

Abstract
Attention-deficit/hyperactivity disorder (ADHD) is accompanied by impairments in cognitive control, such as task-switching deficits. We investigated whether such problems, and their remediation by medication, reflect abnormal reward motivation and associated striatal dopamine transmission in ADHD. We used functional genetic neuroimaging to assess the effects of dopaminergic medication and reward motivation on task-switching and striatal BOLD signal in 23 adults with ADHD, ON and OFF methylphenidate, and 26 healthy controls. Critically, we took into account interindividual variability in striatal dopamine by exploiting a common genetic polymorphism (3′-UTR VNTR) in the DAT1 gene coding for the dopamine transporter. The results showed a highly significant group by genotype interaction in the striatum. This was because a subgroup of patients with ADHD showed markedly exaggerated effects of reward on the striatal BOLD signal during task-switching when they were OFF their dopaminergic medication. Specifically, patients carrying the 9R allele showed a greater striatal signal than healthy controls carrying this allele, whereas no effect of diagnosis was observed in 10R homozygotes. Aberrant striatal responses were normalized when 9R-carrying patients with ADHD were ON medication. These pilot data indicate an important role for aberrant reward motivation, striatal dopamine and interindividual genetic differences in cognitive processes in adult ADHD.
Keywords: attention-deficit/hyperactivity disorder, cognition, DAT1 genotype, dopamine, functional magnetic resonance imaging, human, methylphenidate, reward, striatum, task-switching
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is characterized by symptoms of inattention, impulsivity and/or hyperactivity (American Psychiatric Association, 1994, 2013). Although originally considered a childhood disorder, ADHD persists into adulthood in many cases, and affects between 2.5 and 4.9% of the adult population (Kooij et al., 2005; Kessler et al., 2006; Polanczyk et al., 2007; Simon et al., 2009). A first-line treatment option for ADHD is prescription of psychostimulant medication, primarily the dopamine and noradrenaline transporter blocker methylphenidate.
ADHD is associated with a wide range of cognitive control deficits that span the domains of attention, response inhibition, working memory and task-switching (Barkley, 1997; Bush et al., 1999). Such cognitive control deficits have been attributed most commonly (albeit not exclusively; see Cortese et al., 2012) to (dorsal) prefrontal cortex dysfunction (Dickstein et al., 2006; Cubillo et al., 2010; Dibbets et al., 2010; McCarthy et al., 2014). Accordingly, effects of methylphenidate on cognitive control deficits in ADHD are considered to reflect action (i.e. increasing synaptic levels of dopamine and noradrenaline) in the prefrontal cortex (Aron et al., 2003; Berridge et al., 2006; Schmeichel et al., 2013; for a review, see Arnsten and Li, 2005). In addition to cognitive control deficits, ADHD is accompanied by processing deficits in the domains of reward and motivation (Sergeant et al., 2003; Sonuga-Barke, 2003; Scheres et al., 2007; Furukawa et al., 2014). Unlike the cognitive control deficits, these reward-related deficits are often attributed to changes in the ventral striatum (Ströhle et al., 2008; Plichta et al., 2009; Hoogman et al., 2011, 2013; Carmona et al., 2012; Volkow et al., 2012; Plichta and Scheres, 2014), as is the modulation of reward-related processing by methylphenidate (Dodds et al., 2008). Indeed, besides acting on noradrenaline transporters, methylphenidate acts by blocking dopamine transporters (DAT), which are more abundant in the striatum than in the prefrontal cortex (Volkow et al., 1995; Ciliax et al., 1999).
The observation that both cognitive control deficits and reward-related deficits contribute towards ADHD concurs with the dual pathway model of ADHD, according to which two subtypes of ADHD exist with different developmental pathways, underpinned by different neural circuits and modulated by different branches of the dopamine system (Sonuga-Barke, 2002, 2003, 2005; for more recent models, see Durston et al., 2011; de Zeeuw et al., 2012). More specifically, disturbances in the executive mesocortical dopamine circuit, encompassing the dorsal striatum, dorsomedial thalamus and dorsolateral prefrontal cortex, underlie cognitive deficits in ADHD, whereas motivational deficits are grounded in disturbances in the mesolimbic reward circuit, including the ventral striatum and the orbitofrontal cortex. Here, we approach the issue from a different angle by asking whether cognitive task-related processing deficits and their remediation by methylphenidate reflect indirect modulation of motivation and reward-related processing in the striatum rather than direct modulation of prefrontal processing. This question is grounded in current neuroanatomical and neurochemical models that emphasize a hierarchical arrangement of spiralling striatonigrostriatal loops, allowing directional interaction between motivational and cognitive circuits (Haber et al., 2000; Haber, 2003; Ikeda et al., 2013). Furthermore, it concurs generally with a large body of work showing that striatal dopamine is important not just for motor control but also for cognitive functioning (e.g. Cools et al., 1984). It also follows directly from work showing that methylphenidate-induced changes in striatal dopamine release can contribute towards cognitive (attentional) symptoms in ADHD (Glow and Glow, 1979; Volkow et al., 2012). The hypothesis also concurs with observations that cognitive deficits in children with ADHD can be remediated by increases in motivation (Konrad et al., 2000; Slusarek et al., 2001; Uebel et al., 2010), although inconsistent findings have also been reported (Oosterlaan and Sergeant, 1998; Desman et al., 2008; Shanahan et al., 2008; Karalunas and Huang-Pollock, 2011). None of these studies, however, speak to the neural mechanisms of such motivational effects and their modulation by methylphenidate.
