

Abstract
Many clinical studies involving anti-tumor agents neglect to consider how these agents are metabolized within the host and whether the creation of specific metabolites alters drug therapeutic properties or toxic side effects. However, this is not the case for the anthracycline class of chemotherapy drugs. This review describes the various enzymes involved in the one electron (semi-quinone) or two electron (hydroxylation) reduction of anthracyclines, or in their reductive deglycosidation into deoxyaglycones. The effects of these reductions on drug anti-tumor efficacy and toxic side effects are also discussed. Current evidence suggests that the one electron reduction of anthracyclines augments both their tumor toxicity and their toxicity towards the host, in particular their cardiotoxicity. In contrast, the two electron reduction (hydroxylation) of anthracyclines strongly reduces their ability to kill tumor cells, while augmenting cardiotoxicity through their accumulation within cardiomyocytes and their direct effects on excitation/contraction coupling within the myocytes. The reductive deglycosidation of anthracyclines appears to inactivate the drug and only occurs under rare, anaerobic conditions. This knowledge has resulted in the identification of important new approaches to improve the therapeutic index of anthracyclines, in particular by inhibiting their cardiotoxocity. The true utility of these approaches in the management of cancer patients undergoing anthracycline-based chemotherapy remains unclear, although one such agent (the iron chelator dexrazoxane) has recently been approved for clinical use.
Keywords: Anthracyclines, anti-tumor effects, cardiotoxicity, deoxyaglycone, hydroxylation, metabolites, optimization, semi-quinone, therapeutic index
A). INTRODUCTION
Since their introduction in the 1960’s, anthracyclines have been used in the treatment of many neoplastic diseases, including both solid tumors and hematological cancers. They are planar molecules consisting of a rigid hydrophobic tetracycline ring, with a daunosamine sugar attached through a glycosidic bond [1]. The quinone and hydroquinone substitutions on two of four planar rings are important in their metabolism [1]. Used as a single agent or as part of a regimen, the anthracyclines are key components of neoadjuvant, adjuvant, curative, or palliative treatments for several types of malignancies. Derived from the pigment-producing Streptomyces peucetius bacterium, doxorubicin (DOX) and daunorubicin (DNR) are two naturally occurring anthracyclines [2]. Due to their success in treating cancers from various tissue types, a significant amount of effort has been placed into creating and characterizing novel anthracyclines. This has resulted in the development of approximately 2000 anthracycline analogs. A number of these analogs are now in widespread clinical use, including idarubicin, epirubicin, carminomycin, pirarubicin, aclarubicin, valrubicin and zorubicin [3, 4]. As with other chemotherapy agents, the clinical success of anthracyclines is compromised by innate or acquired resistance to these agents [5] and by their significant toxic side effects in cancer patients, in particular cardiotoxicity [6, 7]. Consequently, all anthracyclines have an associated maximum recommended cumulative dose in an effort to avoid congestive heart failure.
DNR and DOX, the first anthracycline antibiotics to be isolated over the past 50 years, are among the most effective antineoplastic agents currently used in the treatment of human cancers. DNR is used mainly to treat acute lymphoblastic or myeloblastic leukemias, while DOX has efficacy against both solid and non-solid tumors. The latter is widely used for the treatment of breast cancer, Wilms’ tumors, soft tissue sarcomas, leukemias, Hodgkin’s disease, non-Hodgkin’s lymphomas and several other cancers [8]. Although differing from DOX by a single hydroxyl group, this alteration in structure gives DNR distinct reaction kinetics [9]. Nevertheless, the use of both DOX and DNR is limited by their toxic side-effects within the host, including necrosis of tissue at the injection site, mucositis, alopecia, nausea, vomiting, stomatitis, and cumulative cardiotoxicity. Consequently, the maximum recommended cumulative doses for DNR and DOX are set at 550 mg/m2 and 450-550 mg/m2, respectively [10, 11].
Epirubicin (EPI) is obtained by an axial-to-equatorial epimerization of the 4’-hydroxyl group of DOX (Fig. 1). It is currently widely used to treat carcinomas of the breast, stomach, gut, endometrium, lung, ovary, esophagus, and prostate (as well as soft tissue sarcomas) [12]. While EPI has almost equivalent antitumor activity to that of DOX, it possesses different pharmacokinetic and metabolic characteristics. For example, EPI is more glucuronidated, which facilitates excretion in bile and urine. It therefore has a greater margin of safety and has almost double the recommended cumulative dosing of DOX (900-1000 mg/m2 for EPI) [13].
Fig. (1).
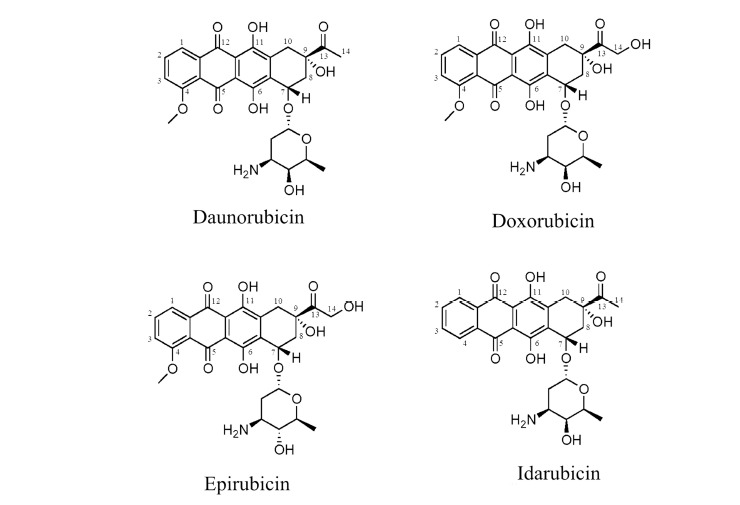
Chemical structures of Daunorubicin (DNR), Doxorubicin (DOX), Epirubicin (EPI) and Idarubicin (IDA).
Idarubicin (IDA), an analog of DNR, lacks the C-4 methoxy group and has been shown to have improved activity for the treatment of acute myelogenous leukaemia. It is also found to be active against multiple myeloma, non–Hodgkin's lymphoma, and breast cancer [12]. IDA is the only anthracycline that can be administered orally or through intravenous injection. The absence of a methoxy group in IDA’s structure (Fig. 1) results in a longer half-life than DNR and significantly enhances lipophilicity. This results in more rapid cellular uptake, superior DNA-binding capacity, and consequently greater cytotoxicity compared to DOX and DNR [14]. Comparative information regarding the half-life and toxicities of the above anthracyclines is presented in Table 1.
Table 1.
Comparison of the four major anthracyclines used in clinical oncology and their differences in clinical use, pharmacokinetics, and toxicities.
| Drugs | Maximum Recommended Dose | Half-Life (hr) | Side Effects |
|---|---|---|---|
| Daunorubicin | 550 mg/m2 | 14-20 | Cardiotoxocity, mucositis, myelosuppression, nausea, vomiting, alopecia |
| Doxorubicin | 450 - 600 mg/m2 | 1-3 | Cardiotoxicity, muscositis, nausea, vomiting |
| Epirubicin | 900-1000 mg/m2 | 33 | Myelosuppression, cardiotoxicity, nausea, vomiting, mucositis |
| Idarubicin | >160 mg/m2 | 12-27 | Cardiotoxicity, myelosuppression muscositis, nausea, vomiting, alopecia |
The mechanism by which anthracyclines enter cells is still not completely known, but is thought to involve their passive diffusion through the plasma membrane, followed by their selective transport into the nucleus by binding to proteasomes [15]. Once in the nucleus, the anthracyclines dissociate from proteasomes and bind to DNA due to their higher affinity for the latter macromolecule [16, 17]. Anthracyclines are thought to inhibit the proliferation of rapidly dividing cells through multiple mechanisms, including their ability to intercalate between and cross-link DNA strands, to alkylate DNA, and to inhibit topoisomerase II [18-20]. These actions are also highly effective in preventing DNA unwinding and strand separation, thereby blocking DNA replication and transcription [19]. In addition, the ability of anthracyclines to generate highly reactive free radicals can result in abundant damage to DNA and to the plasma membrane through lipid oxidation [21-24]. In addition to their cytostatic and cytotoxic effects against tumor cells, anthracyclines have been documented to accumulate in other tissues such as the liver, heart, white blood cells and bone marrow contributing to their systemic toxic side effects [25].
In humans, it is estimated that approximately 50% of DOX is eliminated from the body without any change in its structure, while the remainder of the drug is processed through three major metabolic pathways [25]. Metabolism of anthracyclines occurs through hydroxylation, semiquinone formation or deoxyaglycone formation, which can result in the formation of metabolites that either augment or suppress the anticancer properties of anthracyclines [26, 27].
Hydroxylation of anthracyclines at the C-13 carbonyl group, more commonly referred to as the two electron reduction, results in the formation of secondary alcohol metabolites that have been implicated in anthracycline-induced cardiotoxicity [14]. This major pathway of anthracycline biotransformation is mediated by a heterogenous family of cytosolic NADPH-dependent carbonyl (CBR) and aldo-keto (AKR) reductases (collectively referred to as carbonyl reducing enzymes) that catalyze the formation of daunorubicinol (DNROL), doxorubicinol (DOXOL), epirubicinol (EPIOL) and idarubicinol (IDAOL) from their parent drugs [28]. The AKRs are the primary enzymes involved in DOX hydroxylation in the human heart, whereas the CBRs play more of a role in DNR hydroxylation [29]. The hydroxylation reactions occur in all cell types as the enzymes involved are ubiquitous, and have also been studied extensively in red blood cells, liver and kidney [30].
The one electron reduction of anthracyclines is catalyzed by cytochrome P-450 reductase (CPR), NADH dehydrogenase (NDUFS), nitric oxide synthase (NOS), and xanthine oxidase, leading to the conversion of the quinone moiety of anthracycline drugs to a semiquinone radical [17]. Although this radical is stable under anoxic conditions, in the presence of oxygen, the semiquinone radical is readily re-oxidized to regenerate the parent quinone and results in the generation of a superoxide anion and hydrogen peroxide, thereby increasing the formation of reactive oxygen species (ROS). The resulting free radicals can cause peroxidation of lipids within cellular membranes, protein aggregation and cell death [31]. This redox cycling of DOX, DNR, and other anthracycline analogues has been observed in the cytoplasm, mitochondria and sarcoplasmic reticulum and has been implicated in the production of toxic aldehydes that are able to escape from the cell and contribute to anthracycline toxicity [8].
The final metabolic pathway, deglycosidation, accounts for approximately 1-2% of anthracycline metabolism [29]. The reductive cleavage of the glycosidic bond and the side chain carbonyl group results in the formation of 7-deoxyaglycones and hydroxyaglycones [32]. It has been reported that this reaction is catalyzed by poorly characterized NADPH-dependent hydrolase- and reductase-type glycosidases [NADPH quinone oxidoreductases (NQO1) and NADPH-cytochrome P450 reductase (CPR)] along with the involvement of xanthine dehydrogenase (XDH) [29]. The formation of hydroxyaglycones results from NADPH-dependent enzymes that are present in the cytosol, whereas the 7-deoxyaglycone formation may be initiated by microsomal or mitochondrial oxidoreductases [32]. Several studies report that the anthracycline aglycones may generate ROS, but the resulting 7-deoxyaglycone metabolites have been shown to possess no cytotoxic activity [33]. The aglycones produced have a higher lipophilicity than the parent anthracyclines, and are thought to intercalate into mitochondrial membranes [34]. Moreover, studies suggest that the anthracycline aglycones may cause myocardial damage due to their prominent oxidizing properties that divert more electrons to oxygen in the mitochondria [35]. While anthracycline metabolic pathways and metabolites vary from tissue to tissue and between in vitro and in vivo conditions, the two electron reduction (hydroxylated) product is generally the predominant metabolite for anthracyclines, with considerably lower percentages of the algycone, 7-deoxy aglycone and 7-deoxy hydroxylated aglycone products [33, 36].
Having summarized the various pathways by which anthracyclines are metabolized in humans, this review will describe how this metabolism can affect the above-described biochemical and biological properties of this important class of chemotherapy agents. We will focus particularly on how the metabolism of anthracyclines affects their ability to combat the growth of tumors and to produce toxic side effects in patients. In addition, we will explore how variations in anthracycline metabolism within cancer patients and their tumors can impact treatment efficacy and discuss recent strategies to improve the therapeutic index of anthracyclines. A schematic summary of the major points of this review is provided in Figure 2.
Fig. (2).
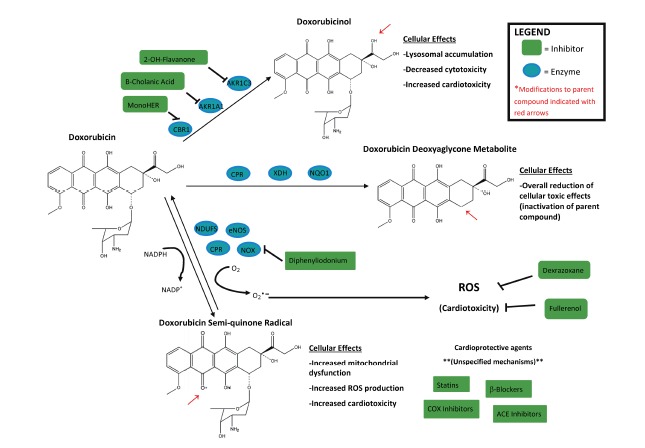
Main pathways of intracellular doxorubicin (DOX) biotransformation in mammalian cells, including catalytic enzymes involved in its metabolism and their inhibitors. The downstream effects of the metabolites are also listed. Similar pathways are involved in metabolism of other anthracyclines.
