

Abstract
Many forms of solid tumor have a characteristic feature known as hypoxia, which describes a low or non-existent presence of oxygen in the cellular microenvironment. This decrease in oxygen causes activation of the hypoxia inducible factor (HIF) pathway, which activates the transcription of many genes that cause cell proliferation, metastasis, increased glycolysis and angiogenesis. Increased HIF expression has been linked with poor patient prognosis, increased malignancy, and therapeutic resistance. Previous work in our lab has identified 1 and 2 as inhibitors of the HIF pathway, specifically as disrupters of the p300-HIF-1α complex formation. A library of sulfonamide analogs has been designed and synthesized with the intent of examining the SAR of this series of compounds and improving potency and physicochemical properties as compared with lead compounds 1 and 2. At the end, we have achieved a thorough understanding of the structural features critical for future optimization work. 2009 Elsevier Ltd. All rights reserved.
Keywords: Hypoxia Inducible Factor inhibitors, structure-activity relationship, molecular docking
Graphical abstract
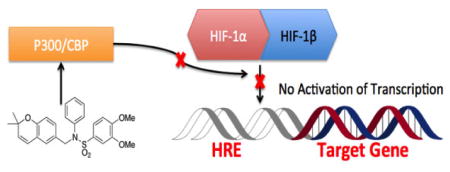
Hypoxia is a condition in which the availability of oxygen to a tissue is reduced from normal physiological levels. Many types of solid tumors display regions with low partial oxygen pressure or even anoxia due to abnormal vasculature and reduced blood flow.1 Under these conditions, the hypoxia inducible factor (HIF) pathway is activated and leads to the transcriptional activation of a number of genes that mediate adaptive responses to hypoxia, including cell metabolism, cell motility, and angiogenesis.2 One of the main transcription factors driving these responses is hypoxia-inducible factor 1 (HIF-1). HIF-1 is a basic helix-loop-helix motif heterodimeric transcription factor comprised of two subunits: HIF-1α and HIF-1β. Under normoxic conditions, human HIF-1α is hydroxylated by the enzyme prolyl hydroxylase 2 (PHD2) on two Proline residues (402 and 564) that mediate the interaction of the HIF-1α oxygen degradation domain with the von Hippel-Lindau tumor suppressor protein (VHL), which serves as an E3 ubiquitin ligase and marks the protein for degradation by the proteasome.3
Under hypoxic conditions, HIF-1α is not dihydroxylated, which abrogates VHL binding and prevents its degradation; thus PHD2 acts as an oxygen sensor to stabilize HIF. Under hypoxia, HIF-1α binds to HIF-1β, and the heterodimer translocates to the nucleus, where it binds to specific DNA sequences in the promoters of hypoxia-regulated genes. Following HIF binding to these hypoxia response elements (HRE), p300/CBP histone acetyl transferases are recruited, which opens the chromatin, recruits the RNA polymerase machinery and activates the transcription of HIF target genes, including VEGF (vascular endothelial growth factor), EPO (erythropoietin), GLUT1 (glucose transporter 1), LDH-A (lactate dehydrogenase), and NOS (nitric oxide synthase).4 The activation of the HIF-1 pathway is associated with several types of cancer and is also related to low success rates of various treatment methods.5 Therefore, inhibition of the HIF-1 pathway is recognized as a viable approach to the development of anti-cancer agents.
Toward the goal of finding small-molecule inhibitors of the HIF pathway, an HRE-alkaline phosphatase assay was designed to screen a library of 10,000 compounds from a 2,2-dimethylbenzopyran combinatorial library.6 The HRE-alkaline phosphatase assay used human glioblastoma cells (LN229-HRE-AP cell line) stably transfected with an alkaline phosphatase reporter under the control of a minimal CMV promoter and an engineered hypoxia-activated enhancer constituted of 6 copies of the HRE (hypoxia response element) from the VEGF gene.7 This initial screening yielded a few promising hits, with the lead compound identified as 1 (Figure 1a) having an IC50 of ~0.6 μM.8 1 was then taken to preliminary in vivo studies, where nude mice were implanted with LN229 glioblastoma cells on their hind flanks. After 1 week, the mice were either injected with 1 (60 mg/kg; 5 days/week) or vehicle (DMSO) for 10–12 weeks until the mice had to be terminated due to large size of tumors in the control group. On average, a 6-fold difference in tumor size was observed between the treatment and control groups, and some of the tumors disappeared completely. The treatment group did not appear to suffer negative side effects from 1 treatment, suggesting that 1 is well tolerated.9
Figure 1.

