

Abstract
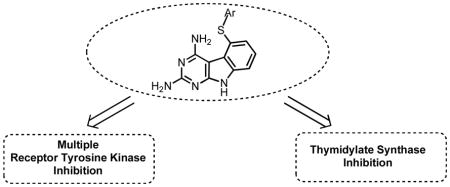
In an effort to optimize the structural requirements for combined cytostatic and cytotoxic effects in single agents, a series of 5-(arylthio)-9H-pyrimido[4,5-b]indole-2,4-diamines 3-7 were synthesized and evaluated as inhibitors of receptor tyrosine kinases (RTKs) as well as thymidylate synthase (TS). The synthesis of these compounds involved the nucleophilic displacement of the common intermediate 5-bromo/5-chloro-9H-pyrimido[4,5-b]indole-2,4-diamine with appropriate aryl thiols. A novel four step synthetic scheme to the common intermediate was developed which is more efficient relative to the previously reported six-step sequence. Biological evaluation of these compounds indicated dual activity in RTKs and human TS (hTS). In the VEGFR-2 assay, compound 5 was equipotent to the standard compound semaxanib and was better than standard TS inhibitor pemetrexed, in the hTS assay. Compounds 3, 6 and 7 were nanomolar inhibitors of hTS and were several fold better than pemetrexed.
Keywords: Multiple receptor Tyrosine kinase inhibitors; Antiangiogenic agents; Pyrimido[4,5-b]indole synthesis; Thymidylate synthase Inhibitors; Combination chemotherapeutic potential in single agents
Graphical Abstract
Angiogenesis – the process of formation of new blood vessels from existing vasculature – is essential for tumor growth and metastasis.1 For angiogenesis, receptor tyrosine kinases (RTKs) play a crucial role.2 RTKs are enzymes that catalyze the transfer of the γ-phosphate of ATP to tyrosine residues of protein substrates. RTK families that are overexpressed in cancer cells include vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR) and platelet derived growth factor receptor (PDGFR) among others.3,4 Agents that circumvent angiogenesis by inhibition of RTKs have established a new paradigm in cancer chemotherapy. RTK inhibitors that function by inhibition of a single RTK are prone to resistance by numerous mechanisms including point mutations in the ATP binding site and upregulation of additional RTKs.5 Consequently multi-RTK inhibition in cancer chemotherapy has emerged as a promising approach and its validity has been highlighted by the approval of several multi-RTK inhibitors including sorafenib, sunitinib, afatinib, cabozanitib and nintedanib (Figure 1). The enzyme thymidylate synthase (TS) utilizes 5,10-methylenetetrahydrofolate as a cofactor to transfer a methyl group to dUMP (deoxyuridine monophosphate).6,7 Because of its vital role in DNA synthesis and cell growth, TS is a viable target for several clinically used cancer chemotherapeutic agents and also for developing anti-opportunistic infection agents.8,9 The fluoropyrimidine, 5-fluorouracil (5-FU) and its derivatives, in particular, capecitabine (Figure 1), have found extensive utility in ovarian, breast, colon, and several other cancers alone and in combinations and are a mainstay in cancer chemotherapy.10,11 TS inhibitors that are antifolates are used clinically, alone or in combination, in a variety of cancers. These include pemetrexed (PMX)12 and in Europe raltitrexed (RTX) (Figure 1).
Figure 1.

Known anticancer agents
Combination cancer chemotherapy is not a novel concept. Recent studies suggest that the combination of separate cytostatic antiangiogenic agents with separate cytotoxic agents is more effective in cancer chemotherapy than either agent alone.13–20 We21 designed single agents that would function by both a cytostatic (antiangiogenic) mechanism and a cytotoxic (antifolate) mechanism. Such single agents are anticipated to circumvent the pharmacokinetic problems of two or more agents and reduce drug-drug interactions. In addition, such agents could be used at lower doses to alleviate toxicity, would be devoid of overlapping toxicities, and delay or prevent tumor cell resistance due to inhibition of multiple mechanisms. Most significantly, providing the cytotoxic agent, by structural design, in the same molecule allows the cytotoxicity to be mainfested as soon as the antiangiogenic effects are operable. A separately dosed cytotoxic agent may miss the timing window and hence circumvent the intent of the combination. Such multi-targeted agents could wield their cytotoxic action as soon as or even during transient tumor vasculature normalization22,23 due to the antiangiogenic effects. Thus such agents, perhaps, do not need to be as potent as conventional, separately dosed cytotoxic agents. Dosing of such an antiangiogenic multi-targeted RTK inhibitor with a built-in cytotoxic mechanism would be equivalent to providing a combination of multi-targeted RTK inhibitors along with a metronomic dosing of a cytotoxic agent. Thus these single agents would be in keeping with the two important mechanisms that suggest the rationale for the success of separate antiangiogenic and cytotoxic chemotherapeutic agents in combination for cancer chemotherapy.22–26 Other advantages of such single agents are in the decreased cost and increased patient compliance which are sometimes as important contributors to chemotherapy failure as resistance, toxicity and lack of efficacy.