B). TWO ELECTRON REDUCTION OF ANTHRACYCLINES (HYDROXYLATION)
Anthracyclines hydroxylated at the C13 position are more polar than the parent compound and have higher water solubility. However, contrary to what might be expected, these hydroxylated metabolites are excreted from certain tissues at slower rates than the parental compound. They are also considerably less potent antineoplastic agents, while having greater cardiotoxicity due to their enhanced ability to accumulate in cardiac tissue relative to the parent compounds [37]. Peters et al. showed that levels of DOXOL in rats administered DOX were greatly enhanced in cardiac muscle compared to other tissues, including liver and skeletal muscle. The reason for this preferential accumulation of DOXOL in cardiac muscle is unknown [38]. With the exception of IDA, the C-13 metabolites for DNR, DOX and EPI are all less cytotoxic than their unhydroxylated forms [39, 40]. IDAOL is equally cytotoxic as IDA [39, 40].
a). Carbonyl Reductases (CBRs)
CBRs have recently been extensively reviewed in their function as drug metabolizing agents by Malatkova and Wsol in 2014 [41]. Thus, for this review, we will only briefly discuss their role in anthracycline metabolism. While AKRs and CBRs are both carbonyl reducing enzymes, this section focuses specifically on the carbonyl reductases CBR1 and CBR3 [41, 42]. These enzymes are found in a wide variety of tissues with a very broad range of substrates. They are generally associated with detoxifying toxic substrates to protect cells and organs [44, 45]. CBR1 is the predominant reductase that hydroxylates DOX in the liver, kidney, and gastrointestinal tract [43]. In a recent study, Kassner and colleagues [43] assessed the relative roles of the AKRs and CBRs in the two electron reduction (hydroxylation) of DOX. Interestingly, the kidney, liver and gastrointestinal tract (which are collectively responsible for the clearance of DOX from the body) express carbonyl reducing activity(ies) with an apparent Km of 140 μM. Enzymes with Km values in this range include carbonyl reductase 1 (CBR1) and AKR1C3. CBR1 was found to be expressed in the above three organs at higher levels than AKR1C3, while the latter exhibited a higher catalytic efficiency. An inhibitor capable of discriminating between the CBR1 and AKR1C3 activities was able to equally block the carbonyl reducing activity of CBR1 and human liver cytosol, but not AKR1C3. This suggests that CBR1 plays the predominant role in the liver’s ability to hydroxylate DOX. As previously discussed, this is distinct from the human heart, where AKRs play the predominant role in the hydroxylation of DOX [29].
b). Aldo-Keto Reductases (AKR)
All members of the AKR superfamily of proteins reduce ketones and aldehydes into secondary and primary alcohols in a NADPH-dependent manner [44]. The superfamily contains more than 140 members, sub-categorized into 15 families. AKR families share a minimum of 40% protein sequence identity, while their sub-families share >60% sequence identity [44]. Individual AKRs are named beginning with AKR, followed by: a number indicating the family of AKRs, a letter denoting the sub-family, and finally a number designating the individual member. The AKR1 family is the largest of the 15 families, and is one of three families including the mammalian AKRs [44]. While the AKR superfamily is comprised of many members, this review focuses primarily on family members that are known to hydroxylate anthracyclines and/or whose expression changes upon selection for anthracycline resistance.
c). AKR1 Family
The AKR1 family contains six sub-families, the largest of which is the AKR1C subfamily. AKR1C members primarily function as steroid metabolizing enzymes, with AKR1C1 to 1C4 sharing >86% sequence homology [44, 45]. AKR1C3, also known as 17β-hydroxy-steroid-dehydrogenase type V (17β-HSD) or 3α-hydroxy-steroid-dehydrogenase type I (3α-HSD), plays a role in the conversion of androstenedione into testosterone and estrone (E1) into estradiol (E2) [45-47]. In addition, AKR1A1, AKR1C1, AKR1C2, and AKR1C3 have been shown to play a role in the metabolism and detoxification of chemotherapy agents [48, 49], with AKR1A1 being specifically identified as the likely AKR involved in the conversion of the anthracycline DOX into DOXOL in cancer patients [50]. A variety of AKR members (AKR1A1, AKR1B1, AKR1B10, AKR1C1, AKR1C4 and AKR7A2) also play a role in the detoxification of reactive aldehydes, some of which may be created by anthracycline-generated ROS [44, 45, 51]. AKRs are differentially expressed in various tissues throughout the body. AKR1A1 is most highly expressed in kidneys, followed by the liver with the lowest expression being in the lungs [44]. AKR1B1, AKR1B10 and the AKR1C isoforms 1 through 4 have been shown to be primarily expressed in the liver, with AKR1C4 being exclusively expressed in this organ. While most studies focus on the AKR1 family of enzymes and their ability to metabolize anthracyclines, it should be noted that AKR7A2 has also been shown to have some metabolic activity towards these substances in a wide variety of organs [52]. While belonging to a different family of enzymes, this ability of AKR7A2 demonstrates the potential that other AKR family members may play yet undiscovered roles in anthracycline metabolism. Due to their roles in xenobiotic detoxification and steroid synthesis, AKR1C isoforms 1 through 3 are also highly expressed in the intestine, mammary glands, prostate, ovary and lungs [44, 53], with AKR1C3 the predominant AKR in mammary glands. The widespread distribution of AKRs in tissues ensures that a substantial amount of the anthracyclines administered to cancer patients is rapidly hydroxylated.
d). Specific Roles of AKR1C2, AKR1C3 and AKR1B10 in Anthracycline Metabolism and Resistance
While several AKRs have been studied for their role in anthracycline metabolism, only a subset specifically affect the biochemical and cytotoxic properties of anthracyclines. Heibein et al. showed recently that while exogenously added DOX localizes to the nucleus of breast tumor cells, exogenous DOXOL accumulates in lysosomes [54]. This suggests that the hydroxylation of this anthracycline prevents the drug from entering the nucleus, possibly due to its poorer affinity for binding to proteasomes. In addition, Heibein et al. observed that DOXOL [54] had significantly less affinity for DNA compared to DOX, which may also account for the former’s lack of localization to the nucleus. These investigators also showed that selection of MCF-7 breast tumor cells for DOX resistance resulted in increased transcription of the AKR1C2, AKR1C3 and AKR1B10 genes [54, 55]. Interestingly, the AKR inhibitor β-cholanic acid was able to restore localization of DOX (but not DOXOL) to the nucleus in DOX-resistant cells [54], suggesting that the inhibitor blocked DOX hydroxylation. Consistent with this view, the intracellular DOX concentration in the DOX-resistant cells increased in the presence of β-cholanic acid [54].
As stated previously, anthracyclines have been shown to be potent inhibitors of DNA topoisomerase II [20] and to induce substantial DNA damage [23] in tumor cells. Interestingly, the hydroxylation of the C-13 carbonyl group in two anthracyclines (DOX and IDA) was found to have little effect on their ability to inhibit topoisomerase II [40]. Moreover, both of these anthacyclines and their alcohol metabolites had a strong ability to induce DNA damage. In fact, in isolated nuclei, the hydroxylated forms of the drugs induced twice the DNA damage as their parental compounds [40]. Given that hydroxylation of most anthracyclines dramatically reduces their cytotoxic effects [39, 40], the above findings suggest that their cytotoxicity (at least in vitro) is not dependent upon their ability to inhibit topoisomerase or to induce DNA damage.
e). Host toxicity
The major toxic side effect of anthracyclines in cancer patients relates to the high cardiotoxic properties of their hydroxylated metabolites, particular for DOX. DOXOL appears to have a direct effect on excitation/contraction coupling in ventricular myocytes [56]. In humans, the hydroxylated anthracyclines induce delayed onset cardiomyopathy, which can manifest itself during the course of treatment or even weeks or months post-treatment [32]. In recent years, studies on the host toxicity of anthracyclines have been largely focussed on the role that carbonyl reductases play in this phenomenon. In one study, mice exhibiting a CBR1 null allele were shown to exhibit substantially reduced cardiotoxicity compared to wildtype mice, when both were administered DOX [57]. In another study, the overexpression of human carbonyl reductase in transgenic mice advanced the development of DOX-induced cardiotoxicity [58]. Mechanistically, it is generally believed that the anthracyclines become metabolized within cardiomyocytes by carbonyl reducing enzymes into their alcohol metabolites. These metabolites are not easily expelled from the myocytes, where they increasingly activate calcium release from calcium release channels [59]. This leads to a general dysfunction in the cardiomyocyte which leads to cell death and eventual cardiac myopathy. Interestingly, a subsequent study by Shadle et al. [60] demonstrated that the anthracycline DNR opened calcium release channels in sarcoplasmic reticulum preparations from rabbit atria at a potency 20 times that of the quinone-deficient analogue, 5-iminodaunorubicin. Moreover, neither anthracycline induced free-radical formation, suggesting that DNR impairs the contraction of the myocardium by interference with sarcoplasmic reticulum function via a mechanism not involving free radical formation.
The specific involvement of polymorphisms in CBR genes and patient cardiotoxicity after anthracycline treatment remains controversial. In one study, two polymorphisms in two carbonyl reductase genes (CBR1 1096G>A; CBR3 V244M) were identified as genetic biomarkers associated with susceptibility to cardiac damage from anthracycline treatment in pediatric oncology patients [61]. In contrast, another study found no relationship between any polymorphisms in AKR or CBR genes and cardiotoxicity associated with anthracycline treatment [62]. Despite this discrepancy as to the role of CBR polymorphisms in the cardiotoxicity of anthracyclines, the link between carbonyl reductases and cardiotoxicity resulting from anthracycline treatment is supported by a number of additional recent studies [63-65]. A convincing line of evidence supporting this link is a very recent study showing that the compound 23-hydroxybetulinic acid inhibits carbonyl reductase activity and was able to reduce both the accumulation of DOXOL in mice hearts and DOX-induced cardiotoxicity [66]. There is less support for the role of AKRs in promoting the cardiotoxicity of anthracyclines. For example, DOX is considered to have the greatest effect on cardiotoxicity; however, it has the lowest affinity for AKR enzymes [48], in particular for AKR7A2, which has been shown to have very low specific activities towards DOX and DNR [52]. In contrast, IDA, which has one of the lowest cardiotoxicities of the anthracyclines has one of the highest affinities for metabolism by AKR’s [48].
C). ONE ELECTRON REDUCTION OF ANTHRACYCLINES (SEMIQUINONE FORMATION)
Anthracycline semiquinone formation involves a one-electron reduction of the quinone moiety. This reaction is thought to be catalyzed by a variety of cellular NADH- and NADPH-dependent reductases. The semiquinone metabolites of anthracyclines are generally agreed to be more cytotoxic than the parental molecules, both in combating tumor growth and in their effects on host tissues. The semiquinone anthracycline may elicit its cytotoxic effects by facilitating the alkylation of cellular macromolecules [67] or through the generation of oxygen radicals [68, 69]. Enzymes that are likely to catalyze the one-electron reduction of the anthracycline include NADPH-cytochrome P450 reductase [70], endothelial nitric oxide synthase [71], and NADH dehydrogenase (ubiquinone) Fe-S [72].
a). Host toxicity via Anthracycline Semiquinone Metabolites
Along with their efficacy in treating a variety of human neoplasms, the anthracyclines are known to cause various hematological toxicities, in particular neutropenia associated with DOX treatment [73]. Along with myelosuppresion, DOX treatment can also cause symptoms such as nausea, vomiting, and cardiac arrhythmias, all of which are clinically manageable [74]. It is currently unclear whether these effects are dependent on the reductive conversion of anthracyclines to their semiquinone form; however, cardiotoxicity is thought to be particularly dependent on the formation of the semiquinone metabolite [71, 72].
At the subcellular level, anthracycline-induced cardiomyopathy is thought to be associated with the drug’s ability to induce mitochondrial dysfunction [68, 69]. As with other muscle tissues, cardiomyocytes rely on mitochondria for ~90% of their ATP production [75]. In fact, mitochondria make up about 20-40% of the cardiomyocyte volume, which is greater than that of skeletal muscle [76]. The presence of particular reductive enzymes within heart mitochondria may account for the ability of anthracycline-containing regimens to induce cardiotoxicity in cancer patients. Anthracyclines and their semiquinone metabolites have been well studied for their often irreversible damage to the heart, which can become apparent between four and twenty years after the completion of chemotherapy [77].
i). NADH dehydrogenase (Ubiquinone) Fe-S [NDUFS]
NADH dehydrogenase (ubiquinone) Fe-S is associated with complex I of the respiratory chain within the inner mitochondrial membrane. It typically catalyzes the transfer of electrons from NADH to coenzyme Q10 [78]. This high molecular weight iron-sulfur protein complex (~1000 kDa) consists of 45 subunits, seven of which are encoded by mitochondrial DNA and the remainder by the nuclear genome [79]. Anthracycline semiquinone formation is particularly favorable in the membranes of heart mitochondria. A study by Nohl et al. found that mitochondria from the heart easily shuttle single electrons to DOX to promote semiquinone formation. In contrast, they found that liver mitochondria are ineffective in producing semiquinones from DOX [72]. Nohl et al. further suggested that NADH dehydrogenase of complex I catalyzes DOX semiquinone formation in heart mitochondrial membranes and that this enzyme is absent in liver mitochondria. The semiquinone metabolite can be reoxidized non-enzymatically to produce superoxide radicals that can lead to DOX aglycone semiquinone formation. The latter metabolite, due to its increased lipophilicity, can accumulate in the inner mitochondrial membrane where it disrupts other electron carriers of the respiratory chain [72].
ii). Endothelial Nitric Oxide Synthase (eNOS)
Endothelial nitric oxide synthase (eNOS) is a membrane-bound enzyme located in coronary endothelial cells [80]. Typically, this enzyme oxidizes the amino acid L-arginine to produce L-citrulline and the nitric oxide radical NO•. This process involves the transfer of electrons from NADPH, FAD, among other electron donors, as well as cofactors including Ca2+ and the Ca2+-binding protein calmodulin [81, 82]. The depletion of one or more of these substrates or cofactors can impair NO• synthesis [83]. eNOS accounts for most of the NO• production in the heart, and regulation of its activity controls the amounts of nitric oxide produced [80]. In the cardiovascular system, the basal release of NO• is necessary for healthy vasodilatory tone [84]. Production of this vasodilator regulates blood pressure and vascular flow to tissues, including the brain, heart and lungs [84]. Supporting the role of eNOS in anthracycline semiquinone formation, Vasquez-vivar et al. demonstrated that DOX binding to the reductase domain of eNOS resulted in a one-electron reduction of DOX to DOX semiquinone in a NADPH-dependent manner [71]. They also showed that this metabolite was able to reduce oxygen independently of the enzyme, producing superoxide [O2-] [71].
