Plaque electrodes (1 cm2) were then placed on each sides of the leg, and eight 200-V/cm pulses of 20 ms were applied to the injected muscle at 2 Hz. Ten mice were injected with the pCMV/14-3-3 γ construct in the left TA muscle and with the control plasmid pcDNA3 in the right TA muscle.
RNA Preparation and Real-Time Quantitative RT-PCR. Seven days after the injection, the muscles were microdissected under a fluorescent binocular to isolate cyan fluorescent protein-positive fibers. Total muscle RNA was extracted by using the RNeasy Mini RNA extraction kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. RT-PCR was performed by using QuantiTect SYBR Green RT-PCR Kit (Qiagen) in the presence of 1 μM of the respective forward and reverse primers. The sequences of the primers used for real time quantitative RT-PCR are provided in Supporting Materials and Methods. All mRNAs measurements of synapse-specific genes were normalized to the presynaptic ErbB3 mRNA, except for the fiber-wide expressed muscle creatine kinase, which was normalized to the GAPDH mRNA.
Analysis of NMJ Morphology. Fifteen days after the injection of the 14-3-3 γ plasmid, the injected muscles were fixed in 3.7% formaldehyde (Sigma) in PBS for 30 min. Cyan fluorescent protein-positive fibers were microdissected, permeabilized in PBS plus 0.5% Triton for 30 min, and then incubated with the α-bungarotoxin (α-BTX)–rhodamine isothiocyanate conjugate (Molecular Probes) to localize the AChR. At least 20 junctions were analyzed for each condition under a confocal microscope (LSM 510, Zeiss).
Results
The 14-3-3 γ Protein Is Part of the MuSK Complex in Torpedo Electrocyte Postsynaptic Membrane. To identify new partners of MuSK, we performed cross-linking experiments with the noncleaveable, heterobifunctional reagent succinimidyl 4-(p-maleimidophenyl)butyrate in purified AChR-rich membranes from Torpedo electric tissue according to Burden et al. (23). Fig. 1a shows that in addition to MuSK (97 kDa), two major cross-linked products of 140 and 125 kDa were identified by Western blot with the anti-MuSK antibody 2847 (10). The polypeptides associated with MuSK in the 140-kDa crosslink products were previously identified by MALDI-TOF MS as the C-terminal domain of MAGI-1c (10) and ColQ (21). The protein associated with MuSK in the 125-kDa cross-linked product was analyzed by MALDI-TOF MS after immunopurification of the MuSK complex and SDS/PAGE separation of the crosslink products. The comparison of experimental m/z of tryptic peptides with masses of theoretical tryptic peptides of proteins, listed in SwissProt database by using profound software, are conducted to identify the 14-3-3 γ protein with a coverage map of 18% (Fig. 1b) and rat MuSK (coverage of 6%). The identified peptides are listed in Supporting Materials and Methods. The higher coverage obtained for 14-3-3 compared with MuSK reflects the remarkable degree of sequence conservation reported within the 14-3-3 family of proteins (reviewed in ref. 14). Interestingly, the γ isoform identified here is the only 14-3-3 isoform significantly expressed in rat muscle (19).
Fig. 1.

MALDI-TOF MS analysis of the MuSK cross-link products. (a) Western blots of cross-linked proteins from Torpedo AChR-rich membranes hybridized with anti-MuSK antibodies revealed two 125-kDa and 140-kDa cross-linked products in addition to MuSK (97 kDa). (b) Coverage maps of experimental tryptic peptides from the 125-kDa cross-linked product compared with theoretical tryptic peptides from databases obtained with profound software disclosed coverages of 6% with rat MuSK (Upper) and 18% with the rat 14-3-3 γ protein (Lower).
