

Abstract
Heart rhythms arise from electrical activity generated by precisely timed opening and closing of ion channels in individual cardiac myocytes. Opening of the primary cardiac voltage-gated sodium (Nav1.5) channel initiates cellular depolarization and the propagation of an electrical action potential that promotes coordinated contraction of the heart. The regularity of these contractile waves is critically important since it drives the primary function of the heart: to act as a pump that delivers blood to the brain and vital organs. When electrical activity goes awry during a cardiac arrhythmia, the pump does not function, the brain does not receive oxygenated blood, and death ensues. Perturbations to NaV1.5 may alter the structure, and hence the function, of the ion channel and are associated downstream with a wide variety of cardiac conduction pathologies, such as arrhythmias.
1. Na+ CHANNELS: INa THE FAST VOLTAGE-GATED Na+ CHANNEL
Overview
Voltage-gated sodium (Nav) channels are transmembrane proteins of approximately 260 kDa that underlie rapid depolarization to initiate action potentials in electrically excitable cells, such as neurons and cardiac myocytes. They are topologically organized into primary and auxiliary subunits, α and β (Fig. 1). The primary α subunit contains pore-forming and voltage-sensing domains (VSDs) that control the gating and ion selection functions of the channel. The auxiliary β subunits modulate gating, regulate expression, and also play a role in the oligomerization of individual channels (Isom, 2001; Namadurai et al., 2015; Yu & Catterall, 2003).
Figure 1. Membrane topology of NaV1.5.
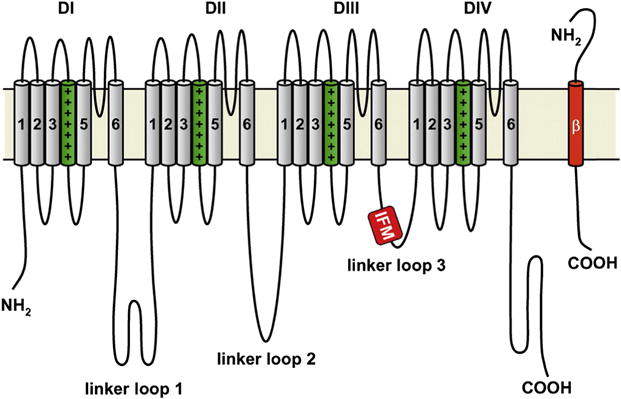
Illustration of the four domains (DI–DIV) of the α subunit along with an associated β subunit. The transmembrane S1–S4 helices that comprise the voltage sensing domains are depicted with the charged S4 helix highlighted in green (dark gray in print versions). The S5 and S6 helices form the central pore domain. The DIII–DIV inactivation loop is highlighted with the critical hydrophobic isoleucine, phenylalanine, and methionine (IFM) sequence. The N-terminal and C-terminal domains of each subunit also labeled. Copyright Hugues Abriel, Oxford University Press, 2007. http://dx.doi.org/10.1016/j.cardiores.2007.07.019
NaV channels can adopt at least three structurally distinct states: open, closed, and inactivated. In the absence of an electrical stimulus, at the cell membrane resting potential, NaV channels reside in a functionally deactivated (closed) state. Upon membrane depolarization, they activate (open) but rapidly become inactivated (nonconducting) again (Yellen, 1998). The functional distinction between deactivated and inactivated states is that channels deactivate during cellular hyperpolarization, while an inactivated channel occurs in response to cellular depolarization, rendering the channel unavailable to open and conduct ions. The relative residency in the closed and inactivated states is critical because the upstroke of the action potential depends on the availability of the channels to open (Kleber & Rudy, 2004). Even a subtle depolarization from the cell resting potential may promote channel inactivation that may reduce cellular excitability. Recovery from inactivation (RFI) occurs only after membrane repolarization, during which the channel recovers from the inactivated state and in a separate transition, deactivates to a closed available state. The time and voltage dependence of NaV channel transitions between discrete channel states depends on a complex interplay of structural, chemical, genetic, and electrical factors that determine channel function (Chen-Izu et al., 2015; Herren, Bers, & Grandi, 2013; Marionneau & Abriel, 2015; Rook et al., 2012).
The alpha (α) subunit
There are 10 genes (SCN1A–SCN10A) that encode mammalian NaV α subunit isoforms, each of which exhibits distinct structures related to their distinct physiological function. SCN5A is the gene that encodes the α subunit of the primary cardiac isoform (NaV1.5) of the voltage-gated sodium channel. NaV1.5 consists of a continuous chain of four heterologous domains (DI–DIV), each containing six transmembrane spanning segments (S1–S6) organized around a central, ion-selective pore (Fig. 2). A ring of DEKA residues between segments S5 and S6 of each domain forms the ion selectivity filter (SF) (Sun et al., 1997; Yamagishi et al., 2001). Positively charged residues are clustered in the S4 segments that comprise the voltage sensor (Kontis & Goldin, 1997; Stuhmer et al., 1989). In some Nay isoforms, the intracellular linker between domains three and four, DIII/DIV, includes a hydrophobic isoleucine-phenylalanine-methionine (IFM) motif, which acts as a gating mechanism to confer rapid occlusion of the channel pore, resulting in fast inactivation subsequent to channel opening (Stuhmer et al., 1989; West et al., 1992). Other studies also suggest a role for the intracellular DIV carboxy-terminus (C-terminus) in channel inactivation in both the brain and cardiac isoforms (NaV1.1 and NaV1.5, respectively) (Cormier et al., 2002; Mantegazza et al., 2001; Motoike et al., 2004).
Figure 2. Homology model of the NaV1.5 channel pore domain.
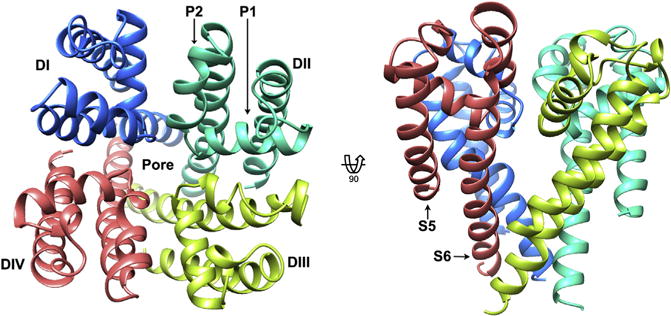
Top view of the extracellular face (left), and transmembrane view (right), of the pore domain of NaV1.5, based on the bacterial template NaVRh (PDB ID code 4DXW). Structure is shown in ribbon representation with each domain colored from DI (blue (dark gray in print versions)) to DIV (red (light gray in print versions)). Pore helices P1 and P2 are labeled in DII, and transmembrane helices S5 and S6 are labeled in DIV for representative purposes. Kevin R. DeMarco.
Additionally, the resolved structure (using cryoelectron microscopy) of the rabbit voltage-gated skeletal calcium channel, CaV1.1, provides new insight into the mammalian architecture of voltage-gated ion channels, as sodium and calcium channels are both four-domain channels closely related among the voltage-gated ion channel superfamily (Wu et al., 2015; Yu et al., 2005). This CaV structure has the potential to serve as a template to inform more accurate homology models of the human NaV channels for use in computational studies (Fig. 3).
Figure 3. Cryoelectron microscopy reveals structural elements of CaV1.1.