Among its many effects on cells, DOX has the ability to induce eNOS gene transcription and increases the activity of the enzyme in bovine aortic endothelial cells [85]. Moreover, an antisense RNA targeting eNOS gene transcripts was able to abrogate DOX-induced apoptosis [85], suggesting that semiquinone formation was associated with DOX cytotoxicity. Similarly eNOS appears to play a role in mediating the toxic side effects of DOX in the host, since Neilan et al. have shown that eNOS gene knock-out in mice protected against DOX-induced cardiac dysfunction, injury and mortality [86]. These investigators further demonstrated that overexpression of eNOS transcripts in cardiomyocytes of mice resulted in greater increases in left ventricular dimensions and larger reductions in systolic function after a single dose of DOX than in eNOS knock-out or wild-type mice. DOX administration led to superoxide production in the hearts of wild-type mice but not in eNOS-deficient mice and DOX-induced superoxide production was even greater in eNOS-overexpressing mice than in wild-type mice [86]. By measuring apoptosis in cardiomyocytes using TUNEL assays, the group further provided evidence that a lack of eNOS protects mice against DOX-induced cardiac dysfunction, at least in part by preventing cardiac cell death via apoptosis.
While the above evidence lends support for eNOS’s role in anthracycline semiquinone-dependent cardiac damage, eNOS [along with inducible nitric oxide synthase (iNOS) and neuronal nitric oxide synthase (nNOS)] play important roles in the regulation of vascular tone by producing nitric oxide [83]. This may be disrupted if anthracyclines like DOX compete with L-arginine as substrates for eNOS. Consistent with this view, as DOX concentrations increase in cells, eNOS activity becomes devoted to semiquinone and superoxide generation rather than nitric oxide production [68, 69]. This would result in vasoconstriction of blood vessels in the heart, which may negatively affect heart health under stress. In rabbits, the level of systemic nitric oxide decreases considerably after the administration of DOX [87]. In humans, endothelial-dependent and -independent vasodilation were found to decrease considerably after DOX treatment (along with a significant decrease in serum nitrate levels [87]), consistent with inhibition of eNOS-mediated oxidation of L-arginine. Redox cycling, a term used when there is no net increase in the semiquinone because it is continuously reoxidized by molecular oxygen, would exacerbate the inhibition of L-arginine oxidation by eNOS in the presence of anthracyclines, since the spontaneous re-oxidation of the semiquinone would provide a continuous replenishment of the quinone substrate. Moens et al. have argued that the uncoupling of eNOS is known to be a major contributor in pressure-overload induced heart failure [88].
The role of iNOS in DOX semiquinone production and DOX-induced cardiotoxicity has been more controversial. It has been suggested that iNOS may elicit cardioprotective effects due to the production of nitric oxide [89], while death-promoting effects may be caused by its facilitation of peroxynitrite formation. Peroxynitrite formation is the result of a reaction between nitric oxide and superoxide radicals [68, 69], which can have DNA damaging effects [90]. The role of nNOS in DOX cellular toxicity is also unclear. One study reported no change in myocardial nNOS transcript levels upon administration of DOX [91]. Nevertheless, it is suggested that the enzyme catalyses the one-electron reduction of DOX to the semiquinone form [92].
Other enzymes such as xanthine dehydrogenase (XDH) [93, 94], NADPH dehydrogenase (NQO1) [94], and NADPH oxidase (NOX) [94] have been implicated in anthracycline-dependent oxygen radical generation, but they may or may not directly catalyse anthracycline semiquinone formation. For example, NQO1 is suggested to contribute to oxygen radical formation during anthracycline treatment [94] and is thus an important factor in treatment-related cardiotoxicity; however, it is known to catalyse two-electron reductions and is even suggested to inhibit one-electron reductions, such as semiquinone formation [95]. The NOXs have also been examined for their roles in DOX-related superoxide production and cardiotoxicity. Gilleron et al. demonstrated that DOX-activated NOXs contributed to superoxide formation and oxidative-stress leading to apoptosis in rat cardiomyoblasts. Moreover, inhibition of NOXs by diphenyliodonium and apocynin strongly reduced DOX-induced oxygen radicals, as well as cell death [96]. Supporting this view, experiments using a Nox2 knock-out model revealed that the left ventricular ejection fraction (8 weeks after DOX treatment) was considerably higher in Nox2-deficient mice compared to wild-type mice [97]. Interestingly, the study also showed that the adverse effects of DOX could be diminished by treatment with the vasodilator losartan [97].
b). Tumor Toxicity via Anthracycline Semiquinone Metabolites
Generally, the semiquinone metabolite of anthracyclines is thought to be more cytotoxic to tumors than the unmodified quinone. Although the ability of DOX to generate reactive oxygen species in tumors has been well demonstrated, several studies suggest that its ability to create covalent modifications or adducts in cellular materials accounts for the enhanced effect of the semiquinone metabolite relative to the quinone [67, 98]. Cummings et al. have suggested that within tumors, DOX metabolism is mostly impacted by NADPH-cytochrome P450 reductase [99].
i). NADPH-Cytochrome P450 Reductase (CPR)
NADPH-cytochrome P450 reductase (CPR) is a membrane bound enzyme localized to the endoplasmic reticulum [100]. It is an essential reductase, as its ability to transfer electrons is required for the functionality of most cytochrome P450 enzymes [101] and heme oxygenases [102]. It is also an important component of xenobiotic metabolism and plasma cholesterol homeostasis [102]. CPR catalyses the conversion of DOX to its semiquinone form and it is suggested that this transformation can occur in a variety of benzanthroquinones, including DNR and DNROL [70]. Bartoszek observed that MCF-7 cells treated with varying doses of DOX in the presence of exogenous purified rat P450 reductase and NADPH were considerably more sensitive (6-fold) to cell killing than cells incubated with drug alone [67]. Additional experiments showed that the potentiation of the drug was abrogated when the experiment was performed by incubating the drug with P450 reductase and NADPH for an hour at physiological temperatures prior to administration to tumor cells. This suggested that the semiquinone species is short-lived, becoming reoxidized back to the quinone form even in the absence of cells [103]. The enhanced biological effect in this study was not associated with altered drug uptake, since the concentration of isotopically labeled DOX in the cell was unaffected by the presence of the reductase [67, 103]. It was also observed that the one-electron reduction of DOX catalysed by P450 reductase took place only under aerobic conditions [103] and involved extensive NADPH consumption [67]. The oxygen requirement in the one-electron reduction was explained using quantum calculations, which showed that the quinone anthracycline (when in complexes with singlet oxygen) is a better electron acceptor than the free quinone [104] [reviewed in 67].
The possible role of P450 reductase, oxygen radical formation, and lipid peroxidation in tumor cell killing by anthracyclines was recently assessed [67]. Malondialdehyde (MDA) levels, a measure of lipid peroxidation in cells, were only slightly (but not significantly) increased in the presence of both DOX and P450 reductase relative to the drug alone. Moreover, the correlation between MDA levels and drug cytotoxicity was quite weak [67]. A variety of radical scavenging agents have been previously shown to affect DOX-induced oxygen radical production [105, 106], but Bartoczek et al. observed that oxygen radical formation was not responsible for tumor cell death in the presence of CPR [103]. These researchers and others further showed that CPR caused a significant increase in the amount of irreversible associations between radiolabeled DOX and both cellular proteins and DNA and that the formation of these adducts correlated with drug cytotoxicity [67, 103]. These findings thus suggested that CPR’s ability to enhance DOX cytotoxicity appeared to be related to the formation of alkylating metabolites rather than augmented redox cycling.
Kostrzewa-Nowak et al. investigated the effects of human liver CPR on reductive activation of DOX during the treatment of human promyelocytic HL60 cells and multi-drug resistant derivatives of these cells overexpressing either P-glycoprotein or MRP-1 [98]. The study involved the reductive activation of DOX by CPR and NADPH extracellularly, followed by treatment of the cells, allowing the metabolite to diffuse into the cells. Their findings indicated that NADPH is a necessary cofactor for CPR-dependent reductive conversion (semiquinone formation) and this formation does not occur at NADPH concentrations below 500 µM. Only at high NADPH concentrations was CPR effective at both reductive conversion of DOX and achieving between a two and three-fold increase in toxicity in both the DOX-sensitive and DOX-resistant human leukemia cell lines [98]. The study was conducted at clinically relevant doses of DOX, since DOX levels can reach 1-2 µM in the plasma of patients receiving treatment [14]. Interestingly, since drug potentiation occurred in cells overexpressing drug transporters capable of exporting DOX out of the cells, it was further proposed by Kostrzewa-Nowak et al. that the reactive metabolite is able to bind to cellular targets and escape MDR protein pumps [98]. These investigators also showed that the addition of superoxide dismutase, an oxygen radical-scavenging enzyme, abrogated reductive conversion of DOX by CPR in the presence of sufficient NADPH [98]. Given that superoxide dismutase is known to scavenge singlet oxygen [107], this is consistent with the proposal by Tempczyk et al. that quinone anthracyclines interacting with singlet oxygen are better electron acceptors than free quinones and that a DOX-singlet oxygen complex may be required for semiquinone formation [104].
As recognized by Kostrzewa-Nowak et al., there is some evidence that contradicts CPR's potential role in DOX activation. Niitsu et al. have also shown an inverse relationship between CPR expression and DOX cytotoxicity [108]. However, Kostrzewa-Nowak et al. address this discrepancy by arguing that the reductive activation of DOX by CPR “could be influenced by many factors such as bioavailability of NADPH, levels of other competing metabolic enzymes, and tissue-specific antioxidant defence systems” [98].
Another study supports the notion that reductive conversion of DOX increases its cytotoxic effect in tumor cells [109]. In this study, two acute lymphoblastic leukemia (ALL) cell lines were characterized for their ability to reduce DOX to the semiquinone. One cell line was sensitive to DOX (EU3-Sens) and the other was resistant (EU1-Res). The levels of transcripts and activities for a variety of enzymes and cofactors required for the reductive conversion of DOX were monitored in the cell lines and it was found that the DOX-sensitive cells exhibited lower intracellular quinone levels and higher NADPH levels than DOX-resistant cells. This is consistent with CPR-dependent reductive conversion of the drug to the semiquinone form. It was also determined that the DOX-resistant cells had higher levels of the superoxide-generating enzyme NADPH oxidase 4 (NOX4) and lower glucose-6-phosphate dehydrogenase (G6PDH) expression. The latter enzyme is responsible for regenerating cellular NADPH. The authors suggest that these changes in NOX4 and G6PDH expression favour the redox cycling and reduced cytotoxicity of DOX in DOX-resistant cells. Interestingly, these investigators conducted similar experiments at lower DOX concentrations (100 nM) and found increased quinone accumulation in the drug-sensitive cell line relative to the drug-resistant cell line [109].
Given all of the above investigations, there is strong evidence for DOX semiquinone formation in both cardiac tissue and tumor tissue and this is associated with its increased cytotoxicity. The mechanism by which the semiquinone metabolite promotes cell death appears to differ considerably between cardiac and tumor tissue. Oxygen radical production resulting from DOX, the semiquinone metabolite, and redox cycling between the two, occurs in cardiomyocytes and is important in cardiotoxicity [105, 110]. In contrast, redox cycling and oxygen radical formation does not appear to contribute to tumor toxicity. Rather, alkylation of cellular targets by the semiquinone metabolite appears to promote increased DOX cytotoxicity [67, 103]. The enzymes responsible for anthracycline semiquinone formation also appear to differ considerably between tumor and host tissues, especially since malignant transformation itself has been shown to be associated with increased levels of CPR [111]. The differences between host and tumor tissue with respect to the enzymes involved in semiquinone formation as well as the mode of toxicity of the semiquinone metabolite may highlight some inherent differences between healthy and malignant tissue, the latter being considerably more genetically unstable. This genetic instability may help to explain why reductive conversion seems to be a requirement for killing tumor tissue but redox cycling may be sufficient for damaging cardiac tissue, as tumor cells may be more adaptable to increased oxygen radical production. Moreover, DOX-resistant tumor cells may have alterations in their cellular physiology that favor redox cycling. These differences in anthracycline metabolism between host and tumor tissue may present unique opportunities to improve tumor killing by anthracyclines, while sparing host toxicities.