The antiangiogenic component is typically targeted to tumor cells and is, under most circumstances, not targeted to normal cells. In contrast, the cytotoxic component is targeted to the tumor cells but not exclusively. Thus the challenge in designing single agents with a cytotoxic component is that the cytotoxic component should be potent enough to kill tumor cells that have been compromised via the antiangiogenic effect but not potent enough to induce serious toxicity to normal cells unaffected by the antiangiogenic effect. One of the most important problems with conventional cytotoxic chemotherapeutic agents is dose limiting toxicities. These single agents should avoid these toxicities as they do not need to be as potent as conventional chemotherapeutic agents.
On the basis of this rationale we21 previously designed, synthesized and evaluated two compounds – 1 and 2 (Figure 1) that each inhibit VEGFR-2 and PDGFR-β for antiangiogenic effects and also inhibit human TS (hTS) for cytotoxic effects in single agents. The inhibitory potency of both these single agents against VEGFR-2, PDGFR-β, and hTS is better than or close to the standard agents used in these assays (Tables 1, 2). In a COLO-205 xenograft mouse model, compound 1 significantly decreased tumor growth (tumor growth inhibition (TGI) = 76% at 35 mg/kg), liver metastases, and tumor blood vessels compared with a standard drug DMBI (Figure 3) and control and thus demonstrated potent tumor growth inhibition, inhibition of metastasis, and antiangiogenic effects in vivo. These compounds afford combination chemotherapeutic potential in single agents.
Table 1.
IC50 Values (μM) of Kinase Inhibition and A431 Cytotoxicity Assay
| Compd # | VEGFR-2 (Flk-1) Kinase Inhibition | EGFR Kinase Inhibition | PDGFR-β Kinase Inhibition | A431 Cytotoxicity |
|---|---|---|---|---|
| 1 | 22.6±4.5 | 15.07±3.1 | 2.8±0.42 | 49.2±4.7 |
| 2 | 56.3±7.1 | 10.41±1.2 | 40.3±5.1 | 14.1±2.0 |
| 3 | 160.3±19.1 | 20.0±3.6 | 90.1±10.2 | 43.1±7.0 |
| 4 | 193.1±22.6 | 167.3±20.1 | 58.2±8.1 | >200 |
| 5 | 10.2 ±2.6 | 4.3±0.62 | >200 | 38.4±5.2 |
| 6 | 182.2±34.2 | 20.8±3.1 | 60.5±7.2 | 67.3±7.2 |
| 7 | >500 | 8.3±1.9 | >200 | 10.3±2.1 |
| Semaxanib | 12.0±2.7 | |||
| CB67645 (24) | 0.2±0.004 | |||
| DMBI | 3.7±0.06 | |||
| Cisplatin | 10.6±2.9 |
Table 2.