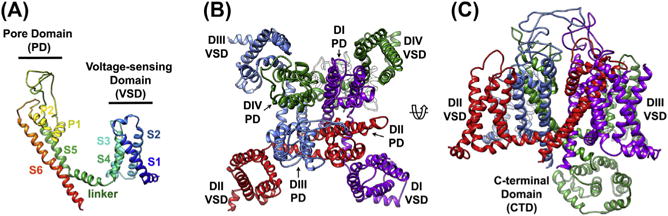
(A) Transmembrane view of a single subunit of CaV1.1 (PDB accession 3JBR) to illustrate the architecture of the pore domain (P1, P2, S5, and S6), voltage-sensing domain (S1–S4), and linker regions common to voltage-gated sodium and calcium channels. The N-terminus is colored blue, and the C-terminus is colored red in ribbon representation. (B) Top view of CaV1.1 colored to highlight the heterotetrameric arrangement of the voltage-sensing and pore domains, with the ion-conducting pore in the center. (C) Transmembrane view of CaV1.1 highlighting the C-terminal domain important for ion channel regulation, as well as a transmembrane fenestration important for local anesthetic ingress. Kevin R. DeMarco.
Subunit β
There are only four genes (SCN1B–SCN4B) that encode the mammalian NaV β subunits, all of which are expressed in cardiac tissue (Maier et al., 2004). They are membrane proteins consisting of a single transmembrane segment connecting an extracellular Immunoglobin (Ig) domain to an intracellular C-terminus (Brackenbury & Isom, 2011; Cusdin, Clare, & Jackson, 2008). Studies have shown that subunits β1 and β3 have the highest sequence homology out of the four and that they are noncovalently bound to α subunits, while subunits β2 and β4 bind to α subunits through a disulfide bond (Chen et al., 2012; Namadurai et al., 2015). Recently, the X-ray crystal structures of the human β3 and β4 subunits have been resolved (Gilchrist et al., 2013; Namadurai et al., 2014), and evidence suggests that the presence of membrane-embedded glutamic acid residues in the helical region of β3 may promote oligomerization (into dimers and trimers) which may play a role in channel localization (Morgan et al., 2000; Namadurai et al., 2015). β4 subunits, on the other hand, lack these residues and have been implicated in “resurgent current” in hippocampal neurons (Lewis & Raman, 2011; Namadurai et al., 2015). Resurgent current arises from Na channel RFI and reopening during repolarization, which results in a transient inward current during the action potential. The proposed mechanism posits that the β4 subunit competes with the inactivation particle upon depolarization, forming a blocked state of the channel that is distinct from the fast-inactivated state. Repolarization favors removal of the β4 particle, resulting in a channel that briefly conducts before inactivating or deactivating, depending on the membrane potential (Lewis & Raman, 2014). Cardiac cells have also been shown to exhibit resurgent currents, termed nonequilibrium gating, although the specific molecular mechanism is not clear, β4 subunits are expressed in cardiac myocytes (Clancy et al., 2003; George, 2009; Moreno & Clancy, 2012).
2. ACTIVATION OF THE CARDIAC Na+ CHANNEL NaV1.5
Activation of the NaV family of ion channels depends fundamentally on voltage sensing, whereby changes in the membrane potential promote changes to channel structure. Under resting conditions NaV channels are closed, but open in response to local membrane depolarization caused by a capacitive current stimulus. If a sufficient number of channels open, then permeation of sodium ions occurs through the channel pore down the electrochemical gradient into the cell, depolarizing it and thus beginning a positive feedback loop in which more sodium channels open in response to the changing membrane potential. This promotes even further depolarization, which amplifies the electrical signal and drives the cellular action potential (Catterall, 2010; Huxley & Kendrew, 1952).
2.1 Structure and mechanism of the NaV voltage-sensing domains
The S1–S4 helices of each asymmetric domain (DI–DIV) of the α subunit comprise the VSD of the NaV. The amphiphilic S4 helix contains an alternating motif of a positively charged residue followed by two hydrophobic residues. This well-conserved “310-helical” arrangement serves as the voltage-sensing element of activation in each homologous domain (Schwaiger et al., 2011; Yarov-Yarovoy et al., 2012). The charged residues on S4 are hypothesized to cause an outward motion of the helix, toward the extracellular side of the membrane, which in turn induces conformational changes within the pore (Armstrong, 1981; Capes et al., 2013; Payandeh et al., 2011; Yarov-Yarovoy, Baker, & Catterall, 2006). This is the “helical screw” model, which is supported both by experimental evidence as well as extensive studies involving structural modeling of the NaV VSD (Catterall, 1986, 2000, 2010; Yarov-Yarovoy et al., 2006, 2012). In this model, the S4 voltage sensor motion is coupled to pore opening and closing via the S4–S5 linker (Long, Campbell, & Mackinnon, 2005). The combination of outward and rotating movement of the S4 segment has been predicted to impose a gating movement of the S4–S5 linker that is roughly orthogonal to the transmembrane orientation of the pore-forming helices in the bacterial NaChBac VSD. Moreover, investigation into charge-neutralized mutants of the mammalian NaV1.4 sodium channel has found that S4, in all four domains, are involved in pore gating, albeit to different extents (Chanda & Bezanilla, 2002; Yarov-Yarovoy et al., 2012). In particular, charges on DIV S4 seem to have a larger effect on inactivation than activation, whereas the S4 helices in DI–DIII seem to influence activation (Chahine et al., 1994; Chanda & Bezanilla, 2002; Kontis, Rounaghi, & Goldin, 1997; Stuhmer et al., 1989). This may imply that the DIV voltage sensor is tightly coupled to fast inactivation (Chanda & Bezanilla, 2002).
An integral component in the helical screw model of NaV activation is the existence of negative countercharges that stabilize and shield the positively charged intramembrane S4 voltage-sensing helix as it responds to membrane depolarization. To overcome the energetically unfavorable translocation of charges over a hydrophobic barrier, the S4 helix of the VSD has been predicted, in molecular modeling studies, to favor one or more intermediate steps (Chanda & Bezanilla, 2002). This has been verified for the active states of the bacterial NachBac channel in which disulfide crosslinking and mutant cycle studies have identified molecular interactions between the S4 helix and surrounding S1–S3 helices that facilitate activation (DeCaen et al., 2008, 2009, 2011; Paldi & Gurevitz, 2010; Yarov-Yarovoy et al., 2012). Structurally, the role of countercharges in mammalian sodium channels have been studied using a combination of molecular modeling and molecular dynamics simulations and are predicted to involve three intermediate states of S4 translocation (Gosselin-Badaroudine et al., 2012). Functional investigation into mammalian VSDs using disulfide cross-linking has not been possible because these channels contain native cysteine residues that eliminate the possibility of intuiting disulfide bonding for any specific pair. However, mutagenesis studies of the VSDs of NaV1.4 demonstrate that reversing the charge of the S1–S3 countercharges had domain-specific outcomes including inhibition of activation in DI–DIII but destabilization of fast inactivation in DIV (Groome & Winston, 2013). While these studies thus far do not include the specific structural aspects of the NaV1.5 VSDs, they provide valuable insight into the intricacies of activation of NaV channels.
2.2 Naturally occurring mutations can cause delayed NaV1.5 activation
Isolated cardiac conduction disease (ICCD) is observed on the ECG in a widening of the QRS complex that indicates delays in ventricular excitation (Grant et al., 2002; Tan et al., 2001). Mutations in NaV1.5 have been shown to cause ICCD and typically result from a depolarizing shift of the Na+ channel activation curve (Grant et al., 2002; Tan et al., 2001). This shift most likely results from a reduction in the rate of channel activation or decreased channel sensitivity to the voltage required for activation. Mutant channels may require a longer time to reach depolarized membrane potentials at which the maximum INa occurs. The lag in activation of ICCD mutant Na+ channels results in a reduction of the action potential upstroke velocity, which is a primary determinant of conduction velocity, leading to an increase in the amount of stimulus current required to excite coupled tissue (Rohr, Kucera, & Kleber, 1998; Shaw & Rudy, 1997; Tan et al., 2001). Computational modeling supports the hypothesis that a depolarizing shift in the NaV1.5 activation and corresponding reduced INa produces right ventricular conduction delay and a transmural activation gradient, which is a mechanism that is consistent with the findings of a study investigating the G514C mutation linked to ICCD (Tan et al., 2001).