D). REDUCTIVE DEGLYCOSIDATION OF ANTHRACYCLINES (DEOXYAGLYCONE FORMATION)
The third, and minor, route of anthracycline metabolism involves the reductive deglycosidation of anthracyclines to 7-deoxyaglycone metabolites. The 7-deoxyaglycone form of an anthracycline is essentially the drug with the sugar moiety removed. The formation of 7-deoxyaglycone requires a two-electron reduction and absolutely requires an anaerobic environment and NADPH [27]. It is generally believed that the formation of the 7-deoxyaglycone metabolite inactivates the parent molecule. However, the 7-deoxyaglycone metabolite has been implicated in the production of ROS by intercalating with the inner mitochondrial membrane due to the increase in lipid solubility that accompanies the loss of the sugar moiety [14, 112]. This intercalation into membranes has also been thought to cause a form of benign acute toxicity in the human myocardium via ROS production [32], although the cardiotoxicity is not to the extent of that observed for the parent molecule. For DOX, the 7-deoxyaglycone metabolite can be produced from both the parent DOX molecule and also from the metabolite DOXOL. Considering that the four main anthracyclines (DOX, EPI, DNR, and IDA) share very similar structures it can be inferred that the formation of 7-deoxyaglycone would occur in similar ways for all four anthracyclines. Formation of the 7-deoxyaglycone metabolite occurs via three enzymes: NAD(P)H Quinone Oxidoreductase 1 (NQO1), Cytochrome P450 Reductase (CPR), and Xanthine Dehydrogenase (XDH).
a). NQO1
NAD(P)H Quinone Oxidoreductase 1 (NQO1), also known as DT-diaphorase, is an NADH- or NADPH-dependent enzyme [99]. The main role of NQO1 is to detoxify quinones via a two-electron reduction [27]. Other roles that NQO1 fulfills include the maintenance of antioxidants [113], the stabilization of p53 [114, 115], acting as a superoxide scavenger [116] and a 20S proteasome gatekeeper [117]. However, NQO1’s ability to perform a two-electron reduction of anthracyclines to form the deoxyaglycone metabolite depends heavily upon the physiological environment at the tumor site [99], with the deoxyaglycone only forming under hypoxic conditions [118]. In this way the harmful semiquinone metabolite is bypassed in favour of the inactive deoxyaglycone metabolite. NQO1 has also been implicated in the formation of semiquinone radicals of anthracyclines in an aerobic environment [27, 94], noting, however, that the deoxyaglycone metabolite is the predominant metabolite under anaerobic conditions [99]. Interestingly, NQO1 is predominantly found in the cytosol of cells [99], with the highest levels of the enzyme found in cardiovascular tissues and the liver [119].
NQO1 expression is induced by many xenobiotics via the Keap1/Nrf2/ARE pathway [120]. While Nrf2 is a critical transcription factor involved in cellular protection from toxic xenobiotics, it is kept at low levels under unstressed conditions by Keap1 [121]. The promoter region of the NQO1 gene contains multiple antioxidant response elements (AREs) and a xenobiotic response element (XRE) [119, 120] that bind Nrf2 and regulate NQO1 expression. Nrf2 has been implicated in chemotherapy drug resistance in vitro. Increased Nrf2 expression and activity have been associated with increased NQO1 levels and DOX resistance in both A2780 ovarian tumor cells and MCF-7 breast cancer cells [54, 122, 123]. While inactivation of Nrf2 in these cell lines can restore DOX cytotoxicity [124], it has been shown in rabbits that continuous treatment with anthracyclines does not elevate Nrf2 levels, while NQO1 levels are down-regulated [125].
Recently, a number of NQO1 polymorphisms have been identified and their relationship to patient survival after anthracycline chemotherapy was assessed. One such NQO1 polymorphism (C609T) results in a Pro187Ser substitution, which results in the increased ubiquitination and degradation of the protein [126]. Due to the degradation of NQO1, there is an impairment in ROS detoxification and reduced survival after chemotherapy [127]. If a patient is homozygous for the T allele (Ser substitution), the enzyme activity appears to be reduced to only 2% of the wild type enzyme (homozygous for the C allele) [126, 128, 129]. Although the T allele has not been found to be significantly related to patient survival at all common cancer sites [127, 130, 131], there is a clear trend toward an increased overall cancer risk associated with this polymorphism [130]. Moreover, this lack of significance could be attributed to the small sample size associated with the low frequency T allele [130, 131]. Interestingly, inhibition of NQO1 by dicoumarol was found to be associated with an increase in DOX-induced cardiotoxicity, suggesting NQO1 may play a protective role against DOX-induced cardiotoxicity [132].
b). CPR and XDH
Cytochrome P450 Reductase (CPR) is also dependent on NADPH [99]. CPR can catalyze both redox cycling in an aerobic environment and 7-deoxyaglycone formation in an anaerobic environment [99]. It has been shown that DOX is stoichiometrically converted to the inactive 7-deoxyaglycone metabolite by CPR, and, similar to NQO1, this formation was completely abolished in an aerobic environment [99]. CPR is found predominantly in the liver; however, it can be found in other organs such as the lung or the kidney [133]. Interestingly, the anthracyclines DNR and DOX have been shown to suppress the activity of CPR in rabbit hepatocytes in vitro [134].
Xanthine Dehydrogenase (XDH) is less effective than both NQO1 and CPR at generating 7-deoxyaglycone metabolites of anthracyclines [99]. Consequently, its role in this process is less studied. XDH has also been implicated in the formation of the semiquinone radical [94].
E). Strategies to Improve Clinical Response and/or Reduce Clinical Toxicity to Anthracyclines
As mentioned previously, one of the most prevalent and serious side effects that limits the use of anthracyclines is their cardiotoxicity. This is particularly the case for breast cancer patients undergoing anthracycline-based adjuvant or neoadjuvant chemotherapy [135]. To reduce the incidence of cardiac toxicity, longer infusion rates are employed to reduce peak plasma levels of anthracyclines. Their cumulative doses are also closely monitored. Nevertheless, these strategies do not completely eliminate the risk of cardiotoxicity [136]. Reduced cardiotoxicity with anthracyclines using liposomal formulations of DOX or DNR to selectively target tumor tissue has shown some efficacy in a clinical setting [137]. Another method for preventing anthracycline-induced cardiotoxicity involves the use of pharmacological cardioprotective agents, such as dexrazoxane [138], ascorbic acid [139], and an engineered bivalent neuregulin [140]. To date, the only agent in this class that has been approved for use and has shown significant clinical efficacy is the iron chelator dexrazoxane (DEX) [138, 141]. While other pharmacological cardioprotective agents have been tested in an in vitro setting, their use clinically is uncertain at this time.
Two approaches have been used to improve the therapeutic index of anthracyclines. One involves altering the metabolism of the drugs in order to enhance their cytotoxic effects on tumors by enhancing their therapeutic mechanism of action. This could include nucleic acid intercalation, topoisomerase II inhibition, iron mediated generation of ROS, and other processes that could lead to improved killing of tumors in vivo [14]. A second approach is to reduce the systemic toxicity of anthracyclines by further chemical modification of anthracyclines or by selectively inhibiting off target effects of the drug and its metabolites, such as cardiotoxicity. Examples of these approaches are illustrated below.
a). Modulators to enhance the tumor cytotoxicity of anthracyclines
i). AKR and CR Inhibitors
As mentioned previously, the 13-hydroxylation of anthracyclines by AKRs substantially reduces their tumor cytotoxicity [37, 39, 40]. Thus, blocking the formation of hydroxylated metabolites could improve the efficacy of anthracyclines. Inhibiting AKRs with agents such as 2-hydroxyflavanone [48] or beta-cholanic acid [54, 55] have been shown to increase anthracycline cytotoxicity in anthracycline-resistant cell lines overexpressing AKRs, but it is unclear whether a similar improvement in clinical response to anthracyclines would be realized in patients with tumors that have intrinsic or acquired resistance to these agents. The semisynthetic flavonoid 7-mono-O-(β-hydroxyethyl)-rutoside, commonly known as monoHER, is known to effectively inhibit CBR1. However, unlike the AKR inhibitors, MonoHER has been documented in a phase II clinical trial to protect metastatic cancer patients from DOX-induced cardiotoxicity [142]. This may be through its ability to reduce production of the cardiotoxic metabolite DOXOL. MonoHER has also been shown to potentiate the cytotoxicity of DOX in human liposarcoma cells by reducing NF-κB activation and promoting DOX-induced apoptosis [143]. In vitro and in vivo experiments have shown that monoHER does not interfere with the anti-tumor effect of DOX. Interestingly, high doses of monoHER (>1500 mg/m2) can augment DOX’s anti-tumor effect, while considerably lower doses are required to achieve monoHER’s cardioprotective effect [68].
b). Strategies to Reduce the Systemic Toxicity of Anthracyclines
i). Iron Chelators (Dexrazoxane)
Iron is known to potentiate the toxicity of anthracyclines by transforming relatively safe species like O2 •− and H2O2 into much more reactive and toxic hydroxyl radical (OH•) or iron-peroxide complexes that have the capacity to damage DNA, proteins, and lipids [8, 144]. It has also been proposed that the redox cycling of the quinone moiety would allow anthracyclines to increase cellular levels of iron by mobilizing ferritin, a ubiquitous intracellular protein that stores and releases iron [145]. In addition, doxorubicin treatment has been shown to result in the preferential accumulation of iron inside the mitochondria of cardiomyocytes [146]. This results in amplification of iron-mediated oxidative stress [147, 148]. Moreover, doxorubicin-induced DNA strand breaks and changes in gene expression that lead to defective mitochondrial biogenesis and ROS formation (and subsequently cardiotoxicity) appear to be reduced in mice having cardiomyoctyes possessing Top2b deletion mutations, suggesting that cardiotoxicity by doxorubicin involves the action of topoisomerase IIβ [149]. Perhaps one of the best pieces of evidence that iron plays a pivotal role in the ROS-mediated toxicity of DOX comes from numerous studies showing that the iron chelator dexrazoxane (DEX) can effectively block anthracycline cardiotoxicity [138]. It has been repeatedly shown to mitigate anthracycline toxicity, and is approved for clinical use [150-152]. Marty et al have shown in a randomized phase III study of 164 breast cancer patients that in comparison to patients receiving an anthracycline alone, patients treated with both an anthracycline and DEX experienced significantly fewer and less severe episodes of congestive heart failure, without affecting the tumor response rate [116]. Other antioxidants have also been shown to reduce both the ROS generation and toxicity of anthracyclines, but their current use remains limited [138].
ii). Statins, Beta Blockers, ACE inhibitors, and COX inhibitors
Recent studies have shown that several statins such as lovastatin [153], beta blockers such as Nebivolol [154], ACE inhibitors (e.g Enalapril) [155, 156] and COX inhibitors demonstrate notable cardioprotective effects, when used in conjunction with DOX [157]. Interestingly, in a recent review of randomized trials and observational studies, where a prophylactic intervention was compared with a control arm in patients with a normal ejection fraction and no past history of heart failure, the authors demonstrated that prophylactic treatment with DEX, a beta-blocker, a statin, or angiotensin antagonists all can reduce cardiotoxicity [157]. Statins have been suggested as an alternative cardioprotective strategy for anthracycline treatments [158]. In mice, DOX increases the cardiac mRNA levels of B-type natriuretic peptide, interleukin-6 and connective tissue growth factor, while lovastatin appeared to counteract these anthracycline-induced cardiac stress responses [159]. Zofenopril, an ACE inhibitor, inhibits cardiotoxicity in rats; however, it is unclear whether the mechanism is direct ACE inhibition or another off target cardioprotective effect [160]. The same can be said for statins, beta blockers and cyclooxygenase-2 inhibitors. The mechanisms by which these agents prevent cardiotoxicity are not well understood, limiting their use and study as agents to combat anthracycline cardiotoxicity [154]. Moreover, no randomized studies have been published comparing the cardioprotective efficacy of statins, beta blockers, ACE inhibitors, or COX inhibitors relative to DEX.
iii). β-glucuronidase-Mediated Release of Anthracycline Prodrugs
Another mechanism for reducing the systemic toxicity of anthracyclines, including cardiotoxicity, is to restrict drug action to tumor sites. For example, a series of glucuronide prodrugs have been synthesized that render DOX nontoxic to host tissues, since the hydrophilic glucuronide group added to DOX prevents entry across hydrophobic cellular membranes. However, the high levels of β-glucuronidase in tumors then permits the release of cytotoxic DOX and tumor-specific cell killing [161-163]. While such prodrugs have shown promise in pre-clinical models [164, 165], none, to our knowledge, have yet to enter clinical trials in humans. This may be due to wide variations in β-glucuronidase levels amongst patient tumors, necessitating screening of patient tumors for high β-glucuronidase expression [166].
iv). Fullerenol
The chemical modification of fullerenes (specifically, the polyhydroxylation of C60 nanoparticles) results in C60(OH)x structures with differing degrees of antioxidant activity in an aqueous environment [167]. One particular fullerene (fullerenol) was able to protect rat hearts from DOX-induced cardiotoxicity [168]. It has been proposed that fullerenol protects cardiomyocytes by acting as a ROS scavenger and/or by removing free iron through the formation of a fullerenol-iron complex [168]. Previous studies have shown that application of DOX to Wistar rats causes damage to the heart and baroreceptors, which results in diastolic dysfunction characterized by increased left ventricle end-diastolic pressure [169]. Fullerenol attenuated these DOX-induced heart disturbances [168]. Fullerenol was also able to successfully treat DOX-induced nephrotoxicity [170], pulmotoxicity [171], and hepatotoxicity [172] in rats.
v). Synthesis of Non-Cardiotoxic Anthracyclines
An alternate strategy for combating anthracycline-induced cardiotoxicity is to synthesize anthracyclines with reduced capacity for cardiotoxicity. For example, a prodrug of DOX (aldoxorubicin) can be administered at significantly higher concentrations in patients, without acute cardiotoxicity. Moreover, recent findings further suggest that aldoxorubicin exhibits stable levels in blood without accumulation in body compartments such as the heart. This may explain why the drug is significantly less cardiotoxic than DOX [173]. The DOX analog GPX-150, modified at two sites to reduce the formation of cardiotoxic metabolites or ROS, has shown promise in a recent phase I trial, where it was administered safely to patients with acceptable toxicity and no cardiotoxocity [174]. Another anthracycline with protein kinase C-activating properties (AD 198) has also shown significant anti-tumor activity. However, unlike DOX, little ventricular damage was observed in mice administered the agent [175].