Here, we aimed to assess whether cognitive task-related processing deficits in adult ADHD can be a function of reward-related striatal functioning using functional MRI (fMRI). To index reward effects on cognitive task-related processing, we used a rewarded task-switching paradigm that we established previously to be particularly sensitive to changes in striatal dopamine transmission (Aarts et al., 2010, 2011, 2012, 2014a, 2014b).
One major challenge for studies aiming to isolate dopaminergic drug effects is that such dopaminergic drug effects vary considerably across different individuals as a function of (genetically determined) baseline levels of dopamine (Verheij and Cools, 2008; Cools and D'Esposito, 2011; van Holstein et al., 2011). Previous work suggests the possibility that the effects of methylphenidate emerge only when taking into account such interindividual differences (Clatworthy et al., 2009), for example by exploiting known common polymorphisms in dopamine genes. Here, we stratified our sample by interindividual variation in the 40-bp variable number of tandem repeats (VNTR) polymorphism in the 3′ untranslated region (3′-UTR) of the DAT gene (DAT1, SLC6A3). This is based on several lines of evidence, suggesting an important role for the DAT in the pathophysiology of ADHD. The DAT is the main mechanism responsible for clearing extracellular dopamine in the striatum. Thus, genetic variation of the DAT1 gene might lead to interindividual variation in the availability of DATs and subsequently in dopamine levels. Although it has remained inconclusive in the literature as to which allele leads to decreased DAT availability (Costa et al., 2011; Faraone et al., 2014), genetic fMRI studies have consistently shown the 9-repeat (9R) allele to be associated with increased striatal reward responses (Dreher et al., 2009; Forbes et al., 2009; Aarts et al., 2010). Furthermore, methylphenidate exerts its action in the striatum by blocking the DAT (Volkow et al., 1998, 2002); mice that lack the DAT (i.e. DAT1 knockout mice) show ADHD-like behaviour (Giros et al., 1996; Gainetdinov et al., 1999), and several dopaminergic genes, including the DAT1 genotype, have been implicated in ADHD (Faraone et al., 2005; Brookes et al., 2008; Franke et al., 2008; for a review, see Durston et al., 2009; Gizer et al., 2009; Franke et al., 2010).
In summary, in this pilot study, we tested the hypothesis that the effects of reward motivation on task-switching and striatal BOLD signal vary as a function of the DAT1 genotype in adult patients with ADHD, when they were OFF relative to ON their methylphenidate regimen, compared with healthy controls.
Methods
Participants
We present data from 23 patients with ADHD (mean±SE age 35.74±2.36; 14 men) and 26 healthy control participants (mean±SE age 38.08±2.00; 11 men). Patients visited our centre on two occasions: once after intake of methylphenidate and once after withdrawal from methylphenidate. Healthy controls were also tested on two occasions, without any methylphenidate (see the Procedure section).
Initially, we recruited 57 participants (29 patients with ADHD and 28 healthy controls) from an ongoing study on ADHD and genetics, IMpACT-NL (Hoogman et al., 2011, 2013; Onnink et al., 2014; http://www.impactADHDgenomics.com), in which they were tested extensively, genotyped and diagnosed (Table 1). Patients were included if they fulfilled the DSM-IV-TR criteria for ADHD in childhood as well as adulthood. All participants were assessed using the Diagnostic Interview for Adult ADHD (Kooij and Francken, 2007). The Structured Clinical Interviews for DSM-IV (SCID-I and SCID-II) were administered. Assessments were carried out by trained professionals (psychiatrists or psychologists). In addition, a quantitative measure of clinical symptoms was obtained using the ADHD rating scale-IV (Kooij et al., 2005). Exclusion criteria for participants were alcohol or substance addiction in the last 6 months, current psychosis, manic episodes, obsessive–compulsive disorder or eating disorders (assessed using SCID-I), full-scale IQ estimate less than 70 (assessed using the Wechsler Adult Intelligence Scale-III), neurological disorders, sensorimotor disabilities and non-Caucasian ethnicity. An additional exclusion criterion for healthy comparison participants was a current or a past neurological or psychiatric disorder according to SCID-I.
Table 1.
Demographics, impulsivity and diagnostic interview for diagnosis×DAT1 group
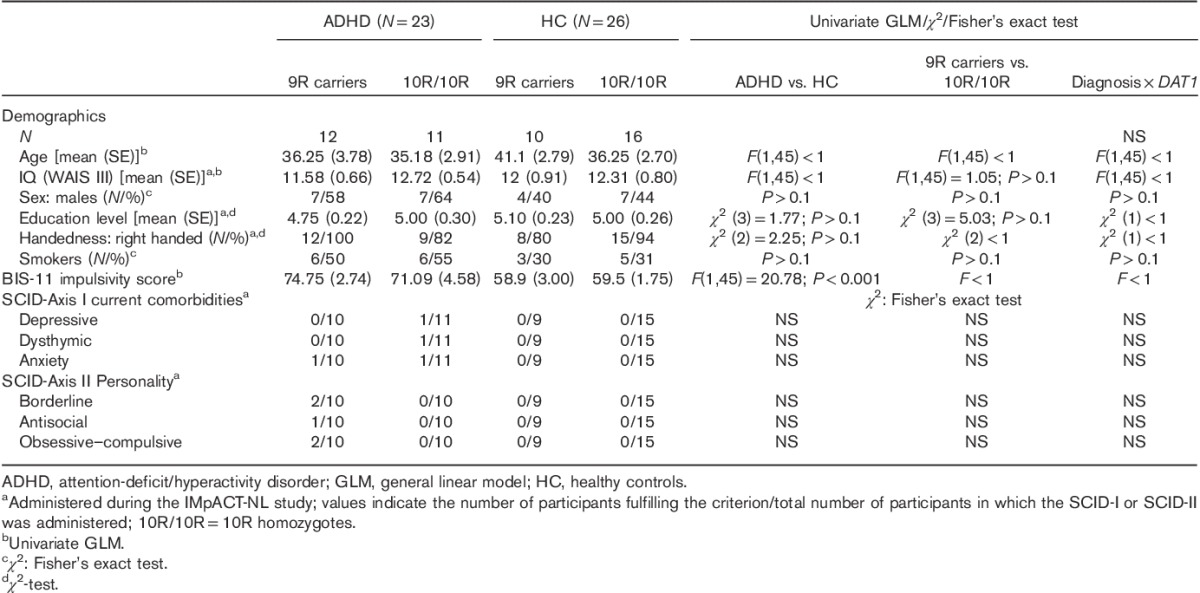
Three patients did not complete the testing sessions. Two patients were excluded because they did not follow instructions on methylphenidate withdrawal and/or intake (see the Procedure section) and one because of excessive head movement. One healthy control participant was excluded from analysis because of the suboptimal quality of the structural data, leading to normalization difficulties, and one for fulfilling the criteria for an ADHD diagnosis according to the ADHD rating scale-IV (Kooij et al., 2005) (see the Neuropsychological assessment section). Hence, 23 patients with ADHD and 26 healthy controls were included in the final analyses.