3. INACTIVATION
Inactivation of NaV1.5 is critically important because the completeness of the inactivation process influences both action potential duration and action potential frequency (restitution) in the myocardium. There are at least three distinct forms of inactivation that have been identified in NaV1.5 channels: fast, intermediate, and slow (Goldin, 2003; Wang et al., 2000).
4. FAST INACTIVATION
Fast inactivation in NaV1.5 is a time-and voltage-dependent process that occurs via a “hinged lid” mechanism in which the cytoplasmic linking region between DIII and DIV binds into the hydrophobic pore, thereby occluding ion conduction (Kellenberger et al., 1997; McPhee et al., 1998; Smith & Goldin, 1997). An NMR solution structure of this linking region, or inactivation particle, has been resolved as a 53 amino acid peptide, consisting of a single alpha-helix structure flanked by two disordered tail regions (Rohl et al., 1999). Several early mutagenesis studies have implicated the importance of a hydrophobic sequence of isoleucine, phenylalanine, and methionine (IFM) within the DIII–DIV linker as critical for sodium channel inactivation: mutation to glutamine (Q) at one or more of the IFM residues either diminished or abolished inactivation (Eaholtz et al., 1999; Eaholtz, Scheuer, & Catterall, 1994; Eaholtz, Zagotta, & Catterall, 1998; Goldin, 2003; Patton et al., 1992; West et al., 1992) Fig. 4.
Figure 4. Solution NMR structure of the DIII–DIV inactivation loop.
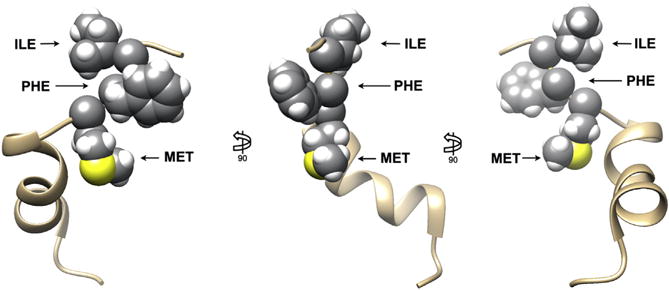
Three views of the putative sodium channel inactivation loop segment (PDB ID code 1BYY). The hydrophobic sequence of residues (IFM), demonstrated to be critical for NaV1.5 inactivation, are highlighted in space-filling representation. The remainder of the segment is shown in ribbon representation. Kevin R. DeMarco.
Experimental studies have implicated an adjacent threonine (T) residue as being important for the binding of the inactivation particle within the mouth of the pore, since its substation with a methionine (M) residue increased by 24% sustained noninactivating currents (Miyamoto et al., 2001). In Nav1.5, cysteine scanning of the IFM sequence has shown it to be critical for stable binding of the inactivation particle to the pore (Deschenes, Trottier, & Chahine, 1999). Additionally, a recent study has shown that a shortened peptide containing the IFM sequence (KIFMK) can not only block inactivation-deficient open Nav1.5 channels (L409C, A410W) but must be cleared from the pore before it can close, as evidenced by tail currents recorded upon repolarization. The same authors also found that an F1760K mutation will eliminate fast inactivation altogether, suggesting that the inactivation particle itself is important for stabilizing the inactivated state of the channel (Wang & Wang, 2005). Hence, there is ample evidence that fast inactivation occurs via a hydrophobic packing of the DIII–DIV linker within the intracellular mouth of the pore. Additionally, site-directed glutamine mutations in the DIII S4–S5 linker have been shown to lead to a positive shift in the voltage dependence of fast inactivation, suggesting that the abolition of hydrophobic residues in that region functionally corresponds to the loss of interaction with the hydrophobic IFM triad (Smith & Goldin, 1997). There is limited consensus on the mechanism of the “hinged lid” hypothesis of fast inactivation, as initial investigation suggested that the glycine and proline residues in the linker (G1484, G1485, and P1512) form the hinge (Kellenberger et al., 1997), but theoretical models in which the inactivation particle forms a hairpin motif centered around G1505 only implicate P1512 in functioning of the hinge (Sirota, Pascutti, & Anteneodo, 2002).
4.1 Structural and functional aspects of fast inactivation gating
A recent study has revealed that fast inactivation in NaV1.4 strictly depends on the motion of the DIV voltage sensor. The authors hypothesize that the opening of the sodium channel actually occurs in two steps: one in which the DI, DII, and DIII voltage sensors open the pore and allow ion permeation and another in which activation of the DIV voltage sensor renders the channel in an intermediate state (S2) and induces a conformational change in the pore itself, which serves as a necessary precursor to the binding of the inactivation particle (Goldschen-Ohm et al., 2013). Using an inactivation-deficient mutant (L435W/L437C/A438W, DI-S6), they argue that the frequent observation of subconductance states in electrophysiological recordings of the inactivation-deficient Nav1.4 channel may indicate a discrete open state that occurs following DIV voltage sensor movement albeit before the fast inactivation particle enters the pore. These secondary conductance states are normally hidden by rapid channel inactivation. The implications for this gating paradigm are significant in that fast inactivation may be a mechanism in which a two-step structural process is translated, functionally, into a single step. In addition, this finding may help to explain conflicting data supporting high-affinity block of both open and inactivated states by local anesthetics (Bennett et al., 1995a; Goldschen-Ohm et al., 2013; Wang et al., 2004). While this study was conducted using WT and mutant forms of NaV1.4, the proposed existence of the preinactivated S2 state should be applicable to NaV1.5 given the high level of homology between both isoforms.
4.2 A role for the carboxy-terminus and allosteric proteins in inactivation
The intracellular C-terminus of the α subunit of NaV1.5 plays an important role in modulating fast inactivation (An et al., 1998; Cormier et al., 2002; Mantegazza et al., 2001; Tateyama et al., 2004). In this channel an uncoupling of the C-terminus from the DIII–DIV inactivation loop has been shown to increase inward current during repolarization leading to delays in cellular repolarization. These studies have suggested that the C-terminus is involved in the structural stabilization of the fast-inactivated state (Motoike et al., 2004). An S1885stop deletion experiment further revealed that the highly charged portion of the C-terminal S6 is likely involved in electrostatic interactions with the inactivation particle (Cormier et al., 2002). One possibility is that the C-terminal stabilization of the DIII–DIV inactivation loop constitutes a mode of intermediate inactivation (Cormier et al., 2002; Kass, 2006; Mantegazza et al., 2001), although there is lack of consensus regarding the precise structural determinants of such an intermediate inactivated state.
Fast inactivation in NaV1.5 can also be modulated by accessory proteins such as fibroblast growth factor homologous factors (FHFs), which are a family of intracellular signaling proteins that interact with the C-terminus. It has been suggested that a tethered FHF-derived N-terminal particle competes for the induction of the intrinsic fast inactivation particle of the NaV at depolarized transitions near the open state (Dover et al., 2010). One study has found that FGF12, an FHF that is the most abundant in human heart, has diminished interaction with a C-terminal H1849R mutant of NaV1.5, resulting most notably in a depolarizing (~ −9 mV) shift in fast (steady state) inactivation, which is a gain-of-function phenotype associated with arrhythmia and long QT syndrome (Musa et al., 2015). Another study has identified the acidic-rich C-terminal region (1773–1832) of NaV1.5 as critical for FGF12 binding, and that the presence of FGF12 modulates the voltage dependence of inactivation, with no effect on activation of the channel (Liu et al., 2003). Congenital long QT syndrome (LQT3) is a disorder characterized by a delay in cardiac cellular repolarization, a condition that can lead to cardiac arrhythmias and sudden death. Other mutations in this C-terminal region, such as E1784K, 1795insD, and Y1795C interfere with fast inactivation and evoke small, sustained currents associated with LQT3 as well (An et al., 1998; Rivolta et al., 2001; Veldkamp et al., 2000).