a) lead compound 1 b) lead compound 2
With 1 as the original lead compound, our laboratory began synthesis of a library of over 100 analogs. This initial library of analogs was screened against a human glioblastoma cell line (LN229-HRE-Lux), with a luciferase reporter under the control of the same hypoxia-inducible promoter as above. From this initial study, N-cyclobutyl-N-((2,2-dimethyl-2H-pyrano[3,2-b]pyridin-6-yl)methyl)-3,4-dimethoxybenzenesulfonamide 2 (Figure 1b) with IC50 of ~0.25 μM was identified as the optimized lead for further study.10 With such promising results from these initial studies, we report herein our further lead optimization and broadening of the library in order to identify more potent compounds and to further develop our SAR (structure-activity relationship).
Lead compounds 1 and 2 were divided into 4 parts (Figure 1): the left-hand core (A, red), the N-substituent (B, green), the right-hand ring (C, blue), and the linker (D, violet). Analogs were designed and synthesized based off of modifications of 3 of these parts, namely the A, B, and C rings. In total, 6 classes of analogs, for a total of 42 compounds, were devised as described in Table 1. Classes 1A–1C are analogs of 1: Class 1A has the A ring modified (3a–g); Class 1B has both the B and C rings modified (4a–f) and features one analogue with a hydrogenated 2,2-dimethylpyranobenzene A ring (4f); Class 1C has modifications to both the B and C rings (5a–n), with two analogues, namely 5m and 5n, that have the hydrogenated 2,2-dimethlpyranobenzene. Classes 2A–2C are analogs of 2: Classes 2A and 2B have, respectively, the A (6a–i) and both B and C (7a–f) regions modified; while 7d and 7e have the hydrogenated A ring.
Table 1.
Classes of compounds synthesized

|
A general 2- or 3-step reaction sequence was followed in the synthesis of the analogs (Scheme 1). First, reductive amination of aldehydes 8 (either commercially available or synthesized from literature procedures, Schemes S1–S2) with various primary amines yielded secondary amines 9. These secondary amines were then sulfonylated with various sulfonyl chlorides to yield sulfonamides 3–7. Some of these sulfonamides were hydrogenated to yield final products.
Scheme 1.

Synthesis of analogs. Reagents and conditions: (a) InCl3, NaBH4, MeOH, room temperature, 20 minutes, 23–92%, or NaBH4, MeOH, room temperature, overnight, and taken directly to the next step; (b) K2CO3, DCM, room; (c) H2, Pd/C, MeOH, overnight, 70–99%.
All 42 of the analogs were evaluated for their inhibitory effects on HIF-1-mediated transcription under hypoxic conditions on human glioma cells LN229-HRE-Lux. Each compound was tested against 2 as positive control and IC50 values were determined. The respective IC50 values for 1 and 2 were 0.59 and 0.28 μM.
Class 1A
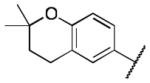
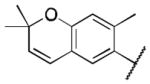
Class 1A analogs were designed to probe the importance of the features of the phenyl A ring to the activity of 1 (Table 2). The first part of the A ring examined was the double bond in the pyran ring. In many cases, double bonds are not preferred in therapeutics because of the possibility of activation and/or metabolism by cytochrome P450 (CYP) monooxygenases in the liver, which can lead to hepatotoxicity in vivo.11 For 3a (IC50 = 0.98 μM), the hydrogenated analog of 1, removal of the double bond resulted in only a small decrease (1.5-fold) in activity, which suggests that the double bond is not essential for HIF-1 inhibition. Next, various substituents were introduced on the phenyl ring. Products 3b–3d (IC50 = 3.3, 3.2, 3.1 μM) had decreased activities by about 5-fold, which suggests a somewhat size constrained binding pocket. Next, the point of attachment of the chromene ring was examined by changing it from carbon 6 to carbon 8, 3e (IC50 = 1.8 μM), which resulted in a 3-fold decrease in activity, confirming attachment at carbon 6 to be important. Finally, two other fused ring systems were synthesized, 3f–3g, with IC50 values greater than 5 μM, thus confirming the need for the chromene moiety in the A ring.
Table 2.
