

Abstract
High-performance liquid chromatography was used in combination with infrared ion spectroscopy for the identification of positional isomers of hydroxy-atorvastatins, the primary metabolites of the drug atorvastatin. The results demonstrate the direct applicability of infrared ion spectroscopy in the field of drug metabolism and, more generally, its promising role in state-of-the-art analytical laboratories for the identification of small molecules buried in complex mixtures. In combination with chromatographic separation, infrared spectroscopy of mass-selected ions provides a promising new route for the identification of the molecular structures of unknown m/z peaks in a mass spectrum. We demonstrate that currently existing experimental protocols allow the measurement of an IR spectrum from less than 10 ng of sample obtained in a collected HPLC fraction.
The study of drug metabolism, i.e., the degradation of a compound in the body, is an important component of the drug development process. Predominantly, different mass spectrometric (MS) techniques are used in combination with liquid chromatography (LC) for the separation and identification of drug metabolites. However, the localization of the exact site of a metabolic biotransformation by mass spectrometric analysis alone is often challenging or impossible since regioisomeric metabolites are frequently indistinguishable on the basis of their MS/MS product ion data, e.g., positional isomers with a site of metabolic modification on an aromatic ring.1 Therefore, nuclear magnetic resonance (NMR) spectroscopy is often needed for the complete identification of a metabolite structure. However, this technique requires large amounts (typically >5–100 μg) of sample in high purity.2 Since the metabolite can often only be extracted from biological (in vitro or in vivo) matrixes, this involves a time-consuming and complex purification process. The latter is currently the main bottleneck for more regular use of NMR for the identification of drug metabolites.1−3
The combination of mass spectrometry and infrared spectroscopy, known as infrared ion spectroscopy (IR-IS), is well established as a powerful tool for the elucidation of molecular structures and conformations of gas phase ions.4−7 Using the combination of state-of-the-art commercial ion trap mass spectrometry platforms8−10 with intense and tunable infrared lasers, such as user facility-based free electron lasers (FELs)11,12 and table top optical parametric oscillators,13−15 an infrared spectrum can be generated for ions at any m/z peak in a mass spectrum.
IR-IS has the potential to become a very valuable alternative for structural elucidation by NMR in many cases.16 Since the technique is based on detection by mass spectrometry, with its well-known selectivity and sensitivity, it can provide full structure information from one simple LC/MS or MS analysis and overcomes the tedious metabolite purification needed for NMR. Since less material is needed for the analysis, also less in vitro and/or in vivo sample needs to be produced, resulting in the use of fewer animals. Also, unstable metabolites that are hard or impossible to extract and purify can be measured using IR ion spectroscopy.
Recent examples of modified commercial ion trap mass spectrometers used for IR-IS measurements enable sensitivity down to the low nanomolar regime and require ∼50 μL of sample solution per IR spectrum.8 This sensitivity thus enables the measurement of IR spectra of ions that are components of collected HPLC fractions by direct infusion electrospray ionization (ESI). Here, we demonstrate the combination of HPLC and IR-IS and apply it for the identification of molecular structures in the field of drug metabolism. Atorvastatin is an extensively studied HMG-CoA reductase inhibitor used as a lipid-lowering drug.17,18 Its primary metabolism pathway is through cytochrome P450 3A4 hydroxylation, giving active ortho- and para-hydroxylated metabolites that in fact account for a significant portion of the HMG-CoA reductase activity.19 Incubation of atorvastatin in human liver microsomes showed two major metabolites that were separated and collected by HPLC and identified as the ortho- and para-hydroxy metabolites on the basis of their IR spectra measured using IR-IS.
Methods
Chemicals and Materials
Atorvastatin was purchased from Sequoia Research Products (Pangbourne, United Kingdom). The metabolites, o- and p-hydroxylated atorvastatin, were obtained from TLC PharmaChem (Ontario, Canada). Methanol and acetonitrile were supplied by Merck (Darmstadt, Germany); formic acid (0.1% in water) was provided by Biosolve (Valkenswaard, The Netherlands), and the dimethyl sulfoxide (DMSO) used during sample preparation was from Merck (Darmstadt, Germany). All LC solvents were HPLC grade.
Sample Preparation
Atorvastatin was incubated at 10 μM for 120 min in human liver microsomes with a final protein concentration of 1 mg/mL. The cell incubations (500 μL) were extracted with DMSO (500 μL), vortexed, sonicated, and centrifuged for 10 min at 16 060g. A 100 μL aliquot of the resulting supernatant was injected for analysis.