4.3 Channelopathies arising from altered fast inactivation
The first naturally occurring mutation that was identified in NaV1.5 is ΔKPQ: a 3 amino acid deletion (lysine, proline, glutamine at positions 1505–1507) in the DIII–DIV linker that was shown to affect fast inactivation and was causally linked to LQT3 (Bennett et al., 1995b). This motif is known to be critical for fast inactivation of the Na+ channel, and the DKPQ mutation results in a small (<1% of peak current amplitude) persistent noninactivating current (“bursting”) during normal cellular repolarization. This mutation was also the first to be modeled in a study of an in silico mutation that linked genotype to phenotype (Clancy & Rudy, 1999). Model simulations showed that delayed ventricular repolarization promotes a substrate for triggered activity via early afterdepolarizations (EADs), which result from reactivation of L-type Ca2+ channels. Since that first study, numerous modeling and simulation studies have been used to predict effects of arrhythmia-linked mutations in genes underlying ion channels and accessory and regulatory proteins (Noble & Rudy, 2001).
4.4 Modulation of fast inactivation by β subunits
Fast inactivation can also be modulated by associated β subunits. In particular, the β3 subunit, which is normally expressed in ventricular and Purkinje cardiac cells, shifts steady state (fast) inactivation leftward, indicating reduced channel availability. When a ΔECD mutation is made in β3, abolition of this gating shift in NaV1.5 and a persistent current is observed, implicating its interference with the DIII–DIV inactivation particle, as has previously been suggested for the β1 subunit (McCormick et al., 1998; Yu et al., 2005).
5. SLOW INACTIVATION
Slow inactivation of NaV1.5 is a process spanning seconds or tens of seconds that can occur during prolonged or repetitive depolarization (Kass, 2004). Slow inactivation is independent of the DIII–DIV linker associated with fast inactivation and is more likely a result of the structural deformation of the pore, such as with C-type inactivation in potassium channels, although the exact mechanism is not yet known (Liu, Jurman, & Yellen, 1996; Vassilev, Scheuer, & Catterall, 1989). Early experiments involving the transfer of all four NaV1.5 P-loop regions into the NaV1.4 transmembrane backbone conferred the cardiac type of slow inactivation (Russ et al., 1996; Vilin et al., 1999). These regions include the DEKA SF, as well as the highly conserved P-loop region that forms the extracellular funnel of the pore domain. For example, a single residue difference between NaV1.4 (V754) and NaV1.5 (I891) in the DII P-loop region caused a higher probability of slow inactivation in the cardiac isoform (Vilin & Ruben, 2001), and a tryptophan mutation (W53C) in the DI P-loop region has been shown to diminish single channel conductance (Tomaselli et al., 1995). Furthermore, a mutation in the DEKA SF itself has been reported to increase the probability of NaV channels entering a slow inactivated state (Hilber et al., 2001). The many regions of the NaV channel that have been implicated in slow inactivation may contribute to an as yet poorly understood coordinated gating process. A study has correlated the onset of slow inactivation in NaV1.4 with immobilization of the VSDs of DI and DII (Silva & Goldstein, 2013), suggesting that slow inactivation may involve additional protein regions and not be solely be due to deformation of the pore domain.
6. RECOVERY FROM INACTIVATION
Repeated regular firing of cardiac action potentials depends crucially on the process of RFI of NaV1.5 channels. RFI is an I process that indicates the fraction of NaV channels that are available for activation. The recovery process occurs on a millisecond timescale, with exponential kinetics that are accelerated with increased hyperpolarization (Bezanilla & Armstrong, 1977; Huxley & Kendrew, 1952). Both antiarrhythmic drugs and naturally occurring mutations have been shown to target and modulate RFI. In a physiological setting, RFI participates in a tightly orchestrated relationship with deactivation, ensuring minimization of leak current during channel repriming (Kuo & Bean, 1994).
6.1 DIV voltage sensor movement is rate limiting
The kinetics of recovery from fast inactivation has been studied in both wild-type NaV1.5 sodium channels and in mutants in which the positive S4 arginine gating charges of each domain of the VSD have been neutralized by mutation to glutamine. Results demonstrate that compared to the wild type, while all NaV1.5 S4 mutants slowly recover, the charge-neutralized DIV mutant exhibited the largest effect, with a pronounced recovery lag of almost 0.5 ms; suggesting that the motion of the DIV VSD may be rate limiting for RFI (Capes et al., 2013). Such findings are consistent with the fact that site-3 toxins, small peptide toxins derived from venoms that bind with high specificity to the extracellular surface of NaV channels inhibit the outward motion of the DIV S4 voltage sensor, consequently inducing a faster RFI (Hanck & Sheets, 2007). During RFI, the voltage sensors of DI–DIII are likely required to deactivate first, closing the channel and preventing conduction, after which the slower inward motion of DIV S4 acts to drive the inactivation particle from its vestibule (Armstrong, 2006; Kuo & Bean, 1994). Furthermore, an accelerated RFI and slower onset of fast inactivation has been found to result from mutations in DIII S1–S2 linker (R1232W) and the DIV S3–S4 linker (T1620M) resulting in idiopathic ventricular fibrillation (Vilin, Fujimoto, & Ruben, 2001), suggesting again an important role for DIV VSD in RFI.
6.2 Modulation of recovery from inactivation by β subunits
RFI of NaV1.5 channels is modulated by at least two of β subunits. One study has shown that the transmembrane helix, plus either the intracellular or extracellular region of β1 is necessary to accelerate its RFI (Fahmi et al., 2001). β1 is known to accelerate RFI, not only in NaV1.5, but also in the skeletal muscle and neuronal isoforms of the channel (Zimmer & Benndorf, 2002). β3 also accelerates RFI of NaV1.5 in oocytes, which may be physiologically significant because β3 is expressed in cardiac ventricular cells and Purkinje fibers (Fahmi et al., 2001).
6.3 Recovery from inactivation channelopathy and disease phenotypes
Examples of LQT-3 mutations that do not result in a classical gain of function as measured during square wave voltage clamp experiments, include D1790G, E1295K, and I1768V. Interestingly, these mutations are not localized to a single region of the channel, rather they are found in varied regions including the C-terminal domain and in the DIII S3–S4 linker region. Studies have shown that these mutations rather result from an acceleration of RFI. Experiments and mathematical simulations have revealed that under nonequilibrium conditions during repolarization, channel reopening results from faster RFI at membrane potentials that facilitate the activation transition (Clancy et al., 2003). Mathematical modeling and simulation approaches predicted that the additional inward current through the defective channels was sufficient to cause prolonged repolarization, consistent with the long QT syndrome phenotype (Clancy et al., 2003).
Additionally, reduction in NaV1.5 have been attributed to changes in RFI that can lead to Brugada syndrome (diagnosed by ST segment elevation on the ECG) by several distinct alterations to ion channel gating, including reduced rates of RFI as well as faster inactivation subsequent to channel opening and protein trafficking defects (Clancy & Rudy, 2002). For example, the 1795insD mutant channel showed a rightward shift in the recovery curve, indicating a delayed RFI in NaV1.5 of 100 ms or more. The resulting loss of current was shown to be sufficient to explain the Brugada phenotype.
7. REGULATION OF CARDIAC NaV1.5
There has been work focused on the regulation of Nay1.5 by post-translational processes (Marionneau & Abriel, 2015). There is also a growing literature on how posttranslational modifications of NaV1.5 are facilitated through the presence of macromolecular complexes that allow selective and specific protein modification to confer functional effects (Abriel, Rougier, &Jalife, 2015). Multiple phosphorylation mechanisms through a variety of kinase pathways as well as glycosylation, arginine methylation, S-nitrosylation, ubiquitylation, and ion homeostatic mechanisms are all implicated as functional regulators of NaV1.5 (Marionneau & Abriel, 2015).