CONCLUDING REMARKS
In the case of anthracyclines, a knowledge of the metabolism of these drugs has provided significant insight into how they exert their anti-tumor effects and their toxic side effects on the host. Some anthracycline metabolites facilitate the ability of these drugs to combat the growth of tumor cells, while others suppress cytotoxicity. The hydroxylation of anthracyclines is of particular interest, since the hydroxylated metabolites have considerably reduced anti-tumor activity, but substantially increased cardiotoxicity. By identifying the precise enzymes that play a role in anthracycline metabolism, it has been possible to identify novel chemical agents that can augment the anti-tumor effects of anthracyclines and/or prevent negative side effects within the host, including cardiotoxicity, nephrotoxicity, and hepatotoxicity. While successful new strategies to improve the therapeutic index of anthracyclines have recently been identified, it will only be through future clinical trials in multiple, independent cohorts of patients that the true efficacy of these strategies will be known. For one such agent (dexrazoxane), its incorporation into standard clinical practice clearly appears to be on the horizon.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Kufe D.W., Pollock R.E., Weichselbaum R.R., Bast R.C., Gansler T.S., Holland J.F., Frei E. Cancer Medicine; 6th Edition ed.; BC Decker, Inc.: Lewiston, NY, : 2003. [Google Scholar]
- 2.Visani G., Isidori A., Minotti G. Anthracycline Cardiotoxicity. In: Viselka J., editor. Cardiomyopathies - From Basic Research to Clinical Management. Rijeka, Croatia: Intech Open Access Publishers; 2012. [Google Scholar]
- 3.Hortobagyi G.N. Anthracyclines in the treatment of cancer. An overview. Drugs. 1997;54(Suppl. 4):1–7. doi: 10.2165/00003495-199700544-00003. [DOI] [PubMed] [Google Scholar]
- 4.Preobrazhenskaya M.N., Tevyashova A.N., Olsufyeva E.N., Huang H-F., Huang H.S. Second Generation Drugs - Derivatives of Natural Anti-Tumor Anthracycline Antibiotics Daunorubicin, Doxorubicin and Carminomycin. J. Med. Sci. 2006;26(4):119–128. [Google Scholar]
- 5.Chien A.J., Moasser M.M. Cellular mechanisms of resistance to anthracyclines and taxanes in cancer: intrinsic and acquired. Semin. Oncol. 2008;35(2) Suppl. 2:S1–S14. doi: 10.1053/j.seminoncol.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Magne N., Chargari C. Re: Therapy Insight: anthracyclines and trastuzumab--the optimal management of cardiotoxic side effects. Nat. Clin. Pract. Oncol. 2008;5(11):E1. doi: 10.1038/ncponc1275. [DOI] [PubMed] [Google Scholar]
- 7.Delemasure S., Vergely C., Zeller M., Cottin Y., Rochette L. Ann. Cardiol. Angeiol. (Paris) 2006;55(2):104–112. doi: 10.1016/j.ancard.2006.02.005. [Preventing the cardiotoxic effects of anthracyclins. From basic concepts to clinical data]. [DOI] [PubMed] [Google Scholar]
- 8.Simunek T., Sterba M., Popelova O., Adamcova M., Hrdina R., Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009;61(1):154–171. doi: 10.1016/s1734-1140(09)70018-0. [DOI] [PubMed] [Google Scholar]
- 9.Kizek R., Adam V., Hrabeta J., Eckschlager T., Smutny S., Burda J.V., Frei E., Stiborova M. Anthracyclines and ellipticines as DNA-damaging anticancer drugs: recent advances. Pharmacol. Ther. 2012;133(1):26–39. doi: 10.1016/j.pharmthera.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Pocket Oncology. Alphen aan den Rijn, The Netherlands: Wolters Kluwer Publishers; 2014. [Google Scholar]
- 11.Barrett-Lee P.J., Dixon J.M., Farrell C., Jones A., Leonard R., Murray N., Palmieri C., Plummer C.J., Stanley A., Verrill M.W. Expert opinion on the use of anthracyclines in patients with advanced breast cancer at cardiac risk. Ann. Oncol. 2009;20(5):816–827. doi: 10.1093/annonc/mdn728. [DOI] [PubMed] [Google Scholar]
- 12.Cortes-Funes H., Coronado C. Role of anthracyclines in the era of targeted therapy. Cardiovasc. Toxicol. 2007;7(2):56–60. doi: 10.1007/s12012-007-0015-3. [DOI] [PubMed] [Google Scholar]
- 13.Fumoleau P., Roche H., Kerbrat P., Bonneterre J., Romestaing P., Fargeot P., Namer M., Monnier A., Montcuquet P., Goudier M.J., Luporsi E. Long-term cardiac toxicity after adjuvant epirubicin-based chemotherapy in early breast cancer: French Adjuvant Study Group results. Ann. Oncol. 2006;17(1):85–92. doi: 10.1093/annonc/mdj034. [DOI] [PubMed] [Google Scholar]
- 14.Minotti G., Menna P., Salvatorelli E., Cairo G., Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004;56(2):185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 15.Kiyomiya K., Matsuo S., Kurebe M. Mechanism of specific nuclear transport of adriamycin: the mode of nuclear translocation of adriamycin-proteasome complex. Cancer Res. 2001;61(6):2467–2471. [PubMed] [Google Scholar]
- 16.Kiyomiya K., Matsuo S., Kurebe M. Proteasome is a carrier to translocate doxorubicin from cytoplasm into nucleus. Life Sci. 1998;62(20):1853–1860. doi: 10.1016/s0024-3205(98)00151-9. [DOI] [PubMed] [Google Scholar]
- 17.Cancer Chemotherapy and Biotherapy: Principles and Practice. Philadelphia, PA: Wolters Kluwer Publishers; 2010. [Google Scholar]
- 18.Sinha B.K., Politi P.M. Anthracyclines. Cancer Chemother. Biol. Response Modif. 1990;11:45–57. [PubMed] [Google Scholar]
- 19.Gewirtz D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999;57(7):727–741. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 20.Hande K.R., Topoisomerase I.I. Inhibitors. Update Cancer Ther. 2008;3:13–26. [Google Scholar]
- 21.Sawyer D.B., Peng X., Chen B., Pentassuglia L., Lim C.C. Mechanisms of anthracycline cardiac injury: can we identify strategies for cardioprotection? Prog. Cardiovasc. Dis. 2010;53(2):105–113. doi: 10.1016/j.pcad.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westman E.L., Canova M.J., Radhi I.J., Koteva K., Kireeva I., Waglechner N., Wright G.D. Bacterial inactivation of the anticancer drug doxorubicin. Chem. Biol. 2012;19(10):1255–1264. doi: 10.1016/j.chembiol.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 23.Muindi J., Sinha B.K., Gianni L., Myers C. Thiol-dependent DNA damage produced by anthracycline-iron complexes. The structure-activity relationships and molecular mechanisms. Mol. Pharmacol. 1985;27(3):356–365. [PubMed] [Google Scholar]
- 24.Mizutani H. Yakugaku Zasshi. 2007;127(11):1837–1842. doi: 10.1248/yakushi.127.1837. [Mechanism of DNA damage and apoptosis induced by anticancer drugs through generation of reactive oxygen species]. [DOI] [PubMed] [Google Scholar]
- 25.Advances in Mitchondrial Medicine: Advances in Experimental Medicine and Biology. Amsterdam, The Netherlands: Springer Publishers; 2012. [Google Scholar]
- 26.Novotna R., Wsol V., Xiong G., Maser E. Inactivation of the anticancer drugs doxorubicin and oracin by aldo-keto reductase (AKR) 1C3. Toxicol. Lett. 2008;181(1):1–6. doi: 10.1016/j.toxlet.2008.06.858. [DOI] [PubMed] [Google Scholar]
- 27.Cummings J., Willmott N., Hoey B.M., Marley E.S., Smyth J.F. The consequences of doxorubicin quinone reduction in vivo in tumour tissue. Biochem. Pharmacol. 1992;44(11):2165–2174. doi: 10.1016/0006-2952(92)90343-h. [DOI] [PubMed] [Google Scholar]
- 28.Menna P., Paz O.G., Chello M., Covino E., Salvatorelli E., Minotti G. Anthracycline cardiotoxicity. Expert Opin. Drug Saf. 2012;11(Suppl. 1):S21–S36. doi: 10.1517/14740338.2011.589834. [DOI] [PubMed] [Google Scholar]
- 29.Mordente A., Minotti G., Martorana G.E., Silvestrini A., Giardina B., Meucci E. Anthracycline secondary alcohol metabolite formation in human or rabbit heart: biochemical aspects and pharmacologic implications. Biochem. Pharmacol. 2003;66(6):989–998. doi: 10.1016/s0006-2952(03)00442-8. [DOI] [PubMed] [Google Scholar]
- 30.Loveless H., Arena E., Felsted R.L., Bachur N.R. Comparative mammalian metabolism of adriamycin and daunorubicin. Cancer Res. 1978;38(3):593–598. [PubMed] [Google Scholar]
- 31.Luo X., Evrovsky Y., Cole D., Trines J., Benson L.N., Lehotay D.C. Doxorubicin-induced acute changes in cytotoxic aldehydes, antioxidant status and cardiac function in the rat. Biochim. Biophys. Acta. 1997;1360(1):45–52. doi: 10.1016/s0925-4439(96)00068-3. [DOI] [PubMed] [Google Scholar]
- 32.Licata S., Saponiero A., Mordente A., Minotti G. Doxorubicin metabolism and toxicity in human myocardium: role of cytoplasmic deglycosidation and carbonyl reduction. Chem. Res. Toxicol. 2000;13(5):414–420. doi: 10.1021/tx000013q. [DOI] [PubMed] [Google Scholar]
- 33.Dessypris E.N., Brenner D.E., Baer M.R., Hande K.R. Uptake and intracellular distribution of doxorubicin metabolites in B-lymphocytes of chronic lymphocytic leukemia. Cancer Res. 1988;48(3):503–506. [PubMed] [Google Scholar]
- 34.Sokolove P.M. Interactions of adriamycin aglycones with mitochondria may mediate adriamycin cardiotoxicity. Int. J. Biochem. 1994;26(12):1341–1350. doi: 10.1016/0020-711x(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 35.Conklin K.A. Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004;3(4):294–300. doi: 10.1177/1534735404270335. [DOI] [PubMed] [Google Scholar]
- 36.Mross K., Mayer U., Hamm K., Burk K., Hossfeld D.K. Pharmacokinetics and metabolism of iodo-doxorubicin and doxorubicin in humans. Eur. J. Clin. Pharmacol. 1990;39(5):507–513. doi: 10.1007/BF00280945. [DOI] [PubMed] [Google Scholar]
- 37.Reszka K.J., Wagner B.A., Teesch L.M., Britigan B.E., Spitz D.R., Burns C.P. Inactivation of anthracyclines by cellular peroxidase. Cancer Res. 2005;65(14):6346–6353. doi: 10.1158/0008-5472.CAN-04-2312. [DOI] [PubMed] [Google Scholar]
- 38.Peters J.H., Gordon G.R., Kashiwase D., Acton E.M. Tissue distribution of doxorubicin and doxorubicinol in rats receiving multiple doses of doxorubicin. Cancer Chemother. Pharmacol. 1981;7(1):65–69. doi: 10.1007/BF00258216. [DOI] [PubMed] [Google Scholar]
- 39.Kuffel M.J., Reid J.M., Ames M.M. Anthracyclines and their C-13 alcohol metabolites: growth inhibition and DNA damage following incubation with human tumor cells in culture. Cancer Chemother. Pharmacol. 1992;30(1):51–57. doi: 10.1007/BF00686485. [DOI] [PubMed] [Google Scholar]
- 40.Ferrazzi E., Woynarowski J.M., Arakali A., Brenner D.E., Beerman T.A. DNA damage and cytotoxicity induced by metabolites of anthracycline antibiotics, doxorubicin and idarubicin. Cancer Commun. 1991;3(6):173–180. doi: 10.3727/095535491820873308. [DOI] [PubMed] [Google Scholar]
- 41.Malatkova P., Wsol V. Carbonyl reduction pathways in drug metabolism. Drug Metab. Rev. 2014;46(1):96–123. doi: 10.3109/03602532.2013.853078. [DOI] [PubMed] [Google Scholar]
- 42.Varatharajan S., Abraham A., Zhang W., Shaji R.V., Ahmed R., Abraham A., George B., Srivastava A., Chandy M., Mathews V., Balasubramanian P. Carbonyl reductase 1 expression influences daunorubicin metabolism in acute myeloid leukemia. Eur. J. Clin. Pharmacol. 2012;68(12):1577–1586. doi: 10.1007/s00228-012-1291-9. [DOI] [PubMed] [Google Scholar]
- 43.Kassner N., Huse K., Martin H.J., Godtel-Armbrust U., Metzger A., Meineke I., Brockmoller J., Klein K., Zanger U.M., Maser E., Wojnowski L. Carbonyl reductase 1 is a predominant doxorubicin reductase in the human liver. Drug Metab. Dispos. 2008;36(10):2113–2120. doi: 10.1124/dmd.108.022251. [DOI] [PubMed] [Google Scholar]
- 44.Jin Y., Penning T.M. Aldo-keto reductases and bioactivation/detoxication. Annu. Rev. Pharmacol. Toxicol. 2007;47:263–292. doi: 10.1146/annurev.pharmtox.47.120505.105337. [DOI] [PubMed] [Google Scholar]
- 45.Rizner T.L., Penning T.M. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids. 2014;79:49–63. doi: 10.1016/j.steroids.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adamski J., Jakob F.J. A guide to 17beta-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2001;171(1-2):1–4. doi: 10.1016/s0303-7207(00)00383-x. [DOI] [PubMed] [Google Scholar]
- 47.Rizner T.L., Smuc T., Rupreht R., Sinkovec J., Penning T.M. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol. Cell. Endocrinol. 2006;248(1-2):126–135. doi: 10.1016/j.mce.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 48.Hofman J., Malcekova B., Skarka A., Novotna E., Wsol V. Anthracycline resistance mediated by reductive metabolism in cancer cells: the role of aldo-keto reductase 1C3. Toxicol. Appl. Pharmacol. 2014;278(3):238–248. doi: 10.1016/j.taap.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 49.Matsunaga T., Yamaguchi A., Morikawa Y., Kezuka C., Takazawa H., Endo S., El-Kabbani O., Tajima K., Ikari A., Hara A. Induction of aldo-keto reductases (AKR1C1 and AKR1C3) abolishes the efficacy of daunorubicin chemotherapy for leukemic U937 cells. Anticancer Drugs. 2014;25(8):868–877. doi: 10.1097/CAD.0000000000000112. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi R.H., Bains O.S., Pfeifer T.A., Grigliatti T.A., Reid R.E., Riggs K.W. Aldo-keto reductase 1C2 fails to metabolize doxorubicin and daunorubicin in vitro. Drug Metab. Dispos. 2008;36(6):991–994. doi: 10.1124/dmd.108.020388. [DOI] [PubMed] [Google Scholar]