All patients had a current prescription of methylphenidate [either immediate-release (Ritalin; N=5; mean±SD 44±22.74 mg/day), or sustained release (Concerta; N=18; mean±SD 48.5±21.19 mg/day), and three of them occasionally took Ritalin in addition to Concerta]. All participants were native speakers of Dutch. Participants were compensated for participation and provided written informed consent in a manner approved by the local ethics committee on research involving human participants (CMO Arnhem-Nijmegen 2009/058, NL27180.091.09).
Procedure
All participants were asked to abstain from alcohol on the day of testing and from nicotine and caffeine at least 1 h before arriving at the centre. The patients were tested once OFF (i.e. withdrawn from Ritalin for 24 h and from Concerta for 48 h before testing) and once ON methylphenidate [i.e. after intake of (mean±SD) 13.15±5.55 mg of Ritalin, the equivalent of (mean±SD) 0.16±0.05 mg/kg body weight of Ritalin, half an hour before arriving at the centre]. Patients using sustained-release methylphenidate were prescribed an equivalent dose [instant dose (mg)=sustained dose (mg)×0.278] of immediate-release methylphenidate by the psychiatrist (J.B.) for 1 day (three doses a day). Three patients using additional medication (one antihistamine and two SSRIs) were asked to take the same dose on both sessions. The order of the ON and OFF session was approximately counterbalanced across participants (Table 2). The healthy control group did not take methylphenidate, but was nevertheless tested twice to rule out order effects. Control data were averaged across the two sessions. Sessions were separated by at least 1 week and both sessions took place at approximately the same time of day. With the exception of medication state, the procedure was identical for both groups and both sessions.
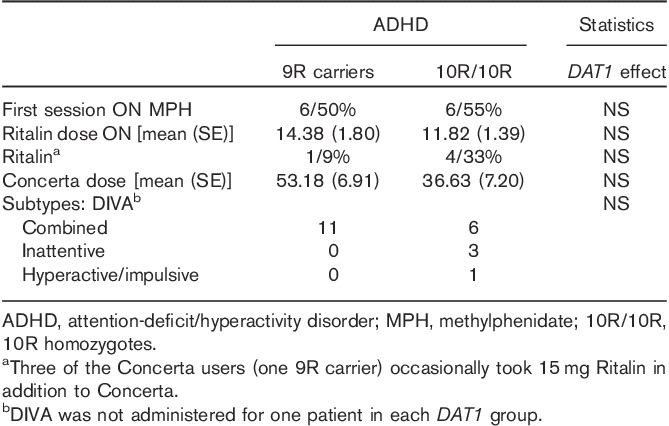
Table 2.
Attention-deficit/hyperactivity disorder characteristics
Cognitive task with reward manipulation
Participants were scanned while performing an established precued task-switching paradigm (Fig. 1) with a reward manipulation (Aarts et al., 2010, 2012, 2014a; van Holstein et al., 2011). The paradigm started ∼60 min after arrival (mean±SD 91.8±16.1 min after drug intake). The task was programmed and presented using the Presentation (R) software (Version 13, www.neurobs.com).
Fig. 1.
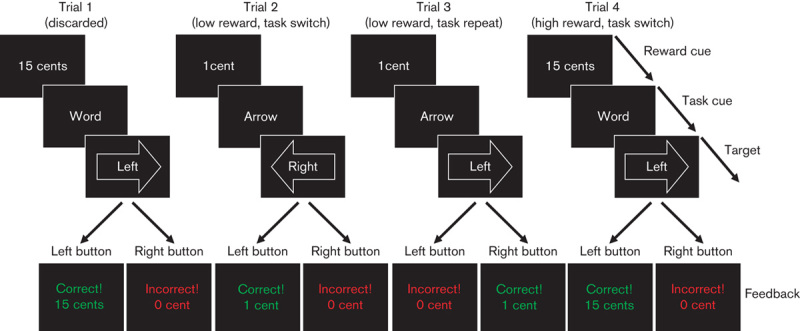
Task-switching paradigm with reward manipulation. Participants were instructed to respond either to the direction indicated by the arrow (i.e. ← or →) or to the direction indicated by the word (i.e. ‘left’ or ‘right’) with a left or a right button press. The task performed on a particular trial either changed compared with the preceding trial (i.e. switch trial; arrow–word or word–arrow) or remained the same (i.e. repeat trial; arrow–arrow, word–word). In addition, we manipulated the amount of anticipated reward (e.g. 1 vs. 15 cents) on a trial-by-trial basis by means of a reward anticipation cue. At the start of each trial, this cue indicated the amount of reward on that trial, providing a correct and sufficiently fast button press (see also Aarts et al., 2010).