7.1 Phosphorylation
One of the key functional modulators of NaV1.5 is phosphorylation by Ca2+/calmodulin-dependent protein kinase II (CaMKII) that results in changes to macroscopic NaV1.5 gating including a reduction in channel availability, increased intermediate inactivation, and slowed RFI (Wagner et al., 2006). Multiple phosphorylation sites in the intracellular loop linking NaV1.5 domains I and II have been identified (S516, S571, and T594) and implicated in the CaMKII-induced changes to INa gating (Ashpole et al., 2012; Hund et al., 2010), although, how each of the three sites confers functional effects is still not clear.
While all of the effects of CaMKII on macroscopic NaV1.5 current result in peak current reduction, CaMKII activation also results in substantial current increases in the late or persistent component of current that can cause NaV1.5 gain of function associated proarrhythmia (Anderson, Brown, & Bers, 2011; Bers & Grandi, 2009; Erickson & Anderson, 2008; Grandi et al., 2007; Horvath et al., 2013; Hund et al., 2008; Ma et al., 2012; Maier, 2011; Wagner et al., 2011). During AP-clamp experiments in canine ventricular myocytes, inhibition of CaMKII has been shown to reduce NaV1.5 late current (Horvath et al., 2013; Maltsev et al., 2008). In addition to phosphorylation by CaMKII, increased in intracellular Ca2+ modifies and promotes NaV1.5 late current (Mori et al., 2000; Wingo et al., 2004) through a variety of mechanisms including activation of CaMKII (Wagner et al., 2006), increased mitochondrial oxidative phosphorylation (Belardinelli, Shryock, & Fraser, 2006), and consequent increased reactive oxygen species (ROS) (Kohlhaas et al., 2010; Liu et al., 2010). Interestingly, an increase NaV1.5 late current can also activate CaMKII, thereby resulting in a positive feedback cycle between NaV1.5 late current and CaMKII (Yao et al., 2011). The effects conferred by CaMKII on both peak and late NaV1.5 current has been suggested to occur through a complex with βIV-spectrin that positions CaMKII to phosphorylate the channel (Hund et al., 2010).
In addition to CaMKII, in rodent heart cells cAMP-dependent protein kinase (PKA) has been shown to phosphorylate serine residues at positions 525 and 528 and confer functional effects such as reducing channel availability, promoting NaV1.5 trafficking, and increasing NaV1.5 macroscopic current (Hallaq et al., 2006; Marionneau & Abriel, 2015; Matsuda, Lee, & Shibata, 1992; Murphy et al., 1996; Tateyama et al., 2003; Zhou et al., 2002). These effects are similar to those observed in expressed NaV1.5 channels following PKC-dependent phosphorylation of S1503, which additionally promotes the NaV1.5 late current (Qu et al., 1994; West et al., 1991).
Other kinases have also been implicated in posttranslational modification of NaV1.5 current. Included are the phosphatidylinositol kinase (PI3K), whose inhibition promotes NaV1.5 late current (Lu et al., 2012), while Fyn and Adenosine Monophosphate-activated Protein Kinases (AMPK) promote NaV1.5 current by increasing channel availability and promoting RFI (Ahern et al., 2005; Light, Wallace, & Dyck, 2003).
7.2 Glycosylation
In additional to kinase modulation of NaV1.5 via phosphorylation, glycosylation of NaV1.5 is an additional presumed mode of posttranslational modification. Multiple putative asparagines have been identified in NaV1.5 as potential N-linked glycosylation sites (Ahmad et al., 2007). However, thus far only asparagines in the ancillary NaVβ1 subunit have been demonstrated to functionally modify the cardiac Na+ current, by causing leftward shifts of the inactivation and activation curves, promoting fast inactivation, and slowing RFI (Ednie et al., 2013; Stocker & Bennett, 2006; Ufret-Vincenty et al., 2001).
7.3 Methylation and ubiquitylation
A set of arginine residues in NaV1.5 located in the DI–DII linker has been shown to undergo arginine methylation (Beltran-Alvarez et al., 2014). The effect of methylation of these residues is to increase the NaV1.5 surface expression and hence current density (Beltran-Alvarez et al., 2013). Exactly the opposite effects on channel function have been shown to result from ubiquitylation, which results in NaV1.5 internalization and reduction in current (van Bemmelen et al., 2004; Laedermann, Decosterd, & Abriel, 2014). The ubiquitylation sites are presently unknown.
7.4 Reactive oxygen species regulation
There are multiple modes of redox regulation of cardiac NaV1.5 through various signaling mechanism, both direct and indirect. A prominent indirect mechanism is by ROS activation of CaMKII, which confers the gain-of-function effects on NaV1.5 late current described earlier (Marionneau & Abriel, 2015). Nitric oxide (generally derived from endothelial nitric oxide synthase (eNOS)), also potentiates NaV1.5 late current, which can occur via direct S-nitrosylation of the channel (Ahern et al., 2000; Cheng et al., 2013). Changes in the NAD(H) balance has been shown to promote mitochondrial ROS production, which leads to a reduction in NaV1.5 peak current, which may provide a link between altered metabolism, excitability, conduction block, and arrhythmic risk (Liu et al., 2013, 2010). Indeed, naturally occurring disease-linked mutations that have been shown to prevent the protein kinase A potentiation of NaV1.5 during oxidative stress, leading to loss of Na channel function and manifestations of Brugada syndrome (Aiba et al., 2014).
8. SUMMARY
Understanding how NaV1.5 structure underlies channel function is continually improving due the development of improved experimental and computational approaches that allow more concrete links between structure and function. Connecting ion channel function to emergent behavior in the heart to cause disease is a critical area of research. The ubiquity of disruption of NaV1.5 channels in cardiac disorders of excitability emphasizes the importance of fundamental understanding of the associated mechanisms and disease processes to ultimately reveal new targets for human therapy.
Acknowledgments
This work was supported by The American Heart Association Pre-doctoral Fellowship (16PRE27260295), Western States Affiliate (KRD) and The American Heart Association (GIAs (15GRNT25700136), Western States Affiliate), and National Institutes of Health NHLBI 1U01HL126273 (CEC).