- 51.Bains O.S., Grigliatti T.A., Reid R.E., Riggs K.W. Naturally occurring variants of human aldo-keto reductases with reduced in vitro metabolism of daunorubicin and doxorubicin. J. Pharmacol. Exp. Ther. 2010;335(3):533–545. doi: 10.1124/jpet.110.173179. [DOI] [PubMed] [Google Scholar]
- 52.O'connor T., Ireland L.S., Harrison D.J., Hayes J.D. Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. Biochem. J. 1999;343(Pt 2):487–504. [PMC free article] [PubMed] [Google Scholar]
- 53.Ji Q., Aoyama C., Chen P.K., Stolz A., Liu P. Localization and altered expression of AKR1C family members in human ovarian tissues. Mol. Cell. Probes. 2005;19(4):261–266. doi: 10.1016/j.mcp.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Heibein A.D., Guo B., Sprowl J.A., Maclean D.A., Parissenti A.M. Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer. 2012;12:381. doi: 10.1186/1471-2407-12-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Veitch Z.W., Guo B., Hembruff S.L., Bewick A.J., Heibein A.D., Eng J., Cull S., Maclean D.A., Parissenti A.M. Induction of 1C aldoketoreductases and other drug dose-dependent genes upon acquisition of anthracycline resistance. Pharmacogenet. Genomics. 2009;19(6):477–488. doi: 10.1097/FPC.0b013e32832c484b. [DOI] [PubMed] [Google Scholar]
- 56.Wang G.X., Wang Y.X., Zhou X.B., Korth M. Effects of doxorubicinol on excitation--contraction coupling in guinea pig ventricular myocytes. Eur. J. Pharmacol. 2001;423(2-3):99–107. doi: 10.1016/s0014-2999(01)01096-2. [DOI] [PubMed] [Google Scholar]
- 57.Olson L.E., Bedja D., Alvey S.J., Cardounel A.J., Gabrielson K.L., Reeves R.H. Protection from doxorubicin-induced cardiac toxicity in mice with a null allele of carbonyl reductase 1. Cancer Res. 2003;63(20):6602–6606. [PubMed] [Google Scholar]
- 58.Forrest G.L., Gonzalez B., Tseng W., Li X., Mann J. Human carbonyl reductase overexpression in the heart advances the development of doxorubicin-induced cardiotoxicity in transgenic mice. Cancer Res. 2000;60(18):5158–5164. [PubMed] [Google Scholar]
- 59.Nagasaki K., Fleischer S. Modulation of the calcium release channel of sarcoplasmic reticulum by adriamycin and other drugs. Cell Calcium. 1989;10(1):63–70. doi: 10.1016/0143-4160(89)90045-6. [DOI] [PubMed] [Google Scholar]
- 60.Shadle S.E., Bammel B.P., Cusack B.J., Knighton R.A., Olson S.J., Mushlin P.S., Olson R.D. Daunorubicin cardiotoxicity: evidence for the importance of the quinone moiety in a free-radical-independent mechanism. Biochem. Pharmacol. 2000;60(10):1435–1444. doi: 10.1016/s0006-2952(00)00458-5. [DOI] [PubMed] [Google Scholar]
- 61.Blanco J.G., Sun C.L., Landier W., Chen L., Esparza-Duran D., Leisenring W., Mays A., Friedman D.L., Ginsberg J.P., Hudson M.M., Neglia J.P., Oeffinger K.C., Ritchey A.K., Villaluna D., Relling M.V., Bhatia S. Anthracycline-related cardiomyopathy after childhood cancer: role of polymorphisms in carbonyl reductase genes--a report from the Children's Oncology Group. J. Clin. Oncol. 2012;30(13):1415–1421. doi: 10.1200/JCO.2011.34.8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lubieniecka J.M., Liu J., Heffner D., Graham J., Reid R., Hogge D., Grigliatti T.A., Riggs W.K. Single-nucleotide polymorphisms in aldo-keto and carbonyl reductase genes are not associated with acute cardiotoxicity after daunorubicin chemotherapy. Cancer Epidemiol. Biomarkers Prev. 2012;21(11):2118–2120. doi: 10.1158/1055-9965.EPI-12-1037. [DOI] [PubMed] [Google Scholar]
- 63.Weiss M. Functional characterization of drug uptake and metabolism in the heart. Expert Opin. Drug Metab. Toxicol. 2011;7(10):1295–1306. doi: 10.1517/17425255.2011.614233. [DOI] [PubMed] [Google Scholar]
- 64.Salazar-Mendiguchia J., Gonzalez-Costello J., Roca J.riza-Sole A., Manito N., Cequier A. Anthracycline-mediated cardiomyopathy: basic molecular knowledge for the cardiologist. Arch. Cardiol. Mex. 2014;84(3):218–223. doi: 10.1016/j.acmx.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Schaupp C.M., White C.C., Merrill G.F., Kavanagh T.J. Metabolism of doxorubicin to the cardiotoxic metabolite doxorubicinol is increased in a mouse model of chronic glutathione deficiency: A potential role for carbonyl reductase 3. Chem. Biol. Interact. 2014 doi: 10.1016/j.cbi.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou F., Hao G., Zhang J., Zheng Y., Wu X., Hao K., Niu F., Luo D., Sun Y., Wu L., Ye W., Wang G. Protective effect of 23-hydroxybetulinic acid on doxorubicin-induced cardiotoxicity: a correlation with the inhibition of carbonyl reductase-mediated metabolism. Br. J. Pharmacol. 2014 doi: 10.1111/bph.12995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bartoszek A. Metabolic activation of adriamycin by NADPH-cytochrome P450 reductase; overview of its biological and biochemical effects. Acta Biochim. Pol. 2002;49(2):323–331. [PubMed] [Google Scholar]
- 68.Octavia Y., Tocchetti C.G., Gabrielson K.L., Janssens S., Crijns H.J., Moens A.L. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012;52(6):1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 69.Octavia Y., Brunner-La Rocca H.P., Moens A.L. NADPH oxidase-dependent oxidative stress in the failing heart: From pathogenic roles to therapeutic approach. Free Radic. Biol. Med. 2012;52(2):291–297. doi: 10.1016/j.freeradbiomed.2011.10.482. [DOI] [PubMed] [Google Scholar]
- 70.Bachur N.R. Anthracycline antibiotic pharmacology and metabolism. Cancer Treat. Rep. 1979;63(5):817–820. [PubMed] [Google Scholar]
- 71.Vasquez-Vivar J., Martasek P., Hogg N., Masters B.S., Pritchard K.A., Jr, Kalyanaraman B. Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry. 1997;36(38):11293–11297. doi: 10.1021/bi971475e. [DOI] [PubMed] [Google Scholar]
- 72.Nohl H., Gille L., Staniek K. The exogenous NADH dehydrogenase of heart mitochondria is the key enzyme responsible for selective cardiotoxicity of anthracyclines. Z. Naturforsch. C. 1998;53(3-4):279–285. doi: 10.1515/znc-1998-3-419. [DOI] [PubMed] [Google Scholar]
- 73.Chand V.K., Link B.K., Ritchie J.M., Shannon M., Wooldridge J.E. Neutropenia and febrile neutropenia in patients with Hodgkin's lymphoma treated with doxorubicin (Adriamycin), bleomycin, vinblastine and dacarbazine (ABVD) chemotherapy. Leuk. Lymphoma. 2006;47(4):657–663. doi: 10.1080/10428190500353430. [DOI] [PubMed] [Google Scholar]
- 74.Lefrak E.A., Pitha J., Rosenheim S., Gottlieb J.A. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32(2):302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 75.Ventura-Clapier R., Garnier A., Veksler V. Energy metabolism in heart failure. J. Physiol. 2004;555(Pt 1):1–13. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marin-Garcia J., Goldenthal M.J., Moe G.W. Abnormal cardiac and skeletal muscle mitochondrial function in pacing-induced cardiac failure. Cardiovasc. Res. 2001;52(1):103–110. doi: 10.1016/s0008-6363(01)00368-6. [DOI] [PubMed] [Google Scholar]
- 77.Steinherz L.J., Steinherz P.G., Tan C.T., Heller G., Murphy M.L. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA. 1991;266(12):1672–1677. [PubMed] [Google Scholar]
- 78.Nakamaru-Ogiso E., Han H., Matsuno-Yagi A., Keinan E., Sinha S.C., Yagi T., Ohnishi T. The ND2 subunit is labeled by a photoaffinity analogue of asimicin, a potent complex I inhibitor. FEBS Lett. 2010;584(5):883–888. doi: 10.1016/j.febslet.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yagi T., Matsuno-Yagi A. The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry. 2003;42(8):2266–2274. doi: 10.1021/bi027158b. [DOI] [PubMed] [Google Scholar]
- 80.Schulz R., Nava E., Moncada S. Induction and potential biological relevance of a Ca(2+)-independent nitric oxide synthase in the myocardium. Br. J. Pharmacol. 1992;105(3):575–580. doi: 10.1111/j.1476-5381.1992.tb09021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lowenstein C.J., Dinerman J.L., Snyder S.H. Nitric oxide: a physiologic messenger. Ann. Intern. Med. 1994;120(3):227–237. doi: 10.7326/0003-4819-120-3-199402010-00009. [DOI] [PubMed] [Google Scholar]
- 82.Minotti G., Di G.M. Microsomal iron-dependent NADPH oxidation: evidence for the involvement of membrane-bound nonheme iron in NADPH oxidation by rat heart microsomes. Arch. Biochem. Biophys. 1990;282(2):270–274. doi: 10.1016/0003-9861(90)90116-g. [DOI] [PubMed] [Google Scholar]
- 83.Giraldez R.R., Panda A., Zweier J.L. Endothelial dysfunction does not require loss of endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2000;278(6):H2020–H2027. doi: 10.1152/ajpheart.2000.278.6.H2020. [DOI] [PubMed] [Google Scholar]
- 84.Moncada S., Higgs A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993;329(27):2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 85.Kalivendi S.V., Kotamraju S., Zhao H., Joseph J., Kalyanaraman B. Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase. Effect of antiapoptotic antioxidants and calcium. J. Biol. Chem. 2001;276(50):47266–47276. doi: 10.1074/jbc.M106829200. [DOI] [PubMed] [Google Scholar]
- 86.Neilan T.G., Blake S.L., Ichinose F., Raher M.J., Buys E.S., Jassal D.S., Furutani E., Perez-Sanz T.M., Graveline A., Janssens S.P., Picard M.H., Scherrer-Crosbie M., Bloch K.D. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation. 2007;116(5):506–514. doi: 10.1161/CIRCULATIONAHA.106.652339. [DOI] [PubMed] [Google Scholar]
- 87.Duquaine D., Hirsch G.A., Chakrabarti A., Han Z., Kehrer C., Brook R., Joseph J., Schott A., Kalyanaraman B., Vasquez-Vivar J., Rajagopalan S. Rapid-onset endothelial dysfunction with adriamycin: evidence for a dysfunctional nitric oxide synthase. Vasc. Med. 2003;8(2):101–107. doi: 10.1191/1358863x03vm476oa. [DOI] [PubMed] [Google Scholar]
- 88.Moens A.L., Leyton-Mange J.S., Niu X., Yang R., Cingolani O., Arkenbout E.K., Champion H.C., Bedja D., Gabrielson K.L., Chen J., Xia Y., Hale A.B., Channon K.M., Halushka M.K., Barker N., Wuyts F.L., Kaminski P.M., Wolin M.S., Kass D.A., Barouch L.A. Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure-overload in mice lacking the beta3-adrenoreceptor. J. Mol. Cell. Cardiol. 2009;47(5):576–585. doi: 10.1016/j.yjmcc.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cole M.P., Chaiswing L., Oberley T.D., Edelmann S.E., Piascik M.T., Lin S.M., Kiningham K.K., St Clair D.K. The protective roles of nitric oxide and superoxide dismutase in adriamycin-induced cardiotoxicity. Cardiovasc. Res. 2006;69(1):186–197. doi: 10.1016/j.cardiores.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 90.Mukhopadhyay P., Rajesh M., Batkai S., Kashiwaya Y., Hasko G., Liaudet L., Szabo C., Pacher P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009;296(5):H1466–H1483. doi: 10.1152/ajpheart.00795.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu B., Li H., Qu H., Sun B. Nitric oxide synthase expressions in ADR-induced cardiomyopathy in rats. J. Biochem. Mol. Biol. 2006;39(6):759–765. doi: 10.5483/bmbrep.2006.39.6.759. [DOI] [PubMed] [Google Scholar]
- 92.Fu J., Yamamoto K., Guan Z.W., Kimura S., Iyanagi T. Human neuronal nitric oxide synthase can catalyze one-electron reduction of adriamycin: role of flavin domain. Arch. Biochem. Biophys. 2004;427(2):180–187. doi: 10.1016/j.abb.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 93.Gustafson D.L., Swanson J.D., Pritsos C.A. Role of xanthine oxidase in the potentiation of doxorubicin-induced cardiotoxicity by mitomycin C. Cancer Commun. 1991;3(9):299–304. doi: 10.3727/095535491820873038. [DOI] [PubMed] [Google Scholar]
- 94.Pawlowska J., Tarasiuk J., Wolf C.R., Paine M.J., Borowski E. Differential ability of cytostatics from anthraquinone group to generate free radicals in three enzymatic systems: NADH dehydrogenase, NADPH cytochrome P450 reductase, and xanthine oxidase. Oncol. Res. 2003;13(5):245–252. doi: 10.3727/096504003108748294. [DOI] [PubMed] [Google Scholar]
- 95. RefSeq NQO1 NAD(P)H dehydrogenase, quinone 1 [ Homo sapiens (human) ]. Hugo Genome Nomenclature Committee, 2008. 2008.