HIF Inhibition by Class 1A analogs
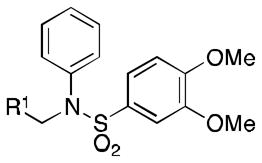
| ||
|---|---|---|
| Name | R1 | IC50[a] |
| 3a |
|
0.98 |
| 3b |
|
3.3 |
| 3c |
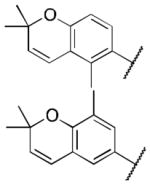
|
3.2 |
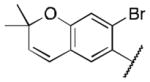
| 3d |
|
3.1 |
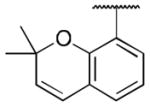
| 3e |
|
1.8 |
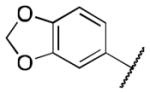
| 3f |
|
>5 |
| 3g |
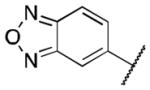
|
>5 |
IC50 values are in micromolar (μM).
Class 1B
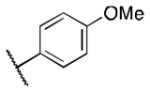
Class 1B analogs were designed to probe the activity of the moieties on the C ring (Table 3). In our previous work, it was determined that removing the 3′-methoxy group from the C ring resulted in little to no loss of activity (4e, IC50 = 0.6 μM).10 We further probed the activity of this site by removing the 4′-methoxy, 4a (IC50 = 1 μM), which resulted in a small 1.7-fold decrease in activity. Next the 4′-monomethoxy was substituted for a 4′-trifluoromethoxy moiety, 4b (IC50 = >5 μM), which resulted in a loss of activity. Other modifications that incorporated electronegative atoms such as fluorine 4c (IC50 = >5 μM) or an electron-withdrawing group like cyano 4d (IC50 = >5 μM) led to a complete loss of activity Thus, the methoxy substituent in the 4′ position seems to be needed for optimal activity while the 3′-methoxy is not. This further supports our model that the 4′-methoxy is likely involved in hydrogen bonding to LYS350 of P300.12 The 4′-monomethoxy compound 4f (IC50 = 3.2 μM), in comparison with its non-hydrogenated form 4e (IC50 = 0.6 μM), decreased in activity by 5-fold. This initial result suggests the role of the double bond A ring is somewhat important for activity.
Table 3.
HIF Inhibition by Class 1B analogues
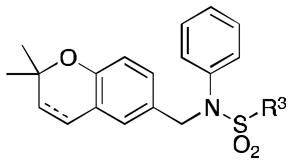
IC50 values are in micromolar (μM).
A ring is hydrogenated 2,2-dimethylpyranobenzene.
Class 1C
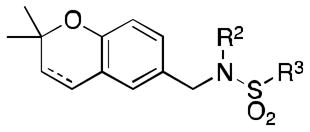
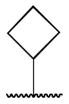
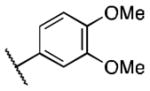
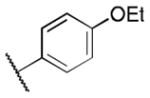
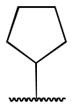
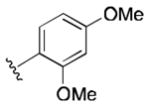
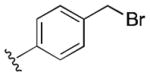
Class 1C analogs were designed to further probe the activity of the B and C rings and to try to introduce more polar groups in order to lower the clogP (Table 4). Although 5k (IC50 = 4 μM) had a clogP of 3.7, it demonstrated a significant loss of activity. These results support our previous computational model, that suggests that the B ring points into a hydrophobic pocket.12 Some of these analogues are hybrid 1/2 compounds, with the A ring from 1 and the B ring from 2 (5a–f). In this combination, only 5a (IC50 = 0.75 μM) demonstrated activity below 1 μM. None of these hybrid compounds were as potent as 2. Analogues 5g–j were developed as analogs that contain the A ring from 1 and an N-cyclopentyl substituent, with varying C rings. Two of these compounds, 5g (IC50 = 1.5 μM) and 5i (IC50 = 3.0 μM) exhibited some activity in the luciferase assay, but with 3- and 6-fold decreases in activity compared to the 3,4-dimethoxysulfonyl compound 5j (IC50 = 0.5 μM), which we reported previously (Ref 10). The activity of 5l (IC50 = >5 μM) decreased in comparison with its non-hydrogenated counterpart 5k (IC50 = 4 μM). However, hybrid 5m (IC50 = 0.39 μM), was more potent than its non-hydrogenated counterpart 5a (IC50 = 0.75 μM). The role of the double bond in the A ring is still unclear and may be studied further in the future.
Table 4.