References
- Abriel H, Rougier JS, Jalife J. Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac death. Circulation Research. 2015;116(12):1971–1988. doi: 10.1161/CIRCRESAHA.116.305017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern CA, et al. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circulation Research. 2005;96(9):991–998. doi: 10.1161/01.RES.0000166324.00524.dd. [DOI] [PubMed] [Google Scholar]
- Ahern GP, et al. Induction of persistent sodium current by exogenous and endogenous nitric oxide. Journal of Biological Chemistry. 2000;275(37):28810–28815. doi: 10.1074/jbc.M003090200. [DOI] [PubMed] [Google Scholar]
- Ahmad I, et al. In silico determination of intracellular glycosylation and phosphorylation sites in human selectins: implications for biological function. Journal of Cellular Biochemistry. 2007;100(6):1558–1572. doi: 10.1002/jcb.21156. [DOI] [PubMed] [Google Scholar]
- Aiba T, et al. A mutation causing Brugada syndrome identifies a mechanism for altered autonomic and oxidant regulation of cardiac sodium currents. Circulation Cardiovascular Genetics. 2014;7(3):249–256. doi: 10.1161/CIRCGENETICS.113.000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An RH, et al. Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha-and beta1-subunits. Circulation Research. 1998;83(2):141–146. doi: 10.1161/01.res.83.2.141. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. Journal of Molecular and Cellular Cardiology. 2011;51(4):468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Sodium channels and gating currents. Physiological Reviews. 1981;61(3):644–683. doi: 10.1152/physrev.1981.61.3.644. [DOI] [PubMed] [Google Scholar]
- Armstrong CM. Na channel inactivation from open and closed states. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(47):17991–17996. doi: 10.1073/pnas.0607603103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashpole NM, et al. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. Journal of Biological Chemistry. 2012;287(24):19856–19869. doi: 10.1074/jbc.M111.322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, Fraser H. Inhibition ofthe late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl. 4):iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Alvarez P, et al. Protein arginine methyl transferases-3 and -5 increase cell surface expression of cardiac sodium channel. FEBS Letters. 2013;587(19):3159–3165. doi: 10.1016/j.febslet.2013.07.043. [DOI] [PubMed] [Google Scholar]
- Beltran-Alvarez P, et al. Identification of N-terminal protein acetylation and arginine methylation of the voltage-gated sodium channel in end-stage heart failure human heart. Journal of Molecular and Cellular Cardiology. 2014;76:126–129. doi: 10.1016/j.yjmcc.2014.08.014. [DOI] [PubMed] [Google Scholar]
- van Bemmelen MX, et al. Cardiac voltage-gated sodium channel NaV1.5 is regulated by Nedd4-2 mediated ubiquitination. Circulation Research. 2004;95(3):284–291. doi: 10.1161/01.RES.0000136816.05109.89. [DOI] [PubMed] [Google Scholar]
- Bennett PB, et al. On the molecular nature of the lidocaine receptor of cardiac Na+ channels. Modification of block by alterations in the alpha-subunit III–IV interdomain. Circulation Research. 1995a;77(3):584–592. doi: 10.1161/01.res.77.3.584. [DOI] [PubMed] [Google Scholar]
- Bennett PB, et al. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995b;376(6542):683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. Journal of Cardiovascular Pharmacology. 2009;54(3):180–187. doi: 10.1097/FJC.0b013e3181a25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. Journal of General Physiology. 1977;70(5):549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Isom LL. Na channel beta subunits: overachievers of the ion channel family. Frontiers in Pharmacology. 2011;2:53. doi: 10.3389/fphar.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capes DL, et al. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. Journal of General Physiology. 2013;142(2):101–112. doi: 10.1085/jgp.201310998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Molecular properties of voltage-sensitive sodium channels. Annual Review of Biochemistry. 1986;55:953–985. doi: 10.1146/annurev.bi.55.070186.004513. [DOI] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67(6):915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahine M, et al. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12(2):281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. Journal of General Physiology. 2002;120(5):629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, et al. Identification of the cysteine residue responsible for disulfide linkage of Na+ channel alpha and beta2 subunits. Journal of Biological Chemistry. 2012;287(46):39061–39069. doi: 10.1074/jbc.M112.397646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, et al. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. Journal of Molecular and Cellular Cardiology. 2013;61:102–110. doi: 10.1016/j.yjmcc.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Izu Y, et al. Na+ channel function, regulation, structure, trafficking and sequestration. Journal of Physiology. 2015;593(6):1347–1360. doi: 10.1113/jphysiol.2014.281428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999;400(6744):566–569. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]
- Clancy CE, Rudy Y. Na+ channel mutation that causes both Brugada and long-QT syndrome phenotypes – a simulation study of mechanism. Circulation. 2002;105(10):1208–1213. doi: 10.1161/hc1002.105183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE, et al. Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation. 2003;107(17):2233–2237. doi: 10.1161/01.CIR.0000069273.51375.BD. [DOI] [PubMed] [Google Scholar]
- Cormier JW, et al. Secondary structure of the human cardiac Na+ channel C terminus – evidence for a role of helical structures in modulation of channel inactivation. Journal of Biological Chemistry. 2002;277(11):9233–9241. doi: 10.1074/jbc.M110204200. [DOI] [PubMed] [Google Scholar]
- Cusdin FS, Clare JJ, Jackson AP. Trafficking and cellular distribution of voltage-gated sodium channels. Traffic. 2008;9(1):17–26. doi: 10.1111/j.1600-0854.2007.00673.x. [DOI] [PubMed] [Google Scholar]
- DeCaen PG, et al. Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(39):15142–15147. doi: 10.1073/pnas.0806486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, et al. Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(52):22498–22503. doi: 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, et al. Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(46):18825–18830. doi: 10.1073/pnas.1116449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes I, Trottier E, Chahine M. Cysteine scanning analysis of the IFM cluster in the inactivation gate of a human heart sodium channel. Cardiovascular Research. 1999;42(2):521–529. doi: 10.1016/s0008-6363(99)00064-4. [DOI] [PubMed] [Google Scholar]
- Dover K, et al. Long-term inactivation particle for voltage-gated sodium channels. Journal of Physiology. 2010;588(Pt 19):3695–3711. doi: 10.1113/jphysiol.2010.192559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaholtz G, et al. Block of brain sodium channels by peptide mimetics of the isoleucine, phenylalanine, and methionine (IFM) motif from the inactivation gate. Journal of General Physiology. 1999;113(2):279–294. doi: 10.1085/jgp.113.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaholtz G, Scheuer T, Catterall WA. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12(5):1041–1048. doi: 10.1016/0896-6273(94)90312-3. [DOI] [PubMed] [Google Scholar]
- Eaholtz G, Zagotta WN, Catterall WA. Kinetic analysis of block of open sodium channels by a peptide containing the isoleucine, phenylalanine, and methionine (IFM) motif from the inactivation gate. Journal of General Physiology. 1998;111(1):75–82. doi: 10.1085/jgp.111.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ednie AR, et al. Expression of the sialyltransferase, ST3Gal4, impacts cardiac voltage-gated sodium channel activity, refractory period and ventricular conduction. Journal of Molecular and Cellular Cardiology. 2013;59:117–127. doi: 10.1016/j.yjmcc.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Erickson JR, Anderson ME. CaMKII and its role in cardiac arrhythmia. Journal of Cardiovascular Electrophysiology. 2008;19(12):1332–1336. doi: 10.1111/j.1540-8167.2008.01295.x. [DOI] [PubMed] [Google Scholar]