Inhibitory concentrations (IC50, μ M) against isolated human and E. coli TS and DHFR
| Compd | TS Inhibitory Activitya | DHFR Inhibitory Activitya | ||
|---|---|---|---|---|
| Humanc | E. colic | Humand | E. colie | |
| 1 | 0.54 | >27 | >33 (17)b | > 33 (35) |
| 2 | 0.39 | >26 | >31 (7) | > 31 (27) |
| 3 | 0.12 | >2.3 (0) | >28 (18) | 28 |
| 4 | 2.3 | >2.3 (20) | >27 (25) | 27 |
| 5 | 1.1 | 2.1 | >27 (0) | >27 (0) |
| 6 | 0.14 | >2.5 (0) | >30 (13) | 31 |
| 7 | 0.48 | >2.4 (13) | >29 (20) | >29 (0) |
| Raltitrexedf | 0.38 | 5.7 | ||
| Pemetrexedg | 9.5 | 76 | ||
| Methotrexate | 0.022 | 0.0066 | ||
| Trimethoprim | 680 | 0.02 | ||
Thymidylate synthase (TS) assay: TS was assayed spectrophotometrically at 30 °C and pH 7.4 in a mixture containing 0.1 M 2-mercaptoethanol, 0.0003 M (6R,S)- tetrahydrofolate, 0.012 M formaldehyde, 0.02 M MgCl2, 0.001 M dUMP, 0.04 M Tris–HCl, and 0.00075 M NaEDTA. This was the assay described by Wahba and Friedkin,44 except that the dUMP concentration was increased 25-fold according to the method of Davisson et al.45 The reaction was initiated by the addition of an amount of enzyme yielding a change in absorbance at 340 nm of 0.016/min in the absence of inhibitor.
Dihydrofolate reductase (DHFR) assay:46 All enzymes were assayed spectrophotometrically in a solution containing 50 μM dihydrofolate, 80 μM NADPH, 0.05 M Tris–HCl, 0.001 M 2-mercaptoethanol, and 0.001 M EDTA at pH 7.4 and 30 °C. The reaction was initiated with an amount of enzyme yielding a change in OD at 340 nm of 0.015/min.
The percent inhibition was determined at a minimum of four inhibitor concentrations within 20% of the 50% point. The standard deviations for determination of 50% points are within ±10% of the value given.
Numbers in parentheses indicate the percent inhibition at the stated concentration.
Kindly provided by Dr. Frank Maley, New York State Department of Health, Albany, NY.
Kindly provided by Dr. J. H. Freisheim, Medical College of Ohio, Toledo, OH.
Kindly provided by Dr. R. L. Blakley, St. Jude Children’s hospital, Memphis TN.
Kindly provided by Dr. Ann Jackman, Institute of Cancer Research, Sutton, Surrey, UK.
Kindly provided by Dr. Chuan Shih, Eli Lilly and Co.
Figure 3.

Standard drugs and control agents
In order to determine a structure activity relationship (SAR) and to optimize 1 and 2, as an initial step, compounds 3-7 were designed with variations in the substitution at the 5-thioaryl portion of the 2,4-diamino pyrimido[4,5-b]indole scaffold. The 2′-naphthyl (3), 1′-naphthyl (4), and 2′,5′-diOMe phenyl (5) substitutions are based on previously known potent RTK inhibitors compounds 8 (VEGFR-1 assay), 9 (chorioallantoic membrane (CAM) assay – a test for angiogenesis), and 10 (VEGFR-2, PDGFR-β assays) respectively.27 The 4′-OMe phenyl (6) was proposed to determine the effect of an electron donating substituent on activity. The 4′-Cl substituted phenyl in 7 was selected on the basis of the multiRTK inhibitory activity (VEGFR-2, VEGFR-1) of vatalanib28 which has a similar substitution pattern.
The synthesis of 1 and 2 involved the nucleophilic displacement of the common intermediate 5-chloro-9H-pyrimido[4,5-b]indole-2,4-diamine 14 (Scheme 1) with appropriate benzene thiols.21 We elected to develop a shorter synthesis than the six step sequence to 14 reported previously.21 The literature is rich with reports wherein 2,4-diamino pyrrolo[2,3-d]pyrimidines are synthesized from appropriately substituted pyrroles and cyclocondensation partners (guanidine/1,1-dimethylguanidine).29–31 Extension of this method to obtain the tricyclic 2,4-diamino pyrimido[4,5-b]indole system by using a suitably substituted indole and the appropriate cyclocondensation partner seemed like a feasible approach. This synthetic strategy required the substituted indole 13 (Scheme 1) which was synthesized in two steps from 2,3-dichloronitrobenzene 11 by a sodium hydride induced displacement with malononitrile to 12, followed by Zn-dust catalyzed reduction. Cyclocondensation of 13 with carbamimidic chloride hydrochloride in methylsulfone at 110–120 °C for 8 hours resulted in an incomplete reaction. Recharging the reaction mixture with carbamimidic chloride hydrochloride and allowing the reaction to proceed for an additional 16 hours (total reaction time = 24 hours) led to completion of the reaction, and 14 was obtained in 45% yield. Thus, a shorter and more efficient four step synthesis to 14 (net yield = 15%) was developed, which is an improvement over the previously reported21 six step synthesis to 14 (net yield = 5%). Synthesis of 3 and 4 was accomplished by SNAr reaction of the aryl chloride 14 with naphthalene thiols 15 and 16 in a microwave apparatus (Biotage initiator®).