HIF Inhibition by Class 1C analogues
| |||
|---|---|---|---|
| Name | R2 | R3 | IC50[a] |
| 5a |
|
|
0.75 |
| 5b |
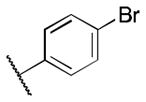
|
>5 | |
| 5c |
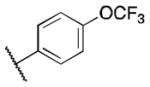
|
>5 | |
| 5d |
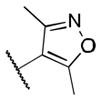
|
>5 | |
| 5e |
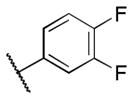
|
4.6 | |
| 5f |
|
4.4 | |
|
| |||
| 5g |
|
|
1.5 |
| 5h |
|
>5 | |
| 5i |
|
3.0 | |
| 5j |
|
0.5 | |
|
| |||
| 5k |
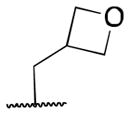
|
|
4.0 |
| 5l[b] |
|
>5 | |
|
| |||
| 5m[b] |
|
|
0.39 |
IC50 values are in micromolar (μM).
A ring is hydrogenated 2,2-dimethylpyranobenzene.
Class 2A
Class 2A analogs were designed to probe the importance of the features of the A ring to the activity of 2 (Table 5). The first strategy was to remove the fused ring feature altogether and determine whether a simple phenyl ring with various substituents would have activity. Unfortunately, only one such compound, 6b (IC50 = 3.9 μM), with a 4-methoxyphenyl A ring had any activity, and this activity was diminished by almost 14-fold from 2. 6a, c–f (IC50 = >5 μM) were considered inactive, which demonstrates the importance of the fused ring system to the activity. However, 6g (IC50 = 1 μM), an analog containing a morpholine substituent, which can increase water solubility, was active. It is highly possible that increased solubility is responsible for the activity of this compound. Next, other fused ring systems that contained oxygen were examined, 6h–i, (IC50 = 1.3, 3.5 μM). Both of these compounds showed decreased activity (by 2.5-fold to 6.0-fold when compared to 2). Overall, no suitable replacement for the A ring of 2 was found.
Table 5.
HIF Inhibition by Class 2A analogs
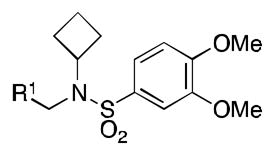
| ||
|---|---|---|
| Name | R1 | IC50[a] |
| 6a |
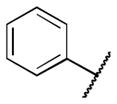
|
>5 |
| 6b |
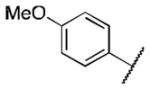
|
3.9 |
| 6c |
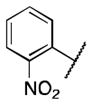
|
>5 |
| 6d |
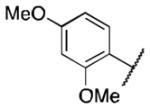
|
>5 |
| 6e |
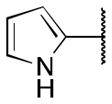
|
>5 |
| 6f |
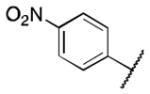
|
>5 |
| 6g |
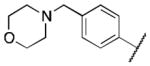
|
1.0 |
| 6h |
|
1.3 |
| 6i |
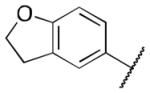
|
3.5 |
IC50 values are in micromolar (μM).
Class 2B
Class 2B analogs (Table 6) were designed to further probe the importance of the B and C rings to the activity of 2. None of the compounds with the conserved reduced A ring of 2 (7a–c) showed improved activity over 2; the best compound 7a (IC50 = 1.8 μM), with the 3′-methoxy removed, still lost potency by 6.4-fold, whereas a similar analog to 1 (4e) did not lose activity. It is interesting that 7d (IC50 = 0.25 μM), the hydrogenated form of 2, demonstrated slightly better activity compared to 2. Additionally, 7e (IC50 = 1.2 μM) demonstrated slightly better activity than its non-hydrogenated counterpart 7a (IC50 = 1.8 μM). This suggests that the double bond in the A ring may not be required for potency, and could possibly be eliminated with no loss of activity for analogs of 2. This suggests that 1 and 2 may bind to different sites or in different poses. Analogue 7f is the only example in this class that does not include a cyclobutyl ring, but instead offers a 3,4-dimethoxybenzeneethyl in the B position. As a result, the activity plummets compared to its fellow class members.
Table 6.
HIF Inhibition by Class 2B analogs
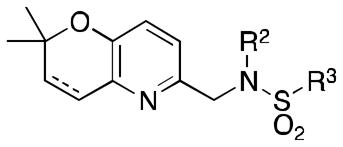
IC50 values are in micromolar (μM).
A ring is hydrogenated 2,2-dimethylpyridinylbenzene.