Identification of the 14-3-3 γ/MuSK Complex at the Membrane. To evaluate the functional relevance of these in vitro data, immunofluorescence experiments were performed on adult rat sternomastoid muscle by using anti-14-3-3 γ antibodies. The 14-3-3 γ protein was concentrated at the NMJ, where it codistributed with AChRs identified by means of α-BTX. A faint staining was also detected at the sarcolemma and in blood vessels (Fig. 2). To study the interaction between MuSK and the 14-3-3 γ protein in vitro, we performed immunofluorescence and immunoprecipitation experiments in COS-7 cells cotransfected with cDNAs encoding the tagged proteins MuSK-myc or MuSK-HA and 14-3-3 γ-GFP. In cotransfected cells, 14-3-3 γ-GFP colocalized with MuSK-myc at discrete sites at the plasma membrane (Fig. 3 a and b). When expressed alone, 14-3-3 γ-GFP disclosed a diffuse cytoplasmic distribution (Fig. 3c). In COS-7 cells cotransfected with 14-3-3 γ-GFP and MuSK-HA, both fusion proteins coimmunoprecipitated (Fig. 3d). As we used a fusion protein (14-3-3 γ-GFP), we showed as a control that transfected GFP did not coprecipitate with MuSK-HA (Fig. 3d). Immunoprecipitation experiments in C2C12 myotubes also indicate that endogeneous MuSK and 14-3-3 γ coimmunoprecipitated (Fig. 3e). This interaction occurred in heterologous COS-7 cells, and treatment of myotubes with agrin before cell lysis did not modify the immunoprecipitation profile (data not shown). It thus seems to be independent of agrin.
Fig. 2.

The 14-3-3 γ protein localizes at the neuromuscular junction. Doublefluorescence experiment shows the accumulation of 14-3-3 γ protein at postsynaptic sites of NMJs evidenced by α-BTX staining of AChRs in cryostat sections of rat sternomastoid muscle fibers. A faint 14-3-3 γ immunolabeling was also detected extrasynaptically. (Scale bar, 20 μm.)
Fig. 3.

Identification of the 14-3-3 γ/MuSK complex. (a and b) In COS-7 cells cotransfected with 14-3-3 γ-GFP and MuSK-myc constructs, the two tagged proteins are colocalized at the plasma membrane (arrows in a; detail in b). (c) Conversely, the 14-3-3 γ protein transfected alone disclosed a cytoplasmic distribution. (d) Immunoprecipitation (IP) experiments of 14-3-3 γ-GFP/MuSK-HA complex with anti-MuSK antibodies after cotransfection in COS-7 cells. Western blotting probed with anti-HA and anti-GFP showed that the 14-3-3 γ-GFP and MuSK-HA coimmunoprecipitated. Control experiments showed that GFP alone did not interact with MuSK-HA. (e) In C2C12 cells, the endogeneous 14-3-3 γ protein coimmunoprecipitated with MuSK. Western blots on lysates indicate the amount of protein of interest in transfected COS cells and in C2C12 myotubes. Control IPs lacking MuSK antibodies were negative. (Scale bar, 10 μm.)
The 14-3-3 γ Represses the Transcription of Synaptic Genes in C2C12 Myotubes. Because a major function of MuSK is to regulate AChR clustering, we used in vitro assays to gain insight into the function of 14-3-3 γ in agrin-elicited AChR aggregation. In a first set of experiments, we looked for the presence of 14-3-3 γ in AChR clusters in C2C12 myotubes by using indirect immunofluorescence microscopy. As shown in Fig. 7a, which is published as supporting information on the PNAS web site, 14-3-3 γ was not detected within AChR clusters. To further explore an eventual role of 14-3-3 γ in AChR clustering, C2C12 myoblasts were transfected with the 14-3-3 γ. After agrin treatment, no significant difference in the number of AChR clusters was observed compared with untransfected myotubes (Fig. 7b). These data suggest that 14-3-3 γ protein is not a major player in the AChR clustering machinery.