Scheme 1.

Reagents and conditions: (a) malononitrile, NaH, DMF, 70–80 °C, 3 h; (b) Zn, HOAc, 50–55 °C, 2 h 30 min; (c) (i) Carbamimidic chloride hydrochloride, methylsulfone, 110–120 °C, 8 h; (ii) recharge with carbamimidic chloride hydrochloride, methylsulfone, 110–120 °C, 16 h; (d) K2CO3, NMP, microwave, 250 °C, 30 min.
SNAr reactions of thiophenols substituted with an electron-withdrawing group (EWG) or an electron-donating group (EDG) were not successful using this method. It has been shown that EWGs on the phenyl nucleus decrease, and EDGs increase the nucleophilicity of sulfur in thiophenols toward 2,4-dichloronitrobenzene.32 Hence the reaction failure with thiophenols bearing EWGs can be attributed to decreased nucleophilicity. The reaction failure of thiophenols bearing EDGs can be surmised to be due to an increased propensity of the thiophenoxide anion to form disulfides33 relative to undergoing an SNAr with 14.
Thus for the synthesis of 5-(substituted phenylthio)-9H-pyrimido[4,5-b]indole-2,4-diamines with thiols bearing electron withdrawing and electron donating substituents the 5-bromo analog (20) (Scheme 2) was judged to be a better substrate since it had been reported that the 5-bromo is a better leaving group under similar conditions.34
Scheme 2.

Reagents and conditions: (a) malononitrile, NaH, THF, reflux, 70–80 °C, 3 h; (b) Zn, HOAc, 50–55 °C, 2 h, 30 min; (c) (i) methylsulfone, 110–120 °C, 8 h; (ii) recharge with carbamimidic chloride hydrochloride, methylsulfone, 110–120 °C, 12 h; (d) CuI, K2CO3, DMF, 180 °C, 4 h.
The synthesis involved a sodium hydride induced displacement of known 1735 (Scheme 2) with malononitrile to afford 18, followed by a Zn-dust catalyzed reductive cycliztion to 19, and a cyclocondensation to afford 20. Displacement of the 5-bromo of 20 with thiols 21-23 in the presence of CuI, potassium carbonate in DMF under microwave conditions furnished target compounds 5-7 respectively in yields ranging from 42–77%.
The RTK inhibitory activities of compounds 3–7 (Table 1) were evaluated using a phosphotyrosine ELISA in human tumor cells known to express high levels of VEGFR-2, EGFR and PDGFRβ.36,37 Whole cell assays were used for evaluating RTK inhibition as these assays afford more meaningful results for translation to in vivo studies.27,38–41 A431 cancer cells known to over express EGFR were used to determine the effect of compounds on cell proliferation. EGFR has been shown to be a factor in the survival of A431 cells.42 For studying cell-proliferation, CYQUANT®, a DNA intercalating dye that has been shown to provide a linear approximation of cell number was used.43
The IC50 values of RTK inhibition vary under different assay conditions. Hence, we used a standard (control) compound in each of the evaluations. For VEGFR-2, the standard was semaxanib (Figure 3); for EGFR, the standard was CB67645 (24); for PDGFR-β the standard was DMBI; for the cytotoxicity study against the growth of A431 cells in culture the standard was cisplatin. Since the inhibitory activities are determined in cells, a definite structure-activity relationship cannot be determined for 1-7 and RTK inhibition.
In the VEGFR-2 assay, compound 5 with electron donating 2′,5′-diOMe phenyl substitution was the most potent in this series and was equipotent to standard semaxanib (Figure 3). However the electron donating 4′-OMe phenyl substitution in 6 exhibited 15-fold less potency than semaxanib. The 2′-naphthyl substituted 3 and the 1′-naphthyl substituted 4 were 13-fold and 16-fold less potent respectively than semaxanib. Hence bulky 5-position substituents were not tolerated (3, 4). Compound 7 with a 4′-Cl phenyl substitution was inactive. The most potent parent compound 1 with an unsubstituted phenyl was 2-fold less active than 5 in the VEGFR-2 assay.