- 96.Gilleron M., Marechal X., Montaigne D., Franczak J., Neviere R., Lancel S. NADPH oxidases participate to doxorubicin-induced cardiac myocyte apoptosis. Biochem. Biophys. Res. Commun. 2009;388(4):727–731. doi: 10.1016/j.bbrc.2009.08.085. [DOI] [PubMed] [Google Scholar]
- 97.Zhao Y., McLaughlin D., Robinson E., Harvey A.P., Hookham M.B., Shah A.M., McDermott B.J., Grieve D.J. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010;70(22):9287–9297. doi: 10.1158/0008-5472.CAN-10-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kostrzewa-Nowak D., Paine M.J., Wolf C.R., Tarasiuk J. The role of bioreductive activation of doxorubicin in cytotoxic activity against leukaemia HL60-sensitive cell line and its multidrug-resistant sublines. Br. J. Cancer. 2005;93(1):89–97. doi: 10.1038/sj.bjc.6602639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cummings J., Allan L., Willmott N., Riley R., Workman P., Smyth J.F. The enzymology of doxorubicin quinone reduction in tumour tissue. Biochem. Pharmacol. 1992;44(11):2175–2183. doi: 10.1016/0006-2952(92)90344-i. [DOI] [PubMed] [Google Scholar]
- 100.Williams C.H. KAMIN, H. Microsomal triphosphopyridine nucleotide-cytochrome c reductase of liver. J. Biol. Chem. 1962;237:587–595. [PubMed] [Google Scholar]
- 101.Lu A.Y., Junk K.W., Coon M.J. Resolution of the cytochrome P-450-containing omega-hydroxylation system of liver microsomes into three components. J. Biol. Chem. 1969;244(13):3714–3721. [PubMed] [Google Scholar]
- 102.Gu J., Weng Y., Zhang Q.Y., Cui H., Behr M., Wu L., Yang W., Zhang L., Ding X. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J. Biol. Chem. 2003;278(28):25895–25901. doi: 10.1074/jbc.M303125200. [DOI] [PubMed] [Google Scholar]
- 103.Bartoszek A., Wolf C.R. Enhancement of doxorubicin toxicity following activation by NADPH cytochrome P450 reductase. Biochem. Pharmacol. 1992;43(7):1449–1457. doi: 10.1016/0006-2952(92)90201-s. [DOI] [PubMed] [Google Scholar]
- 104.Tempczyk A., Tarasiuk J., Ossowski T., Borowski E. An alternative concept for the molecular nature of the peroxidating ability of anthracycline anti-tumor antibiotics and anthracenodiones. Anticancer Drug Des. 1988;2(4):371–385. [PubMed] [Google Scholar]
- 105.Doroshow J.H., Davies K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. 1986;261(7):3068–3074. [PubMed] [Google Scholar]
- 106.Alegria A.E., Samuni A., Mitchell J.B., Riesz P., Russo A. Free radicals induced by adriamycin-sensitive and adriamycin-resistant cells: a spin-trapping study. Biochemistry. 1989;28(21):8653–8658. doi: 10.1021/bi00447a056. [DOI] [PubMed] [Google Scholar]
- 107.Matheson I.B., Etheridge R.D., Kratowich N.R., Lee J. The quenching of singlet oxygen by amino acids and proteins. Photochem. Photobiol. 1975;21(3):165–171. doi: 10.1111/j.1751-1097.1975.tb06647.x. [DOI] [PubMed] [Google Scholar]
- 108.Niitsu N., Kasukabe T., Yokoyama A., Okabe-Kado J., Yamamoto-Yamaguchi Y., Umeda M., Honma Y. Anticancer derivative of butyric acid (Pivalyloxymethyl butyrate) specifically potentiates the cytotoxicity of doxorubicin and daunorubicin through the suppression of microsomal glycosidic activity. Mol. Pharmacol. 2000;58(1):27–36. doi: 10.1124/mol.58.1.27. [DOI] [PubMed] [Google Scholar]
- 109.Finn N.A., Findley H.W., Kemp M.L. A switching mechanism in doxorubicin bioactivation can be exploited to control doxorubicin toxicity. PLOS Comput. Biol. 2011;7(9):e1002151. doi: 10.1371/journal.pcbi.1002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gille L., Nohl H. Analyses of the molecular mechanism of adriamycin-induced cardiotoxicity. Free Radic. Biol. Med. 1997;23(5):775–782. doi: 10.1016/s0891-5849(97)00025-7. [DOI] [PubMed] [Google Scholar]
- 111.Forkert P.G., Lord J.A., Parkinson A. Alterations in expression of CYP1A1 and NADPH-cytochrome P450 reductase during lung tumor development in SWR/J mice. Carcinogenesis. 1996;17(1):127–132. doi: 10.1093/carcin/17.1.127. [DOI] [PubMed] [Google Scholar]
- 112.Corradi F., Paolini L., De C.R. Ranolazine in the prevention of anthracycline cardiotoxicity. Pharmacol. Res. 2014;79:88–102. doi: 10.1016/j.phrs.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 113.Jaiswal A.K., Burnett P., Adesnik M., McBride O.W. Nucleotide and deduced amino acid sequence of a human cDNA (NQO2) corresponding to a second member of the NAD(P)H:quinone oxidoreductase gene family. Extensive polymorphism at the NQO2 gene locus on chromosome 6. Biochemistry. 1990;29(7):1899–1906. doi: 10.1021/bi00459a034. [DOI] [PubMed] [Google Scholar]
- 114.Asher G., Lotem J., Kama R., Sachs L., Shaul Y. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. USA. 2002;99(5):3099–3104. doi: 10.1073/pnas.052706799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Asher G., Lotem J., Sachs L., Kahana C., Shaul Y. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc. Natl. Acad. Sci. USA. 2002;99(20):13125–13130. doi: 10.1073/pnas.202480499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Siegel D., Gustafson D.L., Dehn D.L., Han J.Y., Boonchoong P., Berliner L.J., Ross D. NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol. Pharmacol. 2004;65(5):1238–1247. doi: 10.1124/mol.65.5.1238. [DOI] [PubMed] [Google Scholar]
- 117.Liao S., Williams-ashman H.G. Enzymatic oxidation of some non-phosphorylated derivatives of dihydronicotinamide. Biochem. Biophys. Res. Commun. 1961;4:208–213. doi: 10.1016/0006-291x(61)90272-8. [DOI] [PubMed] [Google Scholar]
- 118.Brown J.M., Giaccia A.J. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408–1416. [PubMed] [Google Scholar]
- 119.Zhu H., Li Y. NAD(P)H: quinone oxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovasc. Toxicol. 2012;12(1):39–45. doi: 10.1007/s12012-011-9136-9. [DOI] [PubMed] [Google Scholar]
- 120.Nioi P., Hayes J.D. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. 2004;555(1-2):149–171. doi: 10.1016/j.mrfmmm.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 121.Itoh K., Mimura J., Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid. Redox Signal. 2010;13(11):1665–1678. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- 122.Chian S., Li Y.Y., Wang X.J., Tang X.W. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the Nrf2 pathway. Asian Pac. J. Cancer Prev. 2014;15(6):2911–2916. doi: 10.7314/apjcp.2014.15.6.2911. [DOI] [PubMed] [Google Scholar]
- 123.Zhong Y., Zhang F., Sun Z., Zhou W., Li Z.Y., You Q.D., Guo Q.L., Hu R. Drug resistance associates with activation of Nrf2 in MCF-7/DOX cells, and wogonin reverses it by down-regulating Nrf2-mediated cellular defense response. Mol. Carcinog. 2013;52(10):824–834. doi: 10.1002/mc.21921. [DOI] [PubMed] [Google Scholar]
- 124.Zeekpudsa P., Kukongviriyapan V., Senggunprai L., Sripa B., Prawan A. Suppression of NAD(P)H-quinone oxidoreductase 1 enhanced the susceptibility of cholangiocarcinoma cells to chemotherapeutic agents. J. Exp. Clin. Cancer Res. 2014;33:11. doi: 10.1186/1756-9966-33-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jirkovsky E., Popelova O., Krivakova-Stankova P., Vavrova A., Hroch M., Haskova P., Brcakova-Dolezelova E., Micuda S., Adamcova M., Simunek T., Cervinkova Z., Gersl V., Sterba M. Chronic anthracycline cardiotoxicity: molecular and functional analysis with focus on nuclear factor erythroid 2-related factor 2 and mitochondrial biogenesis pathways. J. Pharmacol. Exp. Ther. 2012;343(2):468–478. doi: 10.1124/jpet.112.198358. [DOI] [PubMed] [Google Scholar]
- 126.Siegel D., Anwar A., Winski S.L., Kepa J.K., Zolman K.L., Ross D. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:quinone oxidoreductase 1. Mol. Pharmacol. 2001;59(2):263–268. doi: 10.1124/mol.59.2.263. [DOI] [PubMed] [Google Scholar]
- 127.Geng R., Chen Z., Zhao X., Qiu L., Liu X., Liu R., Guo W., He G., Li J., Zhu X. Oxidative stress-related genetic polymorphisms are associated with the prognosis of metastatic gastric cancer patients treated with epirubicin, oxaliplatin and 5-fluorouracil combination chemotherapy. PLoS One. 2014;9(12):e116027. doi: 10.1371/journal.pone.0116027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kuehl B.L., Paterson J.W., Peacock J.W., Paterson M.C., Rauth A.M. Presence of a heterozygous substitution and its relationship to DT-diaphorase activity. Br. J. Cancer. 1995;72(3):555–561. doi: 10.1038/bjc.1995.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Blanco J.G., Leisenring W.M., Gonzalez-Covarrubias V.M., Kawashima T.I., Davies S.M., Relling M.V., Robison L.L., Sklar C.A., Stovall M., Bhatia S. Genetic polymorphisms in the carbonyl reductase 3 gene CBR3 and the NAD(P)H:quinone oxidoreductase 1 gene NQO1 in patients who developed anthracycline-related congestive heart failure after childhood cancer. Cancer. 2008;112(12):2789–2795. doi: 10.1002/cncr.23534. [DOI] [PubMed] [Google Scholar]
- 130.Lajin B., Alachkar A. The NQO1 polymorphism C609T (Pro187Ser) and cancer susceptibility: a comprehensive meta-analysis. Br. J. Cancer. 2013;109(5):1325–1337. doi: 10.1038/bjc.2013.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hubackova M., Vaclavikova R., Ehrlichova M., Mrhalova M., Kodet R., Kubackova K., Vrana D., Gut I., Soucek P. Association of superoxide dismutases and NAD(P)H quinone oxidoreductases with prognosis of patients with breast carcinomas. Int. J. Cancer. 2012;130(2):338–348. doi: 10.1002/ijc.26006. [DOI] [PubMed] [Google Scholar]
- 132.Lagoa R., Ganan C., Lopez-Sanchez C., Garcia-Martinez V., Gutierrez-Merino C. The decrease of NAD(P)H:quinone oxidoreductase 1 activity and increase of ROS production by NADPH oxidases are early biomarkers in doxorubicin cardiotoxicity. Biomarkers. 2014;19(2):142–153. doi: 10.3109/1354750X.2014.885084. [DOI] [PubMed] [Google Scholar]
- 133.Arinc E. The role of polymorphic cytochrome P450 enzymes in drug design, development and drug interactions with a special emphasis on phenotyping. J. Mol. Catal., B Enzym. 2010;64:120–122. [Google Scholar]
- 134.Schroterova L., Kaiserova H., Baliharova V., Velik J., Gersl V., Kvasnickova E. The effect of new lipophilic chelators on the activities of cytosolic reductases and P450 cytochromes involved in the metabolism of anthracycline antibiotics: studies in vitro. Physiol. Res. 2004;53(6):683–691. [PubMed] [Google Scholar]
- 135.Azim H.A., Jr, Colozza M., Bines J., Piccart M. J. Long-term toxic effects of adjuvant chemotherapy in breast cancer. Ann. Oncol. 2011;22(9):1939–1947. doi: 10.1093/annonc/mdq683. [DOI] [PubMed] [Google Scholar]
- 136.van Dalen E.C., van der Pal H.J., Caron H.N., Kremer L.C. Different dosage schedules for reducing cardiotoxicity in cancer patients receiving anthracycline chemotherapy. Cochrane Database Syst. Rev. 2009;(4):CD005008. doi: 10.1002/14651858.CD005008.pub2. [DOI] [PubMed] [Google Scholar]