Participants had to respond to incongruent arrow–word combinations, either by responding to the direction of the arrow or the direction indicated by the word (‘left’ or ‘right’). A task-cue appeared 400 ms before the target indicating the task (arrow or word) that the participant had to perform on the current trial. Relative to the previous trial, the task either changed unpredictably (from arrow to word or vice versa; switch trial) or remained the same (repeat trial). The critical measure of interest, the switch cost, was calculated by subtracting performance on repeat trials from that on switch trials. In addition, we manipulated reward motivation, to assess the effect of reward on task-switching, by presenting high and low reward cues before the task cue. The reward-cue informed the participants whether 1 cent (low reward) or 15 cents (high reward) could be earned with a correct and sufficiently quick response. Immediately after the response, feedback was provided (e.g. ‘correct! 15 cents’). There was a variable interval of 2–6s between the reward-cue and the task-cues. Participants used their right index and middle fingers to respond on a button box.
On both sessions, the task was practiced twice outside the scanner and once inside the scanner. The first practice block contained 24 trials with the task cue, target and feedback (correct/incorrect). As soon as participants succeeded in completing this block with less than five errors, the second practice block of 24 trials was performed, in which the reward cues were included. The third and final practice block was performed during the acquisition of the anatomical scan. The mean response times (RT) of the correct trials per trial-type (arrow-repeat, arrow-switch, word-repeat, word-switch trials) in this third practice block were used as the response deadline in the main experiment. This ensured equal difficulty across participants and sessions.
The main experiment consisted of 160 trials and lasted∼35 min with a 30 s break after every 32 trials. In the break, the amount of money that the participant had earned thus far was displayed on the screen and participants were told in advance that the total amount would be added to their financial compensation as a bonus.
Neuropsychological assessment
During the first session, participants completed the Barratt Impulsiveness Scale (BIS-11a; Patton et al., 1995), a self-report trait measure of impulsivity. At the beginning of both sessions, participants completed the Bond and Lader (1974) visual analogue scale for a comparison of mood between sessions (16 moods rated on a scale of 0–100, resulting in three mood categories) and an ADHD symptom rating scale (Kooij et al., 2005) to assess self-reported ADHD symptoms. Motor speed was measured using the box completion task (Salthouse, 1996), sustained attention with the digit vigilance or the number cancellation task (Lewis and Kupke, 1977) and verbal fluency with the begin letters D, A and T (Spreen and Benton, 1977).
Genotyping
DNA was isolated from EDTA blood samples. Genotyping of the 40-bp VNTR in the 3′-UTR of SLC6A3/DAT1 was carried out as described previously (Hoogman et al., 2013) at the department of Human Genetics of the Radboud University Medical Centre. In line with previous studies reporting the effect of this variant, we established a group of carriers of the 9R allele (i.e. the risk factor for adult ADHD) and a group homozygous for the 10R allele (Colzato et al., 2010; Rokem et al., 2012) (Table 1). We preselected our participants from a previous sample (Hoogman et al., 2013) to homogenize sample numbers per group (diagnosis×genotype) as much as possible. Therefore, Hardy–Weinberg equilibrium was not considered.
In the ADHD group, 12 individuals were carriers of the 9R allele and 11 individuals were homozygous for the 10R allele (Table 1). In the healthy control group, 10 individuals were carriers of the 9R allele and 16 individuals were homozygous for the 10R allele. We performed a power calculation in G*Power (http://www.gpower.hhu.de) on the basis of the effect sizes obtained in an independent dataset using a similar rewarded task-switching paradigm and the same VNTR in the DAT1 gene in healthy volunteers (Aarts et al., 2010). The power calculation showed that we would need at least eight participants per group (four groups: genotype×diagnosis) to obtain significant effects of genotype on striatal BOLD responses during the rewarded task-switching [effect size=0.78; α=0.05; power (1−β)=0.8]. Currently, our smallest group includes 10 participants.
Functional magnetic resonance imaging data acquisition
Participants were scanned in a 3T MR scanner (Magnetom TrioTim; Siemens Medical Systems, Erlangen, Germany) using an eight-channel head coil. T2*-weighted images were acquired with a gradient echo planar imaging (EPI) sequence (30 axial slices, repetition time=2020 ms, echo time=30 ms, voxel size=3.5×3.5×3 mm, field of view=224 mm, flip angle=80°). All functional images were acquired in a single run. Stimuli were presented on a computer display projected onto a mirror attached to the head coil. The first four volumes were discarded to allow for T1 equilibrium. Before the acquisition of the functional images, a high-resolution T1-weighted MP-RAGE anatomical scan was obtained (192 sagittal slices, repetition time=2300 ms, echo time=3.03 ms, voxel size=1×1×1 mm, field of view=256 mm).
Functional magnetic resonance imaging statistical analyses
Data were preprocessed and analysed using SPM5 (Wellcome Department of Cognitive Neurology, London, UK). First, functional EPI images were spatially realigned and corrected for differences in slice acquisition timing. Structural and functional data were co-registered and normalized to a standard anatomical space (Montreal Neurological Institute) using a unified segmentation procedure (Ashburner and Friston, 2005). The normalized images were smoothed with an isotropic 8-mm full-width-at-half-maximum Gaussian kernel.