In the EGFR assay, compound 5 with electron donating 2′,5′-diOMe phenyl substitution exhibited single digit micromolar inhibition. Compound 5 was the most potent compound in this series, but was 22-fold less active than the standard 24 (Figure 3) in this assay. The next most potent compound – the 4′-Cl phenyl substituted 7 was 40-fold less potent than 24. The 2′-naphthyl substituted 3 and the 1′-naphthyl substituted 4 were 100-fold and 835-fold less potent than 24. This indicates that the presence of a bulky substitution might be tolerated if a 3′, 4′-disubstitution is present on the thiophenyl group (3), and is not tolerated if a 2′, 3′-disubstitution is present on thiophenyl group (4). Compound 6 with an electron donating 4′-OMe phenyl was about 100-fold less active than 24. The most potent lead compound 2 with a 4′-Me phenyl substitution was 2.4-fold less active than 5 in the EGFR assay.
In the PDGFR-β assay, the most potent compounds in the series – the 1′-naphthyl substituted 4 and 4′-OMe phenyl substituted 6 were about 16-fold less active than the standard DMBI (Figure 3). The 2′-naphthyl substituted 3 was 24-fold less active than DMBI. Compounds 5 with a 2′,5′-diOMe phenyl substitution and 7 with a 4′-Cl phenyl substitution were inactive in this assay even at 200 micromolar concentrations. The most potent lead compound 1 with an unsubstituted phenyl was about 21-fold more active than 4 and 6 in the PDGFR-β assay.
The most potent compound in the A431 cytotoxicity assay was the 4′-Cl phenyl substituted 7 which was equipotent to the standard Cisplatin. The electron donating 2′,5′-diOMe phenyl substituted 5, and the 2′-naphthyl substituted 3 were the next most potent compounds and were about 4-fold less active than cisplatin. The electron donating 4′-OMe phenyl substituted 6 was about 6-fold less active than cisplatin. The 1′-naphthyl substituted 4 was inactive even at 200 micromolar concentration. The most potent lead compound 2 with a 4′-Me phenyl substitution was equipotent to 7 in the A431 cytotoxicity assay.
Compounds 3-7 were also evaluated against isolated human, and Escherichia coli (E. coli) TS and DHFR and compared against standard compounds (Table 2). In the hTS assay the analogues were active inhibitors with IC50 values ranging from 0.12 to 2.3 μM. In analogues with electron withdrawing substitutions, remarkably the 4′-Cl- substituted analogue 7 was equipotent with the clinically used classical RTX (Figure 1) and in analogues bearing electron donating substituents, the 4′-OMe- substituted analogue 6 was also remarkably 3- and 79- fold more potent than RTX and PMX respectively. Compound 5 bearing a electron donating 2′,5′-diOMe substitution was only 3-fold less active than RTX. This indicates that the increase in activity of 5 was probably not due to electronic factors but more likely due to favorable binding conformations induced by these substituents on the phenyl ring. The 2′-naphthyl substituted (3) and 1′-naphthyl substituted (4) compounds were 3-fold better than and 6-fold worse than RTX respectively indicating that the requirement for bulk in the 5-position is specific. Relative to the 4′-methyl substituted lead compound 2; the most potent compound 3 was 3-fold more active. Against human DHFR, in general, 3-7 were poorly active.
In summary, five novel 5-(Arylthio)-9H-pyrimido[4,5-b]indole-2,4-diamines 3-7 were designed, synthesized and evaluated as inhibitors of RTKs as well as hTS. Biological evaluation showed that compound 5 had excellent VEGFR-2 inhibitory activity without significant human TS activity. Compounds 3, 6, and 7 had good hTS inhibitory activity but were poor VEGFR-2 inhibitors. Some of the analogues were indeed better TS (3, 6) or RTK (5, 7) inhibitors than the parent analogues 1 and 2, and this provides impetus for a continued search for multitargeted RTK activity along with TS inhibition in single molecules that supersede that of the parent compounds 1 and 2.
Supplementary Material
Figure 2.

Designed pyrimido[4,5-b]indoles 3-7, reported RTK inhibitors 8-10, Vatalanib.