Because the MuSK complex has been shown to be involved in synaptic gene transcription, we next asked whether 14-3-3 γ plays a role in this mechanism. At the NMJ, NRG, a ligand for ErbB receptor tyrosine kinases, is likely to be one nerve-supplied signal that induces expression of AChR genes (reviewed in ref. 24). Activation of synaptic genes by NRG involves the mitogen-activated protein kinases (MAPK), c-Jun N-terminal kinase and the phosphatidylinositol 3-kinase (PI3K) signal transduction pathways (reviewed in ref. 2). Because 14-3-3 proteins are regulators of both MAPK and PI3K pathways, we analyzed the effect of the 14-3-3 γ on ε-AChR and δ-AChR, utrophin (promoter A) and nonsynaptic MCK transcription. Using a reporter gene assay in C2C12 myotubes, we first verified that the synaptic reporter gene constructs were specifically activated by NRG (ref. 25 and Fig. 4). After transiently expression of 14-3-3 γ either in the presence or absence of NRG, a repression of the transcription of each of the synaptic reporter genes was observed. In the presence of NRG, inhibition of transcription was 95%, 75%, and 75% for the ε-AChR, δ-AChR, and utrophin genes, respectively (Fig. 4). We also observed a dose-dependent transcriptional inhibition by the 14-3-3 γ plasmid (from 0.15 to 0.5 μg/ml, Fig. 5). Neither activation by NRG nor repression by 14-3-3 γ was observed with the MCK gene (Fig. 4).
Fig. 5.

MuSK potentiates the inhibitory effect of 14-3-3 γ on ε-AChR reporter gene transcription in vitro. C2C12 myotubes transiently expressing MuSK in the presence of NRG showed no significant variation in ε-AChR reporter gene levels. After transfection with limiting amounts of 14-3-3 γ construct (0.15 μg/ml or 0.3 μg/ml per well), partial inhibitions (≈30% and ≈60%, respectively) of ε-AChR reporter gene transcription were observed. In these conditions, the inhibition of ε-AChR reporter gene transcription was further amplified by cotransfection with MuSK. All data were expressed as the mean ± SEM of three independent experiments. *, Significant statistical difference with P < 0.05 in the t test (see Materials and Methods).
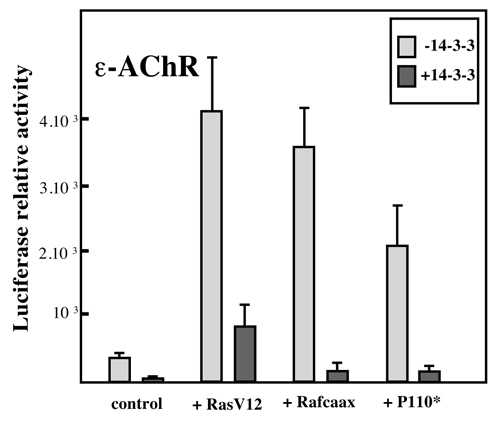
To determine whether the inhibitory effect of 14-3-3 γ on synaptic gene transcription depended on its interaction with MuSK or on the activation of MuSK by agrin, we monitored the effect of the expression of MuSK on the ε-AChR reporter gene transcription in various experimental conditions. As expected from previous studies (26), MuSK expression in C2C12 myotubes did not affect the reporter ε-AChR gene transcription (Fig. 5). However, in the presence of low amounts of 14-3-3 γ (0.15 μg/ml or 0.3 μg/ml cDNA per well that resulted in ≈30% and ≈60% inhibition, respectively), the coexpression of MuSK significantly potentiated (by 400%) the 14-3-3 γ inhibitory effect on the ε-AChR reporter gene. Addition of agrin before monitoring the ε-AChR reporter gene in the presence of NRG did not affect the effect of 14-3-3 γ (data not shown). Finally we observed that the 14-3-3 γ protein strongly inhibits the transcription of the ε-AChR subunit activated by constitutively active Ras, Raf-1, or PI3K (Fig. 8, which is published as supporting information on the PNAS web site). Because 14-3-3 proteins are key regulators of Raf-1 (27, 28) and PI3K (29), respectively, this observation supports the notion that 14-3-3 γ represses synaptic gene transcription by interfering with the MAPK/PI3K signaling pathways activated through NRG/ErbB (refs. 25 and 30 and reviewed in ref. 2).