LC Fractionation
An Acquity i-class UPLC system (Waters, Manchester, UK), equipped with an Acquity FTN sample manager and an Acquity PDA detector, was coupled to a Gilson PrepFC sample collector (Middleton, USA). The separation was performed on a 150 × 3 mm Uptisphere Strategy C18-2 column (Interchim, Montluçon, France) packed with 2.2 μm particles and kept at a temperature of 60 °C. Elution started with a linear gradient at a flow rate of 0.8 mL/min from 95% solvent A (0.1% formic acid in water) and 5% solvent B (acetonitrile/methanol 80/20; v/v%) to 30% solvent A and 70% solvent B over 10 min, increased further to 100% B in 0.1 min, and was kept at 100% B for 2 min. The column eluent first passed through the PDA detector and was subsequently fractionated every 6 s into a 96-well plate. Afterward, the fractions (80 μL) corresponding to the peaks of interest were pooled. The collections corresponded to 57, 106, and 120 ng of peaks A, B, and C, respectively (Figure 1).
Figure 1.
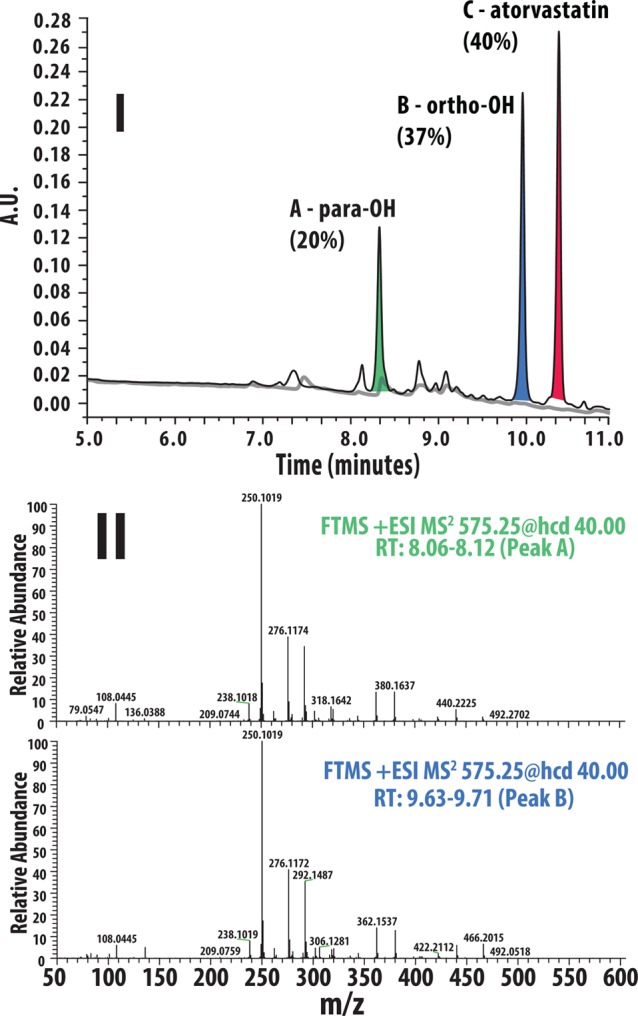
Hydroxy-atorvastatins are separable by HPLC (panel I) but indistinguishable on the basis of their MS/MS spectra (panel II).
Infrared Ion Spectroscopy
IR-IS measurements were performed in a modified quadrupole ion trap mass spectrometer (Bruker, AmaZon Speed ETD) coupled to the beamline of the FELIX IR-FEL. Details of the hardware modifications and of software operation have been previously reported.8,20 Collected HPLC fractions were directly electrosprayed (+ESI) to produce IR/mass spectra. All reference measurements were generated by +ESI from solutions of 10–7 M in 50:50 MeOH/H2O. The FELIX free electron laser generates infrared radiation as 5–10 μs macropulses at a 10 Hz repetition rate having ∼40 mJ pulse energy and a bandwidth of ∼0.4% of the center frequency.
In contrast to classical absorption IR spectroscopy, IR-IS is a form of action spectroscopy in which infrared absorption is monitored by observation of the photodissociation it induces. IR photons are absorbed when they are resonant with a vibration of the trapped ions, resulting in an increase in internal energy. Intramolecular vibrational redistribution of the absorbed energy causes ions to dissociate once they reach the limit of the dissociation channel having the lowest threshold. Typically, after a single IR macropulse, dissociation occurs. Relating the parent and fragment ion intensities (IR yield = ΣI(fragment ions)/ΣI(parent + fragment ions)) as the IR frequency is tuned generates an infrared vibrational spectrum. The yield at each IR point is typically obtained from four to eight averaged mass spectra, and the frequency is calibrated online throughout the measurements using a grating spectrometer.