Acknowledgments
This work was supported, in part, by the National Institutes of Health, Grants AI41743 (A.G.); AI44661 (A.G.), CA89300 (A.G.), and AI47759 (A.G.) from the National Institute of Allergy and Infectious Diseases
Abbreviations
- RTK
receptor tyrosine kinases
- VEGF
vascular endothelial growth factor
- EGFR
epidermal growth factor receptor
- PDGFR
platelet derived growth factor receptor
- c-kit
stem cell factor receptor
- CSF-1R
colony stimulating factor 1 Receptor
- Flt-3
FMS-like tyrosine kinase 3
- hTS
human thymidylate synthase
- 5-FU
5-fluorouracil
- PMX
pemetrexed
- RTX
raltitrexed
- MTX
methotrexate
- TGI
total growth inhibition
- CAM
chorioallantoic membrane
- DMF
dimethyl formamide
- NMP
N-methyl-2-pyrrolidone
- THF
tetrahydrofuran
- TLC
thin layer chromatography
Footnotes
Supplementary data associated (Full details on the enzyme assays, experimental section, synthesis and characterization of compound 3-7) with this article can be found, in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Rahimi N. The ubiquitin-proteasome system meets angiogenesis. Mol Cancer Ther. 2012;11:538–48. doi: 10.1158/1535-7163.MCT-11-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Folkman J. Tumor angiogenesis: therapeutic implications. The New England journal of medicine. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anandappa G, Turner NC. Targeting receptor tyrosine kinases in HER2-negative breast cancer. Current opinion in oncology. 2013;25:594–601. doi: 10.1097/CCO.0000000000000021. [DOI] [PubMed] [Google Scholar]
- 5.Chang J, Wang S, Zhang Z, Liu X, Wu Z, Geng R, Ge X, Dai C, Liu R, Zhang Q, Li W, Li J. Multiple receptor tyrosine kinase activation attenuates therapeutic efficacy of the fibroblast growth factor receptor 2 inhibitor AZD4547 in FGFR2 amplified gastric cancer. Oncotarget. 2015;6:2009–22. doi: 10.18632/oncotarget.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu Rev Biochem. 1995;64:721–62. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 7.Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nature reviews Clinical oncology. 2014;11:282–98. doi: 10.1038/nrclinonc.2014.51. [DOI] [PubMed] [Google Scholar]
- 8.Lehman NL. Future potential of thymidylate synthase inhibitors in cancer therapy. Expert Opin Invest Drugs. 2002;11:1775–1787. doi: 10.1517/13543784.11.12.1775. [DOI] [PubMed] [Google Scholar]
- 9.Zaware N, Sharma H, Yang J, Devambatla RKV, Queener SF, Anderson KS, Gangjee A. Discovery of Potent and Selective Inhibitors of Toxoplasma gondii Thymidylate Synthase for Opportunistic Infections. ACS Medicinal Chemistry Letters. 2013;4:1148–1151. doi: 10.1021/ml400208v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pizzorno GD, RB, Cheng Y-C. Pyrimidine and Purine Antimetabolites. In: Holland JF, Frei E III, editors. Cancer Medicine. B. C. Decker, Inc; Hamilton, London: 2003. pp. 739–744. [Google Scholar]
- 11.Fernandez-Martos C, Nogue M, Cejas P, Moreno-Garcia V, Machancoses AH, Feliu J. The role of capecitabine in locally advanced rectal cancer treatment: an update. Drugs. 2012;72:1057–73. doi: 10.2165/11633870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 12.Genova C, Rijavec E, Truini A, Coco S, Sini C, Barletta G, Dal Bello MG, Alama A, Savarino G, Pronzato P, Boccardo F, Grossi F. Pemetrexed for the treatment of non-small cell lung cancer. Expert Opin Pharmacother. 2013;14:1545–58. doi: 10.1517/14656566.2013.802774. [DOI] [PubMed] [Google Scholar]