The preprocessed fMRI time series were analysed at the first level using an event-related approach in the context of the general linear model (GLM). Our statistical model on the first (participant-specific) level considered the factors reward (high, low), task (arrow, word), task-switching (repeat, switch) and feedback (correct-1 cent, correct-15 cents, error-0 cents, too late-0 cents). This resulted in 21 regressors of interest: two regressors for reward-cues, eight regressors for targets (reward×task×task-switching) and four regressors for feedback. All regressors of interest were modelled as a stick function (duration=0) convolved with a canonical haemodynamic response function. In addition, breaks (duration of 30 s), six motion parameters and their derivatives were modelled as regressors of noninterest. Finally, we included three regressors of noninterest to account for movement-induced intensity changes using the mean time series from the segmented white matter, cerebral spinal fluid and out-of-brain signals (Majdandzić et al., 2007; Verhagen et al., 2008). High-pass filtering (128 s) was applied to the time series of the functional images to remove low-frequency drifts.
At the second level, the reward×task-switching contrast images from the first level were used in three GLMs to assess the effects of reward during task-switching: two models to assess the interaction with the DAT1 genotype and diagnosis (HC vs. ADHD OFF and ADHD ON vs. HC) and one model to test the interaction with the DAT1 genotype and medication (ADHD ON vs. ADHD OFF). Statistical inference (P<0.05) was performed at the cluster level, correcting for multiple comparisons over the search volume (the whole brain). The intensity threshold necessary to determine the cluster-level threshold was set at P less than 0.001, uncorrected. For each effect, we report the t-values at the voxel level, the whole-brain corrected P-values for the cluster (Pcluster) and the size of the cluster (k). In addition, supplementary exploratory analyses were carried out, for which the uncorrected threshold was set to P less than 0.001, and we report the t-values and P-values (Puncorr) at the voxel level.
Behavioural statistical analyses
We excluded the first trial of each block (five trials in total) because these cannot be considered as either repeat or switch trials. All trials to which participants responded (i.e. all trials except response omissions) were included in the analysis, even if the response was too late for a reward to be obtained. For the analysis of the mean RTs, we excluded responses faster than 200 ms. For each participant, we calculated the mean RTs for all the correct responses and the proportion of errors for each of the four conditions, that is reward (high–low)×switching (switch-repeat). To maximize homogeneity of variances between groups and to ensure normal distribution of the data, a natural logarithm (LN) transformation was applied to the mean RTs. The mean proportions of incorrect responses were transformed using the following formula: 2×arcsin√x (Sheskin 2004). Levene’s tests of homogeneity of variances and Shapiro–Wilk tests of normality showed that this transformation was successful in improving variance between groups and the distribution of the data.
Proportions or errors and mean RTs were analysed using a repeated-measures GLM with the within-participant factors reward (high, low), switching (repeat, switch), the between-participant factor DAT1 genotype (9R carriers, 10R homozygotes) and either the between-participant factor diagnosis (ADHD or healthy control) or the within-participant factor medication (ON, OFF). Effects were considered significant when P-values were less than 0.05.
Statistical analysis of mood measures and neuropsychological tests
Mood values were calculated for each session and reduced to three factors: contentedness, alertness and calmness, according to Bond and Lader (1974). Neuropsychological and demographic differences between groups or medication states and their interaction with the DAT1 genotype were tested using SPSS (IBM Corp. IBM SPSS Statistics for Windows, Version 21.0. Armonk, New York, USA) with univariate or repeated measures GLMs or their nonparametric counterparts (Wilcoxon signed rank or Mann–Whitney U-tests, respectively; Table 3). Nonparametric DAT1 genotype×medication interactions were assessed using a Mann–Whitney U-test of the difference between the score OFF and ON medication. Nonparametric DAT1 genotype×diagnosis effects were assessed using the Kruskal–Wallis test (Table 3). An effect was considered significant when P less than 0.05.
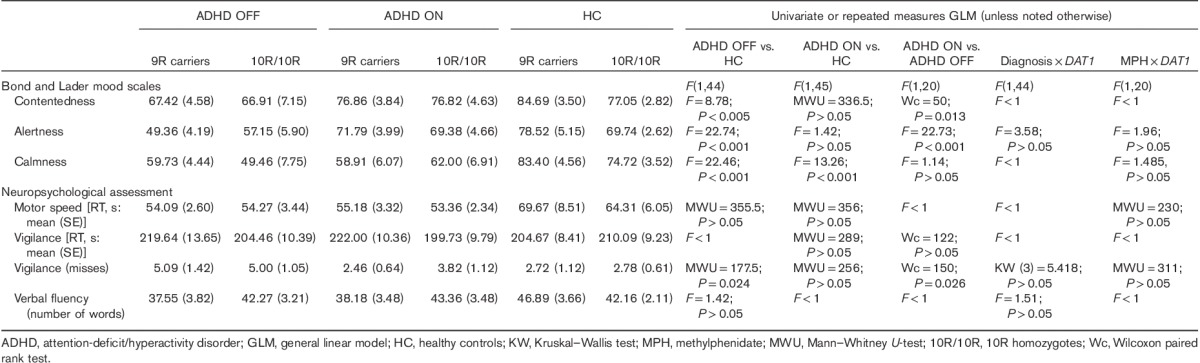
Table 3.