The 14-3-3 γ Represses the Transcription of Synaptic Genes in Muscle Fibers and Alters NMJ Morphology in Vivo. We also evaluated the effect of the transient expression of 14-3-3 γ in vivo in the TA muscle by using in vivo DNA electroporation. In agreement with the in vitro studies, the expression of the 14-3-3 γ induced a strong inhibition of the transcription of several endogenous synaptic genes, including the ε-AChR and α-AChR, utrophin, rapsyn, and musk genes (Fig. 6a). The nonsynaptic MCK gene did not respond to 14-3-3 γ protein, demonstrating the specificity of its effect on synaptic genes. In addition, as shown in Fig. 6b, electroporated muscle fibers expressing the 14-3-3 γ monitored by coexpression of cyan fluoresccent protein displayed severe morphological perturbations of the NMJs compared with the sham transfected and electroporated contralateral control muscles. In cyan fluorescent protein-positive fibers, most junctions (86% vs. 15% in control fibers) disclosed interruptions of the α-BTX-labeled primary gutters (7 ± 2 interruptions per junction) as compared with the control (1.4 ± 0.7). In these fibers, subsynaptic nuclei also appeared more dispersed.
Fig. 6.

The 14-3-3 γ represses the transcription of synaptic genes in muscle fibers and induced a dislocation of the NMJ in vivo. (a) The transcription of the endogenous synaptic genes ε-AChR and α-AChR, utrophin, rapsyn, and musk measured by quantitative RT-PCR was inhibited by expression of 14-3-3 γ. The MCK gene demonstrates the synaptic specificity of the 14-3-3 γ inhibition. (b) In TA muscles injected with the 14-3-3 γ, the AChR-rich postsynaptic specializations at the NMJ visualized by α-BTX disclosed interruptions of the primary gutters as compared with the more continuous “bretzel-like” structure of a control NMJ in sham-transfected fibers. Subneural nuclei stained with 4′,6-diamidino-2-phenylindole also appeared dispersed as compared with control. (Scale bar, 10 μm.)
Discussion
In this work, we identified the adaptor protein 14-3-3 γ as a partner of MuSK in the postsynaptic membrane of Torpedo electrocytes. We show that in C2C12 myotubes as well as in muscle fibers in vivo, expression of 14-3-3 γ repressed specifically synaptic gene transcription, likely by inhibition of the MAPK and PI3K signaling pathways. Moreover, the inhibition by 14-3-3 γ is potentiated by the coexpression with MuSK in vitro. In addition, morphological perturbations of the NMJ were observed in muscle fibers expressing the 14-3-3 γ protein. Our results, together with others (see ref. 13 and references therein), provide evidence that synapse transcription is regulated both in negative and positive ways, even at the level of MuSK. Agrin/MuSK signaling enhances transcription, whereas 14-3-3 γ decreases it. These opposite effects, which might be of interest for synapse differentiation and/or plasticity, likely result from the modulation of signaling pathways downstream of MuSK.
The 14-3-3 proteins participate in regulatory cellular processes by interactions with many specific, although heterogeneous, signal transduction proteins. In fact, 14-3-3 ligands share common binding determinants that mediate the interaction. Several observations pointed out the importance of the phosphorylation of a serine in consensus motives in the regulation of 14-3-3 binding to target proteins. Two consensus 14-3-3 recognition motives: RSxSxP and Rx1–2Sx2–3S, in which at least one of the serine must be phosphorylated were identified in many 14-3-3 ligands (28, 31). Interestingly, these two recognition motives (RSMSP, residues 667–671; and RPSFCS, residues 845–850; in the rat sequence) are present in the C-terminal domain of MuSK. Yet, additional experiments are required to test the hypothesis that these motives are involved in the regulation of 14-3-3 γ/MuSK interaction.