Previously, 1 was computationally observed, as expected, to have several satisfactory interactions on two sites interfering with the p300-HIF-1α complex formation, disrupting the structure of p300 and inhibiting the binding of HIF-1α.12 In the studies presented here, the lowest energy conformation of 1 binds in the same site, making similar interactions with the amino acid residues of interest. For that reason, it is sensible to use this site for comparison of 1 with the remaining analogs previously discussed. In order to gain a better understanding of the effect of molecular interactions between the 1 analogs and p300 on the HIF-1 pathway inhibitory activity demonstrated in vitro, 10 of the analogs were subjected to flexible docking using AutoDock Vina,13 including the previously discovered lead 2. The first model of the NMR structure of p300-CH1/HIF-1α C-TAD protein complex (PDB entry code: 1L3E) was prepared using AutoDock by removal of the HIF-1α chain and Zn ions. The optimal ligand-binding site on p300 was highlighted in a grid box based on the lowest energy conformation of 1. Specifically, the interactions between p300 and 1 are most profound via several key residues that are supported by previous studies.12 The A ring, which consists of a benzopyran moiety, not only has favorable hydrophobic interactions with Ile400 but also has hydrogen bonding between its oxygen and Ser401. As illustrated from Figure 2, the N-phenyl B ring is burrowed into the hydrophobic pocket, interacting with Ile400 and several Leucine residues. Finally, the 3,4-dimethoxybenzene of the D ring interacts with Leu376, Thr380 and Gln352 residues; and Gln352 interacts specifically with the 4-methoxy group of 1, which we have previously reported as an important component for ligand binding in these sulfonamide analogs10. Two active (IC50 value less than 5 μM) ligands were further examined due to their structural similarity to 1 and observed for any favorable or unfavorable interactions that might affect binding and, ultimately, activity in the binding site of p300. These compounds, along with 2, and their respective IC50 values, and calculated binding affinities are summarized in Table 7.
Figure 2.

Molecular docking image of 1 bound to p300
Table 7.
Docking results of selected 1, 2, and selected analogs
IC50 values are in micromolar (μM),
binding affinity values are in kcal/mol.
Qualitative analysis of the docking images reveals some interesting commonality among many of the active analogues. Unsurprisingly, analog 3a, the hydrogenated version of 1, binds very similarly, conserving the Ser401 hydrogen bond and the other major interactions already discussed (Figure 3).
Figure 3.

Molecular docking image of 1 (magenta), 3a (gray), 3e (cyan), and 2 (brown) bound to p300.
Alternatively, 3e is a structural isomer of 1, with the chromene ring attachment point at the 8-position instead of the 6-position. The docking study revealed that the two analogues share many of the same interactions with p300 amino acid residues, but are without the important hydrogen bonding with Ser401 (Figure 3). This could explain the weaker inhibition of 3e (IC50 = 1.8 μM) compared with 1 (IC50 = 0.59 μM), in addition to the lower calculated binding affinity.
Surprisingly, 2 has a low in silico binding affinity compared with 1, even though it has better in vitro activity in the luciferase assay. In the studies performed here, the lowest energy conformation of 2 was longitudinally flipped such that the benzopyran ring interacted with Leu376 and Met379 and formed a hydrogen bond with Thr380 (Figure 3). This leaves the dimethoxyphenyl group to interact with the Ile400 and His349. Having a relatively poor binding affinity of −6.5 kcal/mol, in spite of its extraordinary ability to inhibit HIF transcriptional activity, suggests that 2, and perhaps some of its more closely related analogs, do not bind in specifically the same way 1 does. To further explore this idea, nine of the active analogs (IC50 <5 uM) that are more structurally similar to 2 were subjected to the same molecular docking as discussed above. Seven randomly selected analogues of 2 were docked in the same manner, none of which showed any significant trend between binding affinity and IC50 value (Table S1, Figures S1–18). This suggests that these analogs do not bind with this protein in the same manner as analogues of 1.
In conclusion, 42 analogs were synthesized. As illustrated in Figure 4, a qualitative structure-activity relationship (SAR) was developed. For the A ring, the 2,2-dimethyl chromene with either a N or C in the 5 position is important to the activity. Open ring structures are not well tolerated, with the exception of some 4-position moderately polar substituents. The double bond is not crucial for activity of compounds and can be eliminated with the result of better or only slightly decreased activity. For the B ring, only hydrophobic groups, such as aromatics or small aliphatic rings or chains are acceptable. Introduction of polar moieties in this position dramatically decreases the activity. For the C ring, 3′,4′-dimethoxy is still the best, with the 4′-methoxy more crucial to activity than the 3′-methoxy. Molecular docking studies revealed that analogs more closely related in structure to 1 demonstrated better binding affinity, whereas 2 and other structures with the N-phenyl as the B ring, seem to bind elsewhere. This is based on the poor binding affinities observed when binding is limited to the site of 1. Such information will be important for the synthesis of future analogues.