Mood and neuropsychological assessment
Results
Functional magnetic resonance imaging effects
Main task effects
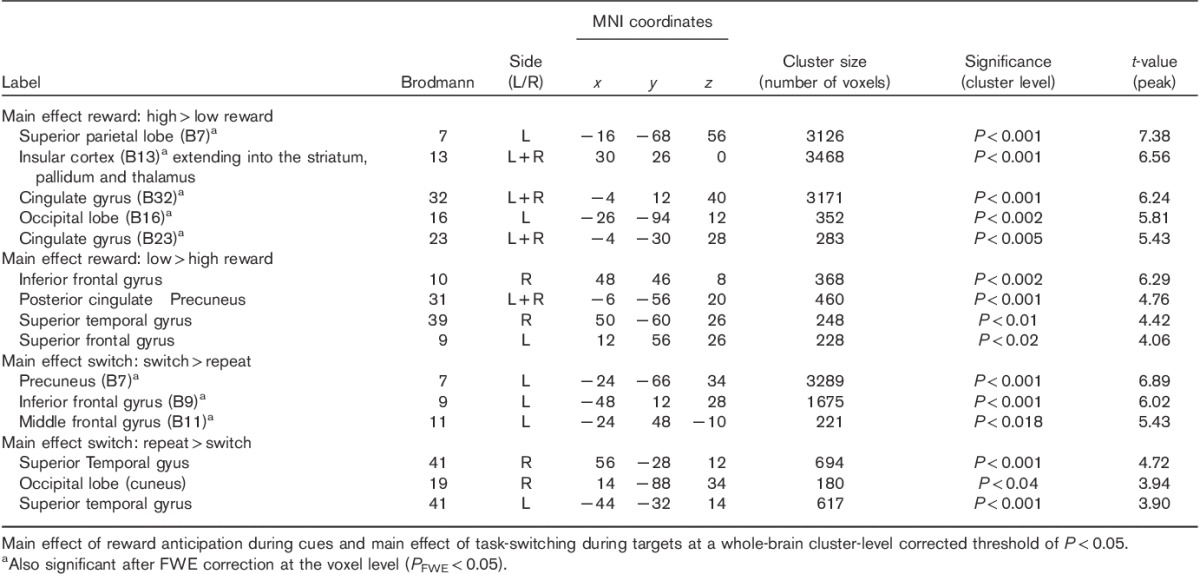
Across groups and sessions, the cue indicating a high reward, compared with the cue indicating a low reward, elicited a robust response in regions in the striatum, the frontal cortex and the occipital cortex (Table 4). There was also a strong main effect of task-switching during the targets, as evidenced by a greater response in the frontal and parietal regions on switch compared with repeat trials (Table 4).
Table 4.
BOLD maxima across all participants
ADHD OFF versus healthy controls
The BOLD signal in the dorsal striatum varied highly significantly as a function of ADHD diagnosis (patients with ADHD OFF their medication versus healthy controls), DAT1 genotype (9R carriers vs. 10R homozygotes) and task (reward×task-switching) (x, y, z=−20, 4, 16; t=4.92; Pcluster<0.001; k=324; Fig. 2a-I). This finding concurs with our hypothesis that the effect of reward on task-switching in the striatum would vary as a function of the DAT1 genotype and diagnosis (healthy controls compared with patients with ADHD). The striatal effect was observed because of a greater task-related signal in patients with ADHD carrying the 9R allele compared with 9R carriers in the healthy control group (reward×task-switching×diagnosis in 9R carriers: x, y, z=−18, 2, 16; t=4.90; Pcluster=0.001; k=333) and greater task-related signal in the 9R-carrying patients with ADHD compared with the 10R homozygous patients with ADHD (reward×task-switching×DAT1 in patients with ADHD OFF medication: x, y, z=−12, −4, 6; t=4.96; Pcluster=0.002; k=295). To illustrate this effect, we extracted the beta values from the cluster in the left dorsal striatum shown in Fig. 2a-I and plotted the results in Fig. 2b. The only other significant neural difference between the ADHD group OFF medication and the healthy control group was observed in the posterior cingulate cortex (reward×task-switching×diagnosis×DAT1: x, y, z=−6, −12, 46; t=5.56; Pcluster<0.001; k=338).
Fig. 2.
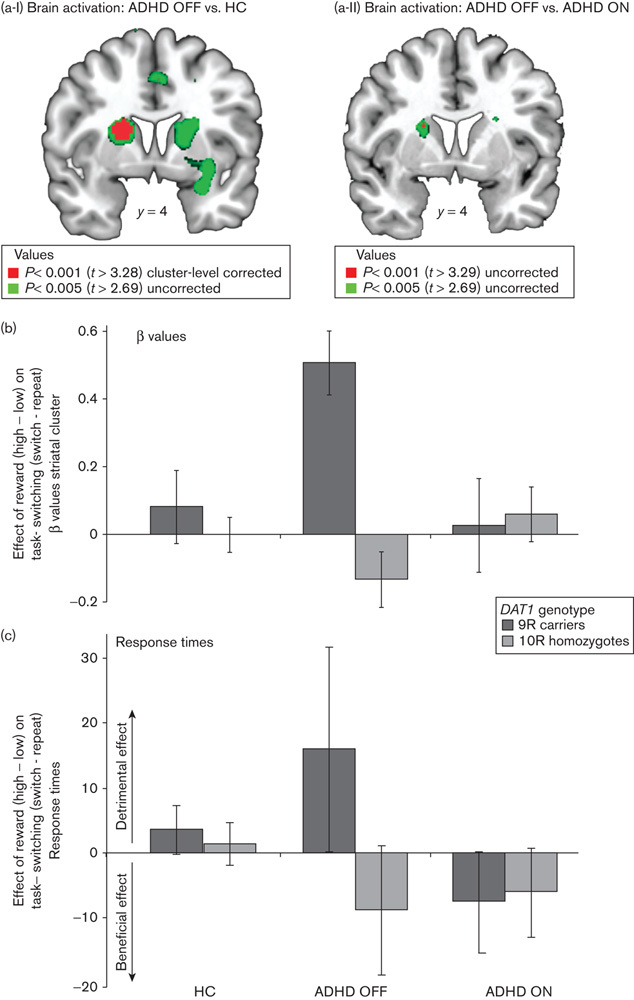
Rewarded task-switching as a function of the DAT1 genotype in patients with attention-deficit/hyperactivity disorder (ADHD) ON and OFF their methylphenidate medication, relative to healthy controls (HC). (a-I) Increased dorsal striatal responses during rewarded task-switching for patients with ADHD OFF methylphenidate relative to healthy controls, as a function of the DAT1 genotype; (a-II) Increased dorsal striatal responses during rewarded task-switching for patients with ADHD OFF methylphenidate relative to when ON methylphenidate, as a function of the DAT1 genotype; (b) The β values from the whole-brain cluster-corrected cluster in the left striatum depicted in (a-I), showing the direction of the effect; (c) The response times during rewarded task-switching. Positive values reflect an increased switch cost (i.e. slower on switch than on repeat trials) for high reward relative to low reward trials, that is a detrimental effect of reward on the switch cost. Error bars represent the SE of the difference between high reward (switch-repeat) minus low reward (switch-repeat).