To gain insight into the function of the 14-3-3 γ protein in synapse formation, we have in a first series of experiments sought a possible implication in AChR clustering in vitro. Our data suggest that 14-3-3 is not involved in this mechanism. This result is in line with the observation that 14-3-3 γ localized tardily to the NMJ, localized after birth (data not shown), and was identified in adult Torpedo tissue. Collectively, our data point to a role of 14-3-3 γ in synapse maintenance or plasticity rather than in development.
In innervated myofibers, two distinct mechanisms contribute to the regulation of synaptic genes transcription: (i) nerveevoked electrical activity represses synaptic genes transcription in extrajunctional areas, and (ii) neurotrophic factors selectively enhances transcription of synaptic genes in the subsynaptic nuclei by the activation of ErbB receptors (reviewed in refs. 2 and 32). Stimulation of ErbB receptors by NRG leads to the activation of MAPK, Erk, and c-Jun N-terminal kinase, which suggested that substrates of MAPK play a pivotal role in regulating synaptic gene expression. These pathways ultimately lead to the phopshorylation of the transcription factor GA binding protein α/β that directs synaptic gene expression (22). More recently, the direct involvement of agrin/MuSK in synaptic gene transcription has been established. Agrin/MuSK seems to activate two converging pathways: the NRG/ErbB pathway that predominates in cultured myotubes, and another one, NRG/ErbB-independent, through MuSK/Rac, which functions in muscle fibers in vivo (13). The 14-3-3 proteins are known to interact with Raf-1 (28). This finding suggests that in myotubes 14-3-3 γ acts downstream of NRG/ErbB pathway, possibly by blocking Raf-1 in an inactive conformation. Taken together, these data provide molecular evidence for a dual control of synaptic gene transcription by MuSK, positively by agrin or negatively by 14-3-3 γ.
We observed that the inhibition of the ε-AChR subunit expression is more effective when 14-3-3 γ and MuSK were coexpressed than when the 14-3-3 γ was expressed alone. This observation suggests that the effect of the 14-3-3 γ protein on ε-AChR gene transcription is related to MuSK. Addition of agrin in the reporter gene assay did not affect the inhibition of the transcription of ε-AChR, indicating that activation of MuSK by agrin is not necessary for 14-3-3 γ effect. Collectively, our data suggest a model of transcriptional regulation of synaptic genes in which MuSK would recruit a pool of cytoplasmic 14-3-3 γ protein, resulting in its local accumulation at postsynaptic sites. At this location, the 14-3-3 γ protein would be positioned close to ErbB receptors and their downstream effectors to regulate their activity. This hypothesis is supported by our observations showing that in muscle fibers, the 14-3-3 γ protein is mostly synaptic and that coexpression of MuSK in COS-7 cells targets the 14-3-3 γ to the plasma membrane. A critical aspect of this model is the possible regulation of the 14-3-3 γ/MuSK interaction by serine phosphorylation. This regulation would implicate the serine phosphorylation of the 14-3-3 recognition motives in the cytoplasmic moiety of MuSK by still unknown serine/threonine kinase(s).
Two negative effectors of ErbB signaling in the postsynaptic membrane of the NMJ have already been identified: the protein phosphatase SHP2, which interacted with ErbB2 after NRG stimulation (33), and Erbin, a partner of ErbB2, which decreased the NRG-activated ε-AChR gene transcription through Ras inactivation (34). Here, we suggest that 14-3-3 γ is another factor regulating the signaling pathways downstream of NRG/ErbB, thereby controlling synaptic gene expression at the NMJ. Remarkably, the expression of the 14-3-3 γ in vivo caused perturbations of the organization of the postsynaptic apparatus. This remodeling might be the consequence of the down-regulation of several key genes, such as musk itself, rapsyn, and utrophin, being critically involved in the organization of the NMJ. This latter observation is in agreement with a recent study showing that in transgenic mice expressing a dominant negative GA binding protein that blocks synaptic gene expression, the morphology of the NMJ is similarly altered (35). In both cases (overexpression of 14-3-3 γ and inhibition of GA binding protein binding), a significant repression of synaptic gene transcription was reported, ultimately leading to perturbations of the postsynaptic apparatus.