- 137.Rafiyath S.M., Rasul M., Lee B., Wei G., Lamba G., Liu D. Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: a meta-analysis. Exp. Hematol. Oncol. 2012;1(1):10. doi: 10.1186/2162-3619-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Galetta F., Franzoni F., Cervetti G., Regoli F., Fallahi P., Tocchini L., Carpi A., Antonelli A., Petrini M., Santoro G. In vitro and in vivo study on the antioxidant activity of dexrazoxane. Biomed. Pharmacother. 2010;64(4):259–263. doi: 10.1016/j.biopha.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 139.Viswanatha Swamy A.H., Wangikar U., Koti B.C., Thippeswamy A.H., Ronad P.M., Manjula D.V. Cardioprotective effect of ascorbic acid on doxorubicin-induced myocardial toxicity in rats. Indian J. Pharmacol. 2011;43(5):507–511. doi: 10.4103/0253-7613.84952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jay S.M., Murthy A.C., Hawkins J.F., Wortzel J.R., Steinhauser M.L., Alvarez L.M., Gannon J., Macrae C.A., Griffith L.G., Lee R.T. An engineered bivalent neuregulin protects against doxorubicin-induced cardiotoxicity with reduced proneoplastic potential. Circulation. 2013;128(2):152–161. doi: 10.1161/CIRCULATIONAHA.113.002203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Marty M., Espie M., Llombart A., Monnier A., Rapoport B.L., Stahalova V. Multicenter randomized phase III study of the cardioprotective effect of dexrazoxane (Cardioxane) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapy. Ann. Oncol. 2006;17(4):614–622. doi: 10.1093/annonc/mdj134. [DOI] [PubMed] [Google Scholar]
- 142.Bruynzeel A.M., Niessen H.W., Bronzwaer J.G., van der Hoeven J.J., Berkhof J., Bast A., Groeningen C. J. The effect of monohydroxyethylrutoside on doxorubicin-induced cardiotoxicity in patients treated for metastatic cancer in a phase II study. Br. J. Cancer. 2007;97(8):1084–1089. doi: 10.1038/sj.bjc.6603994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jacobs H., Bast A., Peters G.J.van, d., Haenen G. R. The semisynthetic flavonoid monoHER sensitises human soft tissue sarcoma cells to doxorubicin-induced apoptosis via inhibition of nuclear factor-kappaB. Br. J. Cancer. 2011;104(3):437–440. doi: 10.1038/sj.bjc.6606065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Minotti G., Cairo G., Monti E. Role of iron in anthracycline cardiotoxicity: new tunes for an old song? FASEB J. 1999;13(2):199–212. [PubMed] [Google Scholar]
- 145.Gammella E., Maccarinelli F., Buratti P., Recalcati S., Cairo G. The role of iron in anthracycline cardiotoxicity. Front. Pharmacol. 2014;5:25. doi: 10.3389/fphar.2014.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ichikawa Y., Ghanefar M., Bayeva M., Wu R., Khechaduri A., Naga Prasad S.V., Mutharasan R.K., Naik T.J., Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 2014;124(2):617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Thomas C.E., Aust S.D. Release of iron from ferritin by cardiotoxic anthracycline antibiotics. Arch. Biochem. Biophys. 1986;248(2):684–689. doi: 10.1016/0003-9861(86)90523-0. [DOI] [PubMed] [Google Scholar]
- 148.Winterbourn C.C., Vile G.F., Monteiro H.P. Ferritin, lipid peroxidation and redox-cycling xenobiotics. Free Radic. Res. Commun. 1991;12-13(Pt 1):107–114. doi: 10.3109/10715769109145774. [DOI] [PubMed] [Google Scholar]
- 149.Zhang S., Liu X., Bawa-Khalfe T., Lu L.S., Lyu Y.L., Liu L.F., Yeh E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012;18(11):1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 150.Sepe D.M., Ginsberg J.P., Balis F.M. Dexrazoxane as a cardioprotectant in children receiving anthracyclines. Oncologist. 2010;15(11):1220–1226. doi: 10.1634/theoncologist.2010-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Langer S.W. Dexrazoxane for the treatment of chemotherapy-related side effects. Cancer Manag. Res. 2014;6:357–363. doi: 10.2147/CMAR.S47238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Limat S., Daguindau E., Cahn J.Y., Nerich V., Brion A., Perrin S., Woronoff-Lemsi M.C., Deconinck E. Incidence and risk-factors of CHOP/R-CHOP-related cardiotoxicity in patients with aggressive non-Hodgkin's lymphoma. J. Clin. Pharm. Ther. 2014;39(2):168–174. doi: 10.1111/jcpt.12124. [DOI] [PubMed] [Google Scholar]
- 153.Seicean S., Seicean A., Plana J.C., Budd G.T., Marwick T.H. Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational clinical cohort study. J. Am. Coll. Cardiol. 2012;60(23):2384–2390. doi: 10.1016/j.jacc.2012.07.067. [DOI] [PubMed] [Google Scholar]
- 154.Georgakopoulos P., Roussou P., Matsakas E., Karavidas A., Anagnostopoulos N., Marinakis T., Galanopoulos A., Georgiakodis F., Zimeras S., Kyriakidis M., Ahimastos A. Cardioprotective effect of metoprolol and enalapril in doxorubicin-treated lymphoma patients: a prospective, parallel-group, randomized, controlled study with 36-month follow-up. Am. J. Hematol. 2010;85(11):894–896. doi: 10.1002/ajh.21840. [DOI] [PubMed] [Google Scholar]
- 155.Hammoud R., Vaccari C.S., Khan B.V. Letter by Hammoud et al regarding article “Prevention of high-dose chemotherapy-induced cardiotoxicity in high-risk patients by angiotensin-converting enzyme inhibition”. Circulation. 2007;115(24):e637. doi: 10.1161/CIRCULATIONAHA.107.690362. [DOI] [PubMed] [Google Scholar]
- 156.Silber J.H., Cnaan A., Clark B.J., Paridon S.M., Chin A.J., Rychik J., Hogarty A.N., Cohen M.I., Barber G., Rutkowsky M., Kimball T.R., Delaat C., Steinherz L.J., Zhao H., Tartaglione M.R. Design and baseline characteristics for the ACE Inhibitor After Anthracycline (AAA) study of cardiac dysfunction in long-term pediatric cancer survivors. Am. Heart J. 2001;142(4):577–585. doi: 10.1067/mhj.2001.118115. [DOI] [PubMed] [Google Scholar]
- 157.Kalam K., Marwick T.H. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: a systematic review and meta-analysis. Eur. J. Cancer. 2013;49(13):2900–2909. doi: 10.1016/j.ejca.2013.04.030. [DOI] [PubMed] [Google Scholar]
- 158.Fogli S., Nieri P., Breschi M.C. The role of nitric oxide in anthracycline toxicity and prospects for pharmacologic prevention of cardiac damage. FASEB J. 2004;18(6):664–675. doi: 10.1096/fj.03-0724rev. [DOI] [PubMed] [Google Scholar]
- 159.Henninger C., Huelsenbeck S., Wenzel P., Brand M., Huelsenbeck J., Schad A., Fritz G. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacol. Res. 2015;91:47–56. doi: 10.1016/j.phrs.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 160.Sacco G., Mario B., Lopez G., Evangelista S., Manzini S., Maggi C.A. ACE inhibition and protection from doxorubicin-induced cardiotoxicity in the rat. Vascul. Pharmacol. 2009;50(5-6):166–170. doi: 10.1016/j.vph.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 161.Murdter T.E., Sperker B., Kivisto K.T., McClellan M., Fritz P., Friedel G., Linder A., Bosslet K., Toomes H., Dierkesmann R., Kroemer H.K. Enhanced uptake of doxorubicin into bronchial carcinoma: beta-glucuronidase mediates release of doxorubicin from a glucuronide prodrug (HMR 1826) at the tumor site. Cancer Res. 1997;57(12):2440–2445. [PubMed] [Google Scholar]
- 162.Houba P.H., Boven E., van der Meulen-Muileman I.H., Leenders R.G., Scheeren J.W., Pinedo H.M., Haisma H.J. A novel doxorubicin-glucuronide prodrug DOX-GA3 for tumour-selective chemotherapy: distribution and efficacy in experimental human ovarian cancer. Br. J. Cancer. 2001;84(4):550–557. doi: 10.1054/bjoc.2000.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Weyel D., Sedlacek H.H., Muller R., Brusselbach S. Secreted human beta-glucuronidase: a novel tool for gene-directed enzyme prodrug therapy. Gene Ther. 2000;7(3):224–231. doi: 10.1038/sj.gt.3301072. [DOI] [PubMed] [Google Scholar]
- 164.Houba P.H., Boven E., van der Meulen-Muileman I.H., Leenders R.G., Scheeren J.W., Pinedo H.M., Haisma H.J. Pronounced antitumor efficacy of doxorubicin when given as the prodrug DOX-GA3 in combination with a monoclonal antibody beta-glucuronidase conjugate. Int. J. Cancer. 2001;91(4):550–554. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1075>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 165.Pinedo G. M., Oosterhoff D., van der Meulen-Muilema IH, Gerritsen W. R., Haisma H. J., Boven E., de Pronounced antitumor efficacy by extracellular activation of a doxorubicin-glucuronide prodrug after adenoviral vector-mediated expression of a human antibody-enzyme fusion protein. Hum. Gene Ther. 2004;15(3):229–238. doi: 10.1089/104303404322886084. [DOI] [PubMed] [Google Scholar]
- 166.Tzou S.C., Roffler S., Chuang K.H., Yeh H.P., Kao C.H., Su Y.C., Cheng C.M., Tseng W.L., Shiea J., Harm I.H., Cheng K.W., Chen B.M., Hwang J.J., Cheng T.L., Wang H.E. Micro-PET imaging of beta-glucuronidase activity by the hydrophobic conversion of a glucuronide probe. Radiology. 2009;252(3):754–762. doi: 10.1148/radiol.2523082055. [DOI] [PubMed] [Google Scholar]
- 167.Wang I.C., Tai L.A., Lee D.D., Kanakamma P.P., Shen C.K., Luh T.Y., Cheng C.H., Hwang K.C. (60) and water-soluble fullerene derivatives as antioxidants against radical-initiated lipid peroxidation. J. Med. Chem. 1999;42(22):4614–4620. doi: 10.1021/jm990144s. [DOI] [PubMed] [Google Scholar]
- 168.Torres V.M., Srdjenovic B., Jacevic V., Simic V.D., Djordjevic A., Simplicio A.L. Fullerenol C60(OH)24 prevents doxorubicin-induced acute cardiotoxicity in rats. Pharmacol. Rep. 2010;62(4):707–718. doi: 10.1016/s1734-1140(10)70328-5. [DOI] [PubMed] [Google Scholar]
- 169.Rabelo E., De A.K., Bock P., Gatelli F.T., Cervo F., Bello K.A., Clausell N., Claudia I.M. Baroreflex sensitivity and oxidative stress in adriamycin-induced heart failure. Hypertension. 2001;38(3 Pt 2):576–580. doi: 10.1161/hy09t1.096185. [DOI] [PubMed] [Google Scholar]
- 170.Injac R., Boskovic M., Perse M., Koprivec-Furlan E., Cerar A., Djordjevic A., Strukelj B. Acute doxorubicin nephrotoxicity in rats with malignant neoplasm can be successfully treated with fullerenol C60(OH)24 via suppression of oxidative stress. Pharmacol. Rep. 2008;60(5):742–749. [PubMed] [Google Scholar]
- 171.Injac R., Radic N., Govedarica B., Perse M., Cerar A., Djordjevic A., Strukelj B. Acute doxorubicin pulmotoxicity in rats with malignant neoplasm is effectively treated with fullerenol C60(OH)24 through inhibition of oxidative stress. Pharmacol. Rep. 2009;61(2):335–342. doi: 10.1016/s1734-1140(09)70041-6. [DOI] [PubMed] [Google Scholar]
- 172.Injac R., Perse M., Cerne M., Potocnik N., Radic N., Govedarica B., Djordjevic A., Cerar A., Strukelj B. Protective effects of fullerenol C60(OH)24 against doxorubicin-induced cardiotoxicity and hepatotoxicity in rats with colorectal cancer. Biomaterials. 2009;30(6):1184–1196. doi: 10.1016/j.biomaterials.2008.10.060. [DOI] [PubMed] [Google Scholar]
- 173.Mita M.M., Natale R.B., Wolin E.M., Laabs B., Dinh H., Wieland S., Levitt D.J., Mita A.C. Pharmacokinetic study of aldoxorubicin in patients with solid tumors. Invest. New Drugs. 2015;33(2):341–348. doi: 10.1007/s10637-014-0183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Holstein S.A., Bigelow J.C., Olson R.D., Vestal R.E., Walsh G.M., Hohl R.J. Phase I and pharmacokinetic study of the novel anthracycline derivative 5-imino-13-deoxydoxorubicin (GPX-150) in patients with advanced solid tumors. Invest. New Drugs. 2015;33(3):594–602. doi: 10.1007/s10637-015-0220-z. [DOI] [PubMed] [Google Scholar]
- 175.Hofmann P.A., Israel M., Koseki Y., Laskin J., Gray J., Janik A., Sweatman T.W., Lothstein L. N-Benzyladriamycin-14-valerate (AD 198): a non-cardiotoxic anthracycline that is cardioprotective through PKC-epsilon activation. J. Pharmacol. Exp. Ther. 2007;323(2):658–664. doi: 10.1124/jpet.107.126110. [DOI] [PubMed] [Google Scholar]