ADHD ON versus healthy controls and ADHD OFF versus ADHD ON
There was no longer an effect of diagnosis when comparing patients with ADHD ON medication with healthy controls, suggesting that the aberrant striatal response was restored by medication. Although a direct comparison of the ON and OFF session (ADHD ON vs. ADHD OFF medication) did not reach significance at our stringent threshold, exploratory analyses confirmed that task-related responses in the same region in the striatum were reduced for patients ON relative to OFF medication depending on the DAT1 genotype (reward×task-switching×DAT1×medication: x, y, z=−20, 4, 16; t=3.63; Puncorr<0.001; Fig. 2a-II and b). This is generally in line with the hypothesis that the effect of methylphenidate and reward motivation on task-switching would vary as a function of the DAT1 genotype and medication status (patients with ADHD ON compared with OFF their medication).
Behavioural effects
Main task effects
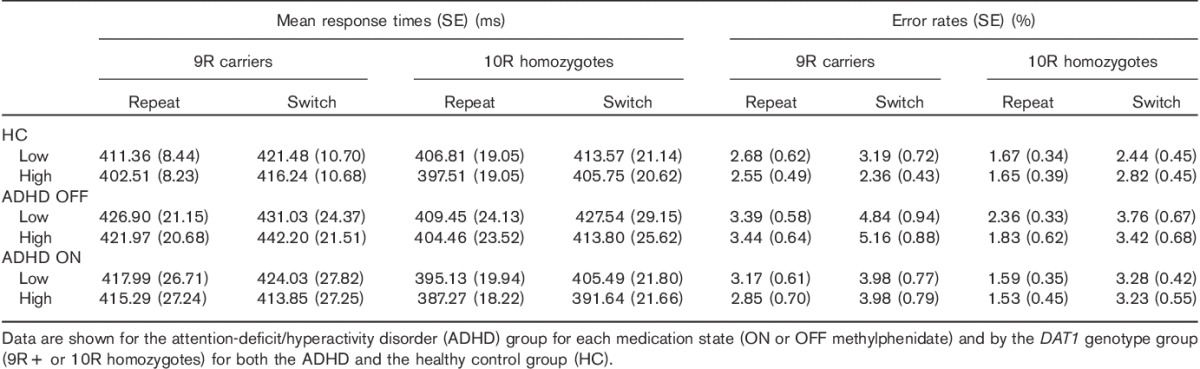
Participants responded more quickly after a high than a low reward (i.e. across groups, irrespective of diagnosis and genotype), as evidenced by a main effect of reward on RTs [F(1,48)=24.36; P<0.001]. Participants also responded more quickly on repeat than switch trials [main effect of task-switching: F(1,48)=24.91; P<0.001]. In addition, participants made more errors on switch than repeat trials [main effect of task-switching F(1,48)=28.67; P<0.001; Table 5].
Table 5.
Means (SE) for response times and error rates (% errors)
Attention-deficit/hyperactivity disorder OFF versus healthy controls
There were no significant differences in RT between the ADHD group OFF medication and healthy controls (Fig. 2c). However, the groups did differ in terms of the reward effect (i.e. low–high reward) on error rates, across switch and repeat trials. This effect depended on the DAT1 genotype [reward×diagnosis×DAT1: F(1,45)=5.56; P=0.023]: irrespective of task-switching, the 10R homozygotes in the ADHD group made fewer errors on high than low reward trials, relative to the 10R homozygotes in the healthy control group [reward×diagnosis in 10R homozygotes: F(1,25)=7.03; P<0.02; Table 5], whereas there was no difference between the 9R carriers in the ADHD group and the healthy 9R group. The critical effect of reward on task-switching errors did not differ between the patients with ADHD OFF medication and the healthy control group as a function of DAT1 genotype [the critical reward×task-switching×diagnosis×DAT1 interaction: error rates F(1,45)<1; RTs F(1,45)=1.92; P>0.1].
Attention-deficit/hyperactivity disorder ON versus healthy controls
There were no significant differences in RT between the ADHD group ON medication and healthy controls. Switch costs in error rates were significantly greater in the ADHD group ON medication than in the healthy control group [task-switching×diagnosis: F(1,45)=6.44; P<0.02]. The critical effect of reward on task-switching did not differ between the patients with ADHD ON medication and the healthy control group as a function of DAT1 genotype [the critical interaction between reward×task-switching×diagnosis×DAT1: error rates F(1,45)=1.37; P>0.1; RTs F(1,45)<1].