Supplementary Material
Acknowledgments
We thank Drs. J. Rossier and V. Labas for expertise in MS; Dr. M. Recouvreur for technical assistance; Drs. S. Burden, W. Hoch, B. J. Jasmin, A. Klippel (Atugen, Berlin), and M. Watanabe (Tokyo Institute of Technology, Yokohama, Japan) for generous gift of constructs and antibodies. This work was supported by the Centre National de la Recherche Scientifique, the Universities Paris 6 and 7, the Association Française contre les Myopathies, the Association pour la Recherche contre le Cancer, the College de France, and the European Communities.
Abbreviations: AChR, acetylcholine receptor; α-BTX, α-bungarotoxin; CMV, cytomegalovirus; MAPK, mitogen-activated protein kinase; MCK, muscle creatine kinase; MuSK, muscle-specific kinase; NMJ, neuromuscular junction; NRG, neuregulin-1; PI3K, phosphatidylinositol 3-kinase; TA, tibialis anterior.
References
- 1.Sanes, J. R. & Licthman, J. W. (2001) Nat. Rev. Neurosci. 2, 791–805. [DOI] [PubMed] [Google Scholar]
- 2.Schaeffer, L., de Kerchove d'Exaerde, A. & Changeux, J.-P. (2001) Neuron 31, 15–22. [DOI] [PubMed] [Google Scholar]
- 3.Burden, S. J. (2002) J. Neurobiol. 53, 501–511. [DOI] [PubMed] [Google Scholar]
- 4.Jennings, C. G. B., Dyer, S. M. & Burden, S. J. (1993) Proc. Natl. Acad. Sci. USA 90, 2895–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valenzuela, D. M., Stitt, T. N., DiStefano, P. S., Rojas, E., Mattsson, K., Compton, D., Nunez, L., Park, J. S., Stark, J. L., Gies, D. R., et al. (1995) Neuron 15, 573–584. [DOI] [PubMed] [Google Scholar]
- 6.Luo, Z. G., Wang, Q., Zhou, J. Z., Wang, J., Luo, Z., Liu, M., He, X., Wynshaw-Boris, A., Xiong, W. C., Lu, B. & Mei, L. (2002) Neuron 35, 489–505. [DOI] [PubMed] [Google Scholar]
- 7.Finn, A. J., Feng, G. & Pendergast, A. M. (2003) Nat. Neurosci. 7, 717–723. [DOI] [PubMed] [Google Scholar]
- 8.Luo, Z. G., Je, H. S., Wang, Q., Yang, F., Dobbins, G. C., Yang, Z. H., Xiong, W. C., Lu, B. & Mei, L. (2003) Neuron 40, 703–717. [DOI] [PubMed] [Google Scholar]
- 9.Mohamed, A. S., Rivas-Plata, K. A., Kraas, J. R., Saleh, S. M. & Swope, S. L. (2001) J. Neurosci. 21, 3806–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strochlic, L., Cartaud, A., Labas, V., Hoch, W., Rossier, J. & Cartaud, J. (2001) J. Cell Biol. 153, 1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuhrer, C., Sugiyama, J. E., Taylor, R. G. & Hall, Z. W. (1997) EMBO J. 16, 4951–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meier, T., Hauser, D. M., Chiquet, M., Laudmann, L., Ruegg, M. A. & Brenner, H. R. (1997) J. Neurosci. 17, 6534–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lacazette, E., Le Calvez, S., Gajendran, N. & Brenner, H. R. (2003) J. Cell Biol. 161, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu, H., Subramanian, R. R. & Masters, S. C. (2000) Annu. Rev. Pharmacol. Toxicol. 40, 617–647. [DOI] [PubMed] [Google Scholar]