- 13.Ma J, Waxman DJ. Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol Cancer Ther. 2008;7:3670–3684. doi: 10.1158/1535-7163.MCT-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleibeuker EA, Ten Hooven MA, Verheul HM, Slotman BJ, Thijssen VL. Combining radiotherapy with sunitinib: lessons (to be) learned. Angiogenesis. 2015;18:385–95. doi: 10.1007/s10456-015-9476-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cesca M, Bizzaro F, Zucchetti M, Giavazzi R. Tumor delivery of chemotherapy combined with inhibitors of angiogenesis and vascular targeting agents. Frontiers in oncology. 2013;3:259. doi: 10.3389/fonc.2013.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall M, Gourley C, McNeish I, Ledermann J, Gore M, Jayson G, Perren T, Rustin G, Kaye S. Targeted anti-vascular therapies for ovarian cancer: current evidence. British journal of cancer. 2013;108:250–8. doi: 10.1038/bjc.2012.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heath VL, Bicknell R. Anticancer strategies involving the vasculature. Nature reviews Clinical oncology. 2009;6:395–404. doi: 10.1038/nrclinonc.2009.52. [DOI] [PubMed] [Google Scholar]
- 18.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 19.Ma J, Waxman DJ. Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol Cancer Ther. 2008;7:3670–84. doi: 10.1158/1535-7163.MCT-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Souza CM, Araujo e Silva AC, de Jesus Ferraciolli C, Moreira GV, Campos LC, dos Reis DC, Lopes MT, Ferreira MA, Andrade SP, Cassali GD. Combination therapy with carboplatin and thalidomide suppresses tumor growth and metastasis in 4T1 murine breast cancer model. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2014;68:51–7. doi: 10.1016/j.biopha.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Gangjee A, Zaware N, Raghavan S, Ihnat M, Shenoy S, Kisliuk RL. Single agents with designed combination chemotherapy potential: synthesis and evaluation of substituted pyrimido[4,5-b]indoles as receptor tyrosine kinase and thymidylate synthase inhibitors and as antitumor agents. J Med Chem. 2010;53:1563–1578. doi: 10.1021/jm9011142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: Targets for anti-angiogenesis and normalization. Microvascular Research. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nature Clinical Practice Oncology. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 24.Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O’Reilly MS, Folkman J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]
- 25.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature (London, U K) 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 26.Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, Bohlen P, Kerbel RS. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105:R15–R24. doi: 10.1172/JCI8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gangjee A, Yang J, Ihnat MA, Kamat S. Antiangiogenic and antitumor agents. Design, synthesis, and evaluation of novel 2-amino-4-(3-bromoanilino)-6-benzylsubstituted pyrrolo[2,3-d]pyrimidines as inhibitors of receptor tyrosine kinases. Bioorg Med Chem. 2003;11:5155–5170. doi: 10.1016/j.bmc.2003.08.034. [DOI] [PubMed] [Google Scholar]
- 28.Bold G, Altmann KH, Frei J, Lang M, Manley PW, Traxler P, Wietfeld B, Brueggen J, Buchdunger E, Cozens R, Ferrari S, Furet P, Hofmann F, Martiny-Baron G, Mestan J, Roesel J, Sills M, Stover D, Acemoglu F, Boss E, Emmenegger R, Laesser L, Masso E, Roth R, Schlachter C, Vetterli W, Wyss D, Wood JM. New Anilinophthalazines as Potent and Orally Well Absorbed Inhibitors of the VEGF Receptor Tyrosine Kinases Useful as Antagonists of Tumor-Driven Angiogenesis. J Med Chem. 2000;43:2310–2323. doi: 10.1021/jm9909443. [DOI] [PubMed] [Google Scholar]
- 29.Piper JR, McCaleb GS, Montgomery JA, Kisliuk RL, Gaumont Y, Sirotnak FM. Syntheses and antifolate activity of 5-methyl-5-deaza analogs of aminopterin, methotrexate, folic acid, and N10-methylfolic acid. J Med Chem. 1986;29:1080–7. doi: 10.1021/jm00156a029. [DOI] [PubMed] [Google Scholar]
- 30.Gangjee A, Zeng Y, McGuire JJ, Kisliuk RL. Synthesis of Classical and Nonclassical, Partially Restricted, Linear, Tricyclic 5-Deaza Antifolates. J Med Chem. 2002;45:5173–5181. doi: 10.1021/jm0202369. [DOI] [PubMed] [Google Scholar]