- Fahmi AI, et al. The sodium channel beta-subunit SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. Journal of Physiology. 2001;537(Pt 3):693–700. doi: 10.1111/j.1469-7793.2001.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AL., Jr Genetic modulation of impaired cardiac conduction: sodium channel beta4 subunit missing in action. Circulation Research. 2009;104(11):1238–1239. doi: 10.1161/CIRCRESAHA.109.199752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist J, et al. Crystallographic insights into sodium-channel modulation by the beta4 subunit. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(51):E5016–E5024. doi: 10.1073/pnas.1314557110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Mechanisms of sodium channel inactivation. Current Opinion in Neurobiology. 2003;13(3):284–290. doi: 10.1016/s0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Goldschen-Ohm MP, et al. Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nature Communications. 2013;4:1350. doi: 10.1038/ncomms2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin-Badaroudine P, et al. Gating pore currents and the resting state of NaV1.4 voltage sensor domains. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(47):19250–19255. doi: 10.1073/pnas.1217990109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi E, et al. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophysical Journal. 2007;93(11):3835–3847. doi: 10.1529/biophysj.107.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AO, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. Journal of Clinical Investigation. 2002;110(8):1201–1209. doi: 10.1172/JCI15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groome JR, Winston V. S1–S3 counter charges in the voltage sensor module of a mammalian sodium channel regulate fast inactivation. Journal of General Physiology. 2013;141(5):601–618. doi: 10.1085/jgp.201210935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallaq H, et al. Quantitation of protein kinase A-mediated trafficking of cardiac sodium channels in living cells. Cardiovascular Research. 2006;72(2):250–261. doi: 10.1016/j.cardiores.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Hanck DA, Sheets MF. Site-3 toxins and cardiac sodium channels. Toxicon. 2007;49(2):181–193. doi: 10.1016/j.toxicon.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herren AW, Bers DM, Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. American Journal of Physiology. Heart and Circulatory Physiology. 2013;305(4):H431–H445. doi: 10.1152/ajpheart.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilber K, et al. The selectivity filter of the voltage-gated sodium channel is involved in channel activation. Journal of Biological Chemistry. 2001;276(30):27831–27839. doi: 10.1074/jbc.M101933200. [DOI] [PubMed] [Google Scholar]
- Horvath B, et al. Dynamics of the late Na+ current during cardiac action potential and its contribution to after depolarizations. Journal of Molecular and Cellular Cardiology. 2013;64(0):59–68. doi: 10.1016/j.yjmcc.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hund TJ, et al. Role of activated CaMKII in abnormal calcium homeostasis and I(Na) remodeling after myocardial infarction: insights from mathematical modeling. Journal of Molecular and Cellular Cardiology. 2008;45(3):420–428. doi: 10.1016/j.yjmcc.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hund TJ, et al. A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. Journal of Clinical Investigation. 2010;120(10):3508–3519. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley HE, Kendrew JC. Extractability of the Lotmar-Picken material from dried muscle. Nature. 1952;170(4334):882. doi: 10.1038/170882a0. [DOI] [PubMed] [Google Scholar]
- Isom LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001;7(1):42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- Kass RS. Sodium channel inactivation goes with the flow. Journal of General Physiology. 2004;124(1):7–8. doi: 10.1085/jgp.200409123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RS. Sodium channel inactivation in heart: a novel role of the carboxy-terminal domain. Journal of Cardiovascular Electrophysiology. 2006;17(Suppl. 1):S21–S25. doi: 10.1111/j.1540-8167.2006.00381.x. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, et al. Molecular analysis of potential hinge residues in the inactivation gate of brain type IIA Na+ channels. Journal of General Physiology. 1997;109(5):607–617. doi: 10.1085/jgp.109.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiological Reviews. 2004;84(2):431–488. doi: 10.1152/physrev.00025.2003. [DOI] [PubMed] [Google Scholar]
- Kohlhaas M, et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121(14):1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis KJ, Goldin AL. Sodium channel inactivation is altered by substitution of voltage sensor positive charges. Journal of General Physiology. 1997;110(4):403–413. doi: 10.1085/jgp.110.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis KJ, Rounaghi A, Goldin AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. Journal of General Physiology. 1997;110(4):391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Bean BP. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12(4):819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Laedermann CJ, Decosterd I, Abriel H. Ubiquitylation of voltage-gated sodium channels. Handbook of Experimental Pharmacology. 2014;221:231–250. doi: 10.1007/978-3-642-41588-3_11. [DOI] [PubMed] [Google Scholar]
- Lewis AH, Raman IM. Cross-species conservation of open-channel block by Na channel beta4 peptides reveals structural features required for resurgent Na current. Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2011;31(32):11527–11536. doi: 10.1523/JNEUROSCI.1428-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, Raman IM. Resurgent current of voltage-gated Na(+) channels. Journal of Physiology. 2014;592(Pt 22):4825–4838. doi: 10.1113/jphysiol.2014.277582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Wallace CH, Dyck JR. Constitutively active adenosine monophosphate-activated protein kinase regulates voltage-gated sodium channels in ventricular myocytes. Circulation. 2003;107(15):1962–1965. doi: 10.1161/01.CIR.0000069269.60167.02. [DOI] [PubMed] [Google Scholar]
- Liu CJ, et al. Modulation of the cardiac sodium channel NaV1.5 by fibroblast growth factor homologous factor 1B. Journal of Biological Chemistry. 2003;278(2):1029–1036. doi: 10.1074/jbc.M207074200. [DOI] [PubMed] [Google Scholar]
- Liu M, et al. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2013;54:25–34. doi: 10.1016/j.yjmcc.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Liu H, Dudley SC., Jr Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circulation Research. 2010;107(8):967–974. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Brown DA, O’Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. Journal of Molecular and Cellular Cardiology. 2010;49(5):728–736. doi: 10.1016/j.yjmcc.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16(4):859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309(5736):903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- Lu Z, et al. Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Science Translational Medicine. 2012;4(131):131ra50. doi: 10.1126/scitranslmed.3003623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, et al. Calmodulin kinase II and protein kinase C mediate the effect of increased intracellular calcium to augment late sodium current in rabbit ventricular myocytes. American Journal of Physiology. Cell Physiology. 2012;302(8):C1141–C1151. doi: 10.1152/ajpcell.00374.2011. [DOI] [PubMed] [Google Scholar]
- Maier LS. CaMKII regulation of voltage-gated sodium channels and cell excitability. Heart Rhythm. 2011;8(3):474–477. doi: 10.1016/j.hrthm.2010.09.080. [DOI] [PubMed] [Google Scholar]
- Maier SK, et al. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation. 2004;109(11):1421–1427. doi: 10.1161/01.CIR.0000121421.61896.24. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, et al. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. American Journal of Physiology. Heart and Circulatory Physiology. 2008;294(4):H1597–H1608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza M, et al. Role of the C-terminal domain in inactivation of brain and cardiac sodium channels. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(26):15348–15353. doi: 10.1073/pnas.211563298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marionneau C, Abriel H. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. Journal of Molecular and Cellular Cardiology. 2015;82:36–47. doi: 10.1016/j.yjmcc.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Matsuda JJ, Lee H, Shibata EF. Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circulation Research. 1992;70(1):199–207. doi: 10.1161/01.res.70.1.199. [DOI] [PubMed] [Google Scholar]
- McCormick KA, et al. Molecular determinants of Na+ channel function in the extracellular domain of the beta1 subunit. Journal of Biological Chemistry. 1998;273(7):3954–3962. doi: 10.1074/jbc.273.7.3954. [DOI] [PubMed] [Google Scholar]
- McPhee JC, et al. A critical role for the S4–S5 intracellular loop in domain IV of the sodium channel -subunit in fast inactivation. Journal of Biological Chemistry. 1998;273(2):1121–1129. doi: 10.1074/jbc.273.2.1121. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, et al. Solution structures of the inactivation gate particle peptides of rat brain type-IIA and human heart sodium channels in SDS micelles. Journal of Peptide Research. 2001;57(3):203–214. doi: 10.1111/j.1399-3011.2001.00817.x. [DOI] [PubMed] [Google Scholar]
- Moreno JD, Clancy CE. Pathophysiology of the cardiac late Na current and its potential as a drug target. Journal of Molecular and Cellular Cardiology. 2012;52(3):608–619. doi: 10.1016/j.yjmcc.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan K, et al. Beta 3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(5):2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, et al. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-sensitivity? Biochemistry. 2000;39(6):1316–1323. doi: 10.1021/bi9912600. [DOI] [PubMed] [Google Scholar]
- Motoike HK, et al. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. Journal of General Physiology. 2004;123(2):155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BJ, et al. cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. Journal of Biological Chemistry. 1996;271(46):28837–28843. doi: 10.1074/jbc.271.46.28837. [DOI] [PubMed] [Google Scholar]