- 15.Tzivion, G., Shen, Y. H. & Zhu, J. (2001) Oncogene 20, 6331–6338. [DOI] [PubMed] [Google Scholar]
- 16.Van Hemert, M. J., de Steensma, H. Y. & van Heusden, G. P. H. (2001) BioEssays 23, 936–946. [DOI] [PubMed] [Google Scholar]
- 17.Watty, A., Neubauer, G., Dreger, M., Zimmer, M., Wilm, M. & Burden, S. J. (2000) Proc. Natl. Acad. Sci. USA 97, 4585–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hopf, C. & Hoch, W. (1998) J. Biol. Chem. 273, 6467–6473. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe, M., Isobe, T., Ichimura, T., Kuwano, R., Takahashi, Y. & Kondo, H. (1993) Mol. Brain Res. 17, 135–146. [DOI] [PubMed] [Google Scholar]
- 20.Luo, Z. J., Zhang, X. F., Rapp, U. & Avruch, J. (1995) J. Biol. Chem. 270, 23681–23687. [DOI] [PubMed] [Google Scholar]
- 21.Cartaud, A., Strochlic, L., Guerra, M., Blanchard, B., Lambergeon, M., Krejci, E., Cartaud, J. & Legay, C. (2004) J. Cell Biol. 165, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaeffer, L., Duclert, N., Huchet-Dynamus, M. & Changeux, J.-P. (1998) EMBO J. 17, 3078–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burden, S. J., DePalma, R. L. & Gottesman, G. S. (1983) Cell 35, 687–692. [DOI] [PubMed] [Google Scholar]
- 24.Fischbach, G. D. & Rosen, K. M. (1997) Annu. Rev. Neurosci. 20, 429–458. [DOI] [PubMed] [Google Scholar]
- 25.Tansey, M. G., Chu, G. C. & Merlie, J. P. (1996) J. Cell Biol. 134, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones, G., Moore, C., Hashemolhosseini, S. & Brenner, H. R. (1999) J. Neurosci. 19, 3376–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark, G. J., Drugan, J. K., Rossman, K. L., Carpenter, J. W., Rogers-Graham, K., Fu, H., Der, C. J. & Campbell, S. L. (1997) J. Biol. Chem. 272, 20990–20993. [DOI] [PubMed] [Google Scholar]
- 28.Fu, H., Xia, K., Pallas, D. C., Cui, C., Conroy, K., Narsimhan, R. P., Mamon, H., Collier, R. J. & Roberts, T. M. (1994) Science 266, 126–129. [DOI] [PubMed] [Google Scholar]
- 29.Munday, A. D., Berndt, M. C. & Mitchell, C. A. (2000) Blood 96, 577–584. [PubMed] [Google Scholar]
- 30.Altiok, N., Altiok, S. & Changeux, J.-P. (1997) EMBO J. 16, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muslin, A. J., Tanner, J. W., Allen, P. M. & Shaw, A. S. (1996) Cell 84, 889–897. [DOI] [PubMed] [Google Scholar]
- 32.Arber, S., Burden, S. J. & Harris, A. J. (2002) Curr. Opin. Neurobiol. 12, 100–103. [DOI] [PubMed] [Google Scholar]
- 33.Tanowitz, M., Si, J., Yu, D. H., Feng, G. S. & Mei, L. (1999) J. Neurosci. 19, 9426–9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang, Y. Z., Zang, M., Xiong, W. C., Luo, Z. & Mei, L. (2003) J. Biol. Chem. 278, 1108–1114. [DOI] [PubMed] [Google Scholar]
- 35.de Kerchove D'Exaerde, A., Cartaud, J., Ravel-Chapuis, A., Seroz, T., Pasteau, F., Angus, L. M., Jasmin, B. J., Changeux, J.-P. & Schaeffer, L. (2002) EMBO Rep. 3, 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}