- 31.Rigby JH, Balasubramanian N. Preparation of highly substituted 2-pyridones by reaction of vinyl isocyanates and enamines. Journal of Organic Chemistry. 1989;54:224–8. [Google Scholar]
- 32.Leandri G, Tundo A. Sulfides and disulfides. XV. Competitive reactions of aryl thiols with 2,4-dinitrochlorobenzene. Annali di Chimica (Rome, Italy) 1955;45:832–41. [Google Scholar]
- 33.Ward ER, Day LA. Reactions of aromatic nitro compounds with alkaline sulfides. IV. Disulfide formation. Journal of the Chemical Society. 1952:398–400. [Google Scholar]
- 34.Gangjee A, Zaware N. Unpublished work. [Google Scholar]
- 35.Zhang L, Fan J, Vu K, Hong K, Le Brazidec JY, Shi J, Biamonte M, Busch DJ, Lough RE, Grecko R, Ran Y, Sensintaffar JL, Kamal A, Lundgren K, Burrows FJ, Mansfield R, Timony GA, Ulm EH, Kasibhatla SR, Boehm MF. 7′-Substituted Benzothiazolothio- and Pyridinothiazolothio-Purines as Potent Heat Shock Protein 90 Inhibitors. J Med Chem. 2006;49:5352–5362. doi: 10.1021/jm051146h. [DOI] [PubMed] [Google Scholar]
- 36.Fong TAT, Shawver LK, Sun L, Tang C, App H, Powell TJ, Kim YH, Schreck R, Wang X, Risau W, Ullrich A, Hirth KP, McMahon G. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59:99–106. [PubMed] [Google Scholar]
- 37.Stockwell BR, Haggarty SJ, Schreiber SL. High-throughput screening of small molecules in miniaturized mammalian cell-based assays involving post-translational modifications. Chem Biol. 1999;6:71–83. doi: 10.1016/S1074-5521(99)80004-0. [DOI] [PubMed] [Google Scholar]
- 38.Smith GK, Wood ER. Cell-based assays for kinase drug discovery. Drug Discovery Today: Technologies. 2010;7:e13–e19. doi: 10.1016/j.ddtec.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Falgueyret JP, Black WC, Cromlish W, Desmarais S, Lamontagne S, Mellon C, Riendeau D, Rodan S, Tawa P, Wesolowski G, Bass KE, Venkatraman S, Percival MD. An activity-based probe for the determination of cysteine cathepsin protease activities in whole cells. Analytical Biochemistry. 2004;335:218–227. doi: 10.1016/j.ab.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Weissman A, Keefer J, Miagkov A, Sathyamoorthy M, Perschke S, Wang FL. Cell-Based Screening Assays. In: John BT, David JT, editors. Comprehensive Medicinal Chemistry II. Elsevier; Oxford: 2007. pp. 617–646. [Google Scholar]
- 41.Zeng XF, Li WW, Fan HJ, Wang XY, Ji P, Wang ZR, Ma S, Li LL, Ma XF, Yang SY. Discovery of novel fatty acid synthase (FAS) inhibitors based on the structure of ketoaceyl synthase (KS) domain. Bioorg Med Chem Lett. 2011;21:4742–4744. doi: 10.1016/j.bmcl.2011.06.075. [DOI] [PubMed] [Google Scholar]
- 42.Schilder RJ, Hall L, Monks A, Handel LM, Fornace AJ, Jr, Ozols RF, Fojo AT, Hamilton TC. Metallothionein gene expression and resistance to cisplatin in human ovarian cancer. Int J Cancer. 1990;45:416–22. doi: 10.1002/ijc.2910450306. [DOI] [PubMed] [Google Scholar]
- 43.Wilson SM, Barsoum MJ, Wilson BW, Pappone PA. Purine nucleotides modulate proliferation of brown fat preadipocytes. Cell Proliferation. 1999;32:131–140. doi: 10.1046/j.1365-2184.1999.32230131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wahba AJ, Friedkin M. Enzymic synthesis of thymidylate. I. Early steps in the purification of thymidylate synthetase of Escherichia coli. J Biol Chem. 1962;237:3794–3801. [PubMed] [Google Scholar]
- 45.Davisson VJ, Sirawaraporn W, Santi DV. Expression of human thymidylate synthase in Escherichia coli. J Biol Chem. 1989;264:9145–8. [PubMed] [Google Scholar]
- 46.Kisliuk RL, Strumpf D, Gaumont Y, Leary RP, Plante L. Diastereoisomers of 5,10-methylene-5,6,7,8-tetrahydropteroyl-D-glutamic acid. J Med Chem. 1977;20:1531–3. doi: 10.1021/jm00221a038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.