Results and Discussion
As detailed above, atorvastatin (10 μM) was incubated in human liver microsomes for 120 min to generate the relevant metabolites which were separated by HPLC and the relevant fractions were collected. Demonstrated in Figure 1, the two isobaric metabolites, while separable by HPLC (panel I), are unidentifiable on the basis of their MS/MS product ion spectra showing identical product ions with identical relative abundances (panel II). The structures of atorvastatin and the two hydroxylated metabolites are illustrated in Figure 2. Using instead the HPLC/IR-IS approach, we are able to first separate the two hydroxylated metabolites using HPLC and then identify them on the basis of their readily distinguishable IR spectra. Using direct infusion +ESI, the ions were generated from the collected HPLC fractions. These results are illustrated in panels I–III of Figure 2. Note that these metabolites are distinguishable by their vibrational signatures in both the 1000–1900 cm–1 spectral region (measured using the FELIX IR FEL) and the 3200–3700 cm–1 spectral region (measured using a table top IR optical parametric oscillator laser source, LaserVision, USA). Using identical experimental (MS and IR laser) conditions, distinction can be made on both the basis of line positions (for example, peak splitting in the regions of ∼1250 and 3650 cm–1) and differences in IR fragment yields (intensities) since both properties of the IR spectra are highly reproducible.
Figure 2.

Panels I and II present IR spectra (colored traces) generated for ions at m/z 575 from LC fractions A and B and the identification of isobaric drug metabolites based on IR spectral match with reference compounds (black traces). Panel III highlights the difference in IR spectra of the two isomeric metabolites. Panel IV contains the IR spectrum measured for unmetabolized atorvastatin (peak C in LC chromatogram presented in Figure 1).
From the HPLC experiments, fractions were collected containing the relevant metabolites at ∼300 nM and IR measurements were obtained in duplicate in both spectral regions from a single collected fraction. Note that these amounts of sample are directly compatible with the standard operating protocols of the experiment8 since the measurement of one IR spectrum typically takes 10–20 min using ESI flow rates of ∼2 μL/min and concentrations ranging from nM to μM (in this case, corresponding to less than 10 ng of sample consumption per IR spectrum). Such experimental conditions compare very favorably against the alternative use of NMR spectroscopy for such an identification, maintaining the sensitivity and selectivity inherent to mass spectrometry while providing orthogonal identification on the basis of retention time, mass-to-charge, and IR spectral fingerprint.
Comparison of the IR-IS spectra from the HPLC fractions to IR-IS spectra of the pure reference compounds recorded under the same conditions (see black traces in panels I and II of Figure 2) shows that identical IR spectra are generated independently of the purity and concentration of the sample due to the selectivity of the MS. A particularly interesting perspective of the current approach is to rely on quantum–chemically calculated IR spectra in place of reference IR measurements, unlocking the potential of reference-free identification of unknowns in cases where model compounds are not commercially or synthetically available.21−23 This is hardly possible from MS/MS or LC data as CID fragmentation propensities and retention times are hard or even impossible to reliably compute. On the other hand, IR spectral predictions from quantum–chemical calculations are rapidly approaching experimental accuracy. Additionally, information on the functional groups contained in an unknown molecule is contained in its IR spectrum. For example, the IR features at approximately 3650 cm–1 in panels I and II of Figure 2 are a strong indication of OH functionality. Identification of specific functional groups in a molecule can be, for example, useful for narrowing down a list of isobaric compounds in a database search. In the case of coeluting isobaric compounds, a composite IR spectrum of all species is obtained. However, if specific spectral contributions from each compound are sufficiently distinct (in terms of frequency), deconvolution is often possible. Moreover, for compounds showing different fragmentation, unique IR spectra can be generated for each isobaric precursor by monitoring specific fragment ions
Currently, only a few laboratories worldwide possess the MS and IR laser infrastructure to conduct routine IR-IS experiments. This is mainly a result of the limited availability of FELs and the low output power and limited tuning ranges of OPOs. However, several FEL facilities (including FELIX) are open to external researchers. Moreover, with the ongoing introduction of new mid-IR laser sources (such as external cavity tunable quantum cascade lasers), implementation of affordable benchtop IR-IS techniques in state-of-the-art analytical laboratories for small molecule identification is quickly becoming a reality. This is especially exciting for online screening approaches in HPLC/MS analyses using a limited IR frequency range.
Acknowledgments
The authors gratefully acknowledge the excellent assistance from the FELIX operators and staff. Financial support for this project was provided by NWO Chemical Sciences under VICI project no. 724.011.002. This work is part of the research program of FOM, which is financially supported by NWO.
The authors declare no competing financial interest.