- Musa H, et al. SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(40):12528–12533. doi: 10.1073/pnas.1516430112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namadurai S, et al. Crystal structure and molecular imaging of the NaV channel beta3 subunit indicates a trimeric assembly. Journal of Biological Chemistry. 2014;289(15):10797–10811. doi: 10.1074/jbc.M113.527994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namadurai S, et al. A new look at sodium channel beta subunits. Open Biology. 2015;5(1):140192. doi: 10.1098/rsob.140192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D, Rudy Y. Models of cardiac ventricular action potentials: iterative interaction between experiment and simulation. Philosophical Transactions of the Royal Society of London Series A-Mathematical Physical and Engineering Sciences. 2001;359(1783):1127–1142. [Google Scholar]
- Paldi T, Gurevitz M. Coupling between residues on S4 and S1 defines the voltage-sensor resting conformation in NaChBac. Biophysical Journal. 2010;99(2):456–463. doi: 10.1016/j.bpj.2010.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton DE, et al. Amino acid residues required for fast Na(+)-channel inactivation: charge neutralizations and deletions in the III–IV linker. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, et al. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, et al. Modulation of cardiac Na+ channels expressed in a mammalian cell line and in ventricular myocytes by protein kinase C. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(8):3289–3293. doi: 10.1073/pnas.91.8.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivolta I, et al. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. Journal of Biological Chemistry. 2001;276(33):30623–30630. doi: 10.1074/jbc.M104471200. [DOI] [PubMed] [Google Scholar]
- Rohl CA, et al. Solution structure of the sodium channel inactivation gate. Biochemistry. 1999;38(3):855–861. doi: 10.1021/bi9823380. [DOI] [PubMed] [Google Scholar]
- Rohr S, Kucera JP, Kleber AG. Slow conduction in cardiac tissue, I – effects of a reduction of excitability versus a reduction of electrical coupling on microconduction. Circulation Research. 1998;83(8):781–794. doi: 10.1161/01.res.83.8.781. [DOI] [PubMed] [Google Scholar]
- Rook MB, et al. Biology of cardiac sodium channel Nav1.5 expression. Cardiovascular Research. 2012;93(1):12–23. doi: 10.1093/cvr/cvr252. [DOI] [PubMed] [Google Scholar]
- Russ U, et al. Effects of the Na+/H+-exchange inhibitor Hoe 642 on intracellular pH, calcium and sodium in isolated rat ventricular myocytes. Pflugers Archiv. 1996;433(1–2):26–34. doi: 10.1007/s004240050244. [DOI] [PubMed] [Google Scholar]
- Schwaiger CS, et al. 3(1)(0)-helix conformation facilitates the transition of a voltage sensor S4 segment toward the down state. Biophysical Journal. 2011;100(6):1446–1454. doi: 10.1016/j.bpj.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue – roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circulation Research. 1997;81(5):727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- Silva JR, Goldstein SA. Voltage-sensor movements describe slow inactivation of voltage-gated sodium channels I: wild-type skeletal muscle Na(V)1.4. Journal of General Physiology. 2013;141(3):309–321. doi: 10.1085/jgp.201210909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirota FL, Pascutti PG, Anteneodo C. Molecular modeling and dynamics of the sodium channel inactivation gate. Biophysical Journal. 2002;82(3):1207–1215. doi: 10.1016/S0006-3495(02)75477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4–S5. Biophysical Journal. 1997;73(4):1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker PJ, Bennett ES. Differential sialylation modulates voltage-gated Na+ channel gating throughout the developing myocardium. Journal of General Physiology. 2006;127(3):253–265. doi: 10.1085/jgp.200509423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer W, et al. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339(6226):597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Sun YM, et al. On the structural basis for size-selective permeation of organic cations through the voltage-gated sodium channel – effect of alanine mutations at the DEKA locus on selectivity, inhibition by Ca2+ and H+, and molecular sieving. Journal of General Physiology. 1997;110(6):693–715. doi: 10.1085/jgp.110.6.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HL, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409(6823):1043–1047. doi: 10.1038/35059090. [DOI] [PubMed] [Google Scholar]
- Tateyama M, et al. Modulation of cardiac sodium channel gating by protein kinase A can be altered by disease-linked mutation. Journal of Biological Chemistry. 2003;278(47):46718–46726. doi: 10.1074/jbc.M308977200. [DOI] [PubMed] [Google Scholar]
- Tateyama M, et al. Structural effects of an LQT-3 mutation on heart Na+ channel gating. Biophysical Journal. 2004;86(3):1843–1851. doi: 10.1016/S0006-3495(04)74251-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli GF, et al. A mutation in the pore of the sodium channel alters gating. Biophysical Journal. 1995;68(5):1814–1827. doi: 10.1016/S0006-3495(95)80358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ufret-Vincenty CA, et al. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. Journal of Biological Chemistry. 2001;276(30):28197–28203. doi: 10.1074/jbc.M102548200. [DOI] [PubMed] [Google Scholar]
- Vassilev P, Scheuer T, Catterall WA. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(20):8147–8151. doi: 10.1073/pnas.86.20.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldkamp MW, et al. Two distinct congenital arrhythmias evoked by a multidys-functional Na+ channel. Circulation Research. 2000;86(9):E91–E97. doi: 10.1161/01.res.86.9.e91. [DOI] [PubMed] [Google Scholar]
- Vilin YY, Fujimoto E, Ruben PC. A novel mechanism associated with idiopathic ventricular fibrillation (IVF) mutations R1232W and T1620M in human cardiac sodium channels. Pflugers Archiv. 2001;442(2):204–211. doi: 10.1007/s004240100529. [DOI] [PubMed] [Google Scholar]
- Vilin YY, et al. Structural determinants of slow inactivation in human cardiac and skeletal muscle sodium channels. Biophysical Journal. 1999;77(3):1384–1393. doi: 10.1016/S0006-3495(99)76987-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilin YY, Ruben PC. Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochemistry and Biophysics. 2001;35(2):171–190. doi: 10.1385/CBB:35:2:171. [DOI] [PubMed] [Google Scholar]
- Wagner S, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. Journal of Clinical Investigation. 2006;116(12):3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, et al. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circulation Research. 2011;108(5):555–565. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DW, et al. Enhanced Na(+) channel intermediate inactivation in Brugada syndrome. Circulation Research. 2000;87(8):E37–E43. doi: 10.1161/01.res.87.8.e37. [DOI] [PubMed] [Google Scholar]
- Wang SY, et al. Block of inactivation-deficient Na+ channels by local anesthetics in stably transfected mammalian cells: evidence for drug binding along the activation pathway. Journal of General Physiology. 2004;124(6):691–701. doi: 10.1085/jgp.200409128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Wang GK. Block of inactivation-deficient cardiac Na(+) channels by acetyl-KIFMK-amide. Biochemical and Biophysical Research Communications. 2005;329(2):780–788. doi: 10.1016/j.bbrc.2005.02.039. [DOI] [PubMed] [Google Scholar]
- West JW, et al. A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science. 1991;254(5033):866–868. doi: 10.1126/science.1658937. [DOI] [PubMed] [Google Scholar]
- West JW, et al. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingo TL, et al. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nature Structural & Molecular Biology. 2004;11(3):219–225. doi: 10.1038/nsmb737. [DOI] [PubMed] [Google Scholar]
- Wu J, et al. Structure of the voltage-gated calcium channel Cav1.1 complex. Science. 2015;350(6267):aad2395. doi: 10.1126/science.aad2395. [DOI] [PubMed] [Google Scholar]
- Yamagishi T, et al. Molecular architecture of the volta re-dependent Na channel: functional evidence for at helices in the pore. Journal of General Physiology. 2001;118(2):171–181. doi: 10.1085/jgp.118.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, et al. Nav1.5-dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. American Journal of Physiology. Cell Physiology. 2011;301(3):C577–C586. doi: 10.1152/ajpcell.00125.2011. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Baker D, Catterall WA. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(19):7292–7297. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, et al. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(2):E93–E102. doi: 10.1073/pnas.1118434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Quarterly Reviews of Biophysics. 1998;31(3):239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yu EJ, et al. Distinct domains of the sodium channel beta3-subunit modulate channel-gating kinetics and subcellular location. Biochemical Journal. 2005;392(Pt 3):519–526. doi: 10.1042/BJ20050518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biology. 2003;4(3):207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, et al. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacological Reviews. 2005;57(4):387–395. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- Zhou J, et al. Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circulation Research. 2002;91(6):540–546. doi: 10.1161/01.res.0000033598.00903.27. [DOI] [PubMed] [Google Scholar]
- Zimmer T, Benndorf K. The human heart and rat brain IIA Na+ channels interact with different molecular regions of the beta1 subunit. Journal of General Physiology. 2002;120(6):887–895. doi: 10.1085/jgp.20028703. [DOI] [PMC free article] [PubMed] [Google Scholar]