Figure 4.

Structure-Activity Relationship of analogs.
Supplementary Material
Acknowledgments
Financial support from the NIH (CA180805, CA116804) is gratefully acknowledged. We also thank the GSU MBD and CDT programs for fellowships to SKZ, and a Department of Education GAANN grant (P200A120122) with Barbara Baumstark as the Principal Investigator in support of JF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncol. 2005;7:134–153. doi: 10.1215/S1152851704001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guillemin K, Krasnow MH. The hypoxic response: Huffing and HIFing. Cell. 1997;89:9–12. doi: 10.1016/s0092-8674(00)80176-2. [DOI] [PubMed] [Google Scholar]
- 3.(a) Maxwell PH, Wiesener MS, Chang G-W, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]; (b) Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]; (b) Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 5.(a) Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM, Semenza GL, van Diest PJ, van der Wall E. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer. 2003;97:1573–1581. doi: 10.1002/cncr.11246. [DOI] [PubMed] [Google Scholar]; (b) Belozerov VE, Van Meir EG. Hypoxia inducible factor-1: A novel target for cancer therapy. Anti-Cancer Drug. 2005;16:901–909. doi: 10.1097/01.cad.0000180116.85912.69. [DOI] [PubMed] [Google Scholar]
- 6.Nicolaou KC, Pfefferkorn JA, Mitchell HJ, Roecker AJ, Barluenga S, Cao GQ, Affleck RL, Lillig JE. Natural Product-like Combinatorial Libraries Based on Privileged Structures. 2. Construction of a 10‚000-Membered Benzopyran Library by Directed Split-and-Pool Chemistry Using NanoKans and Optical Encoding. J Am Chem Soc. 2000;122:9954–9967. [Google Scholar]
- 7.Tan C, de Noronha RG, Roecker AJ, Pyrzynska B, Khwaja F, Zhang Z, Zhang H, Teng Q, Nicholson AC, Giannakakou P, Zhou W, Olson JJ, Pereira MM, Nicolaou KC, Van Meir EG. Identification of a Novel Small-Molecule Inhibitor of the Hypoxia-Inducible Factor 1 Pathway. Cancer Res. 2005;65:605–612. [PubMed] [Google Scholar]
- 8.Tan C, de Noronha RG, Devi NS, Jabbar AA, Kaluz S, Liu Y, Mooring SR, Nicolaou KC, Wang B, Van Meir EG. Sulfonamides as a new scaffold for hypoxia inducible factor pathway inhibitors. Bioorg Med Chem Lett. 2011;21:5528–5532. doi: 10.1016/j.bmcl.2011.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin S, Kaluz S, Devi NS, Jabbar AA, de Noronha RG, Mun J, Zhang Z, Boreddy PR, Wang W, Wang Z, Abbruscato T, Chen Z, Olson JJ, Zhang R, Goodman MM, Nicolaou KC, Van Meir EG. Arylsulfonamide KCN1 Inhibits In Vivo Glioma Growth and Interferes with HIF Signaling by Disrupting HIF-1α Interaction with Cofactors p300/CBP. Clin Cancer Res. 2012;18:6623–6633. doi: 10.1158/1078-0432.CCR-12-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mooring SR, Jin H, Devi NS, Jabbar AA, Kaluz S, Liu Y, Van Meir EG, Wang B. Design and Synthesis of Novel Small-Molecule Inhibitors of the Hypoxia Inducible Factor Pathway. J Med Chem. 2011;54:8471–8489. doi: 10.1021/jm201018g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dansette PM, Macherey A-C. Biotransformations Leading to Toxic Metabolites: Chemical Aspects. In: Wermuth CG, editor. The Practice of Medicinal Chemistry. 3. Amsterdam: Elsevier; 2008. pp. 674–696. [Google Scholar]
- 12.Shi Q, Yin S, Kaluz S, Ni N, Devi N, Mun J, Wang D, Damera K, Chen W, Burroughs S, Mooring S, Goodman M, Van Meir E, Wang B, Snyder J. Binding Model for the Interaction of Anticancer Arylsulfonamides with the p300 Transcription Cofactor. ACS Med Chem Lett. 2012;3:620–625. doi: 10.1021/ml300042k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.