References
- Cuyckens F.; Wassvik C.; Mortishire-Smith R. J.; Tresadern G.; Campuzano I.; Claereboudt J. Rapid Commun. Mass Spectrom. 2011, 25, 3497–3503. 10.1002/rcm.5258. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Monshouwer M.; Fitch W. L. Pharm. Res. 2007, 24, 248–257. 10.1007/s11095-006-9162-7. [DOI] [PubMed] [Google Scholar]
- Lu W.; Xu Y.; Zhao Y.; Cen X. Curr. Drug Metab. 2014, 15, 865–874. 10.2174/1389200216666141230105649. [DOI] [PubMed] [Google Scholar]
- Polfer N. C.; Oomens J. Mass Spectrom. Rev. 2009, 28, 468–494. 10.1002/mas.20215. [DOI] [PubMed] [Google Scholar]
- MacAleese L.; Maitre P. Mass Spectrom. Rev. 2007, 26, 583–605. 10.1002/mas.20138. [DOI] [PubMed] [Google Scholar]
- Oomens J.; Sartakov B. G.; Meijer G.; von Helden G. Int. J. Mass Spectrom. 2006, 254, 1–19. 10.1016/j.ijms.2006.05.009. [DOI] [Google Scholar]
- Fridgen T. D. Mass Spectrom. Rev. 2009, 28, 586–607. 10.1002/mas.20224. [DOI] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Gebhardt C. R.; Oomens J. Rev. Sci. Instrum. 2016, 87, 103108. 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
- Bakker J. M.; Besson T.; Lemaire J.; Scuderi D.; Maître P. J. Phys. Chem. A 2007, 111, 13415–13424. 10.1021/jp074935e. [DOI] [PubMed] [Google Scholar]
- Nosenko Y.; Menges F.; Riehn C.; Niedner-Schatteburg G. Phys. Chem. Chem. Phys. 2013, 15, 8171–8178. 10.1039/c3cp44283g. [DOI] [PubMed] [Google Scholar]
- Lemaire J.; Boissel P.; Heninger M.; Mauclaire G.; Bellec G.; Mestdagh H.; Simon A.; Caer S. L.; Ortega J. M.; Glotin F.; Maitre P. Phys. Rev. Lett. 2002, 89, 273002. 10.1103/PhysRevLett.89.273002. [DOI] [PubMed] [Google Scholar]
- Oepts D.; van der Meer A. F. G.; van Amersfoort P. W. Infrared Phys. Technol. 1995, 36, 297–308. 10.1016/1350-4495(94)00074-U. [DOI] [Google Scholar]
- Martens J. K.; Grzetic J.; Berden G.; Oomens J. Int. J. Mass Spectrom. 2015, 377, 179–187. 10.1016/j.ijms.2014.07.027. [DOI] [Google Scholar]
- Sinha R. K.; Erlekam U.; Bythell B. J.; Paizs B.; Maître P. J. Am. Soc. Mass Spectrom. 2011, 22, 1645–1650. 10.1007/s13361-011-0173-1. [DOI] [PubMed] [Google Scholar]
- Schindler B.; Joshi J.; Allouche A.-R.; Simon D.; Chambert S.; Brites V.; Gaigeot M.-P.; Compagnon I. Phys. Chem. Chem. Phys. 2014, 16, 22131–22138. 10.1039/C4CP02898H. [DOI] [PubMed] [Google Scholar]
- Cismesia A. P.; Bailey L. S.; Bell M. R.; Tesler L. F.; Polfer N. C. J. Am. Soc. Mass Spectrom. 2016, 27, 757–766. 10.1007/s13361-016-1366-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever P. S.; Dahlöf B.; Poulter N. R.; Wedel H.; Beevers G.; Caulfield M.; Collins R.; Kjeldsen S. E.; Kristinsson A.; McInnes G. T.; Mehlsen J.; Nieminen M.; O’Brien E.; Östergren J. Lancet 2003, 361, 1149–1158. 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- Yacoub M.; awwad A. A.; Alawi M.; Arafat T. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2013, 917–918, 36–47. 10.1016/j.jchromb.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Park J. E.; Kim K. B.; Bae S. K.; Moon B. S.; Liu K. H.; Shin J. G. Xenobiotica 2008, 38, 1240–1251. 10.1080/00498250802334391. [DOI] [PubMed] [Google Scholar]
- Martens J.; Grzetic J.; Berden G.; Oomens J. Nat. Commun. 2016, 7, 11754. 10.1038/ncomms11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Oomens J. Anal. Chem. 2016, 88, 6126–6129. 10.1021/acs.analchem.6b01483. [DOI] [PubMed] [Google Scholar]
- Katari M.; Nicol E.; Steinmetz V.; van der Rest G.; Carmichael D.; Frison G. Chem. - Eur. J. 2017, 10.1002/chem.201700340. [DOI] [PubMed] [Google Scholar]
- Polfer N. C. Chem. Soc. Rev. 2011, 40, 2211–2221. 10.1039/c0cs00171f. [DOI] [PubMed] [Google Scholar]