ADHD OFF versus ADHD ON
There was no significant difference between the two medication sessions in RTs or error rates. The critical DAT1 by medication by reward task-switching interaction only trended towards significance for RTs [reward×task-switching×medication×DAT1: F(1,21)=3.23; P=0.087; Fig. 2c], and was absent for error rates [reward×task-switching×medication×DAT1: F(1,21)<1].
In summary, unlike the brain data, the behavioural data did not show any significant effects of diagnosis or medication status and/or genotype on how anticipated reward influences task-switching performance (i.e. reward×task-switching effects). To assess whether the increased BOLD signal in the striatum of 9R-carrying patients with ADHD was accompanied, if anything, by behavioural impairment or improvement, we examined the numerical (marginal trend) pattern in RTs (Fig. 2c). Disentangling this marginally significant effect [reward×task-switching×medication×DAT1: F(1,21)=3.23; P=0.087] showed that 9R-carrying patients OFF medication tended to show a greater switch cost on high than low reward trials compared with these patients ON their medication [reward×task-switching×medication in 9R carriers: F(1,11)=4.40; P=0.06; Fig. 2c]. These data suggest that the increased dorsal striatal responses in patients with ADHD carrying the 9R allele are accompanied, if anything, by a detrimental effect of reward on task-switching that can be remediated by methylphenidate (Fig. 2b and c).
Demographic and neuropsychological data
Table 1 summarizes the demographic and neuropsychological data of the patients with ADHD and healthy controls for the two DAT1 genotype groups. There was no difference between patients and healthy controls, or between the 9R-carrying and 10R homozygous group, in terms of age, IQ, sex, handedness, smoking status and level of education (Table 1), nor an interaction between diagnosis and the DAT1 genotype. As expected, the patients with ADHD scored higher on the Barratt Impulsiveness Scale (mean±SE: 73.00±2.58); that is, they were more impulsive than the healthy controls [mean±SE: 59.27±1.54; t (47)=4.70; P<0.001]. There were no differences in current SCID Axis I disorders or SCID Axis II personality traits as a function of diagnosis, DAT1 genotype or diagnosis×DAT1 genotype.
Counterbalancing of the ON and OFF sessions within the two DAT1 genotype patient groups was successful: there was no difference between the two DAT1 groups in the number of patients being ON medication during the first session. Furthermore, there were no significant differences in the dose of Ritalin or Concerta between the DAT1 genotype groups, nor in the number of patients usually taking either form of methylphenidate, or in their ADHD subtype (i.e. combined, inattentive or hyperactive/impulsive) (Table 2).
Table 3 summarizes the mood and neuropsychological test scores. Most importantly, there were no interactions between the DAT1 genotype and either medication state (ON or OFF) or diagnosis on mood measures or on the neuropsychological test scores. However, patients OFF medication were reportedly less content and less alert than healthy controls, and compared with when they were ON medication (Table 3; contentedness: ADHD ON median 83, range 41.6–95.2; ADHD OFF median 67.16, range 23.2–97.6). In addition, healthy controls reported more calmness than the patients, both ON and OFF medication (Table 3). There were no differences in motor speed (box completion task) on the time to complete the vigilance test (number cancellation RT) or in verbal fluency. We did observe a difference between the ADHD group OFF medication and the healthy control group for missed items on the vigilance test, that is the ADHD group OFF their medication missed more numbers (median 4, range 0–17) relative to the healthy control group (median 2, range 0–11) and relative to when ON medication (median 3, range 0–13). This difference was no longer present when comparing the ADHD group ON medication with the healthy control group (Table 3).
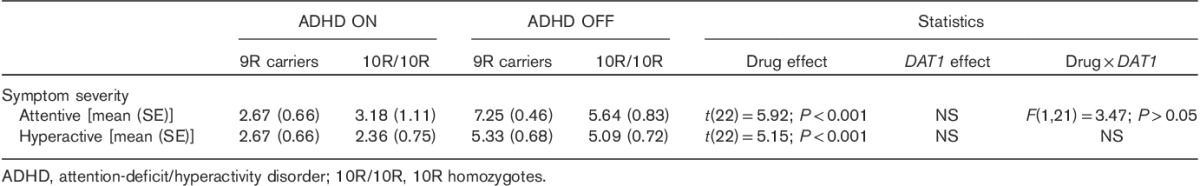
As expected, methylphenidate ameliorated symptom severity (Table 6) both on attentive and on hyperactive symptoms. We did not observe effects of the DAT1 genotype, nor an interaction between the DAT1 genotype and medication status on symptom severity (Table 6).
Table 6.
Self-reported symptom severity
Discussion
We investigated the effects of reward motivation on task-switching in adult patients with ADHD, ON and OFF methylphenidate, relative to a matching healthy control group. Task-related BOLD responses were assessed as a function of interindividual variability in the DAT1 gene. When OFF medication, adults with ADHD showed greater effects of reward on dorsal striatal BOLD responses during task-switching than healthy controls. Critically, this effect was only observed when taking the DAT1 genotype into account, resulting in a strong genotype-by-diagnosis interaction. Specifically, patients carrying the 9R allele showed exaggerated striatal responses relative to healthy controls carrying the same allele as well as relative to patients homozygous for the 10R allele. These aberrant striatal responses were normalized when patients with ADHD were ON medication, such that they no longer differed from those of controls. In short, the present pilot study shows a dysfunctional influence of reward motivation on task-switching in the dorsal striatum of adult patients with ADHD, but only in those carrying the 9R risk allele. These findings, albeit preliminary because of the small sample size, suggest that abnormal cognitive task-related processing in adult ADHD depends critically on interindividual trait differences in striatal dopamine transmission as well as on the motivational state of the individual patient.
The present results indicate the importance of taking into account interindividual variability, as for example indexed by the DAT1 genotype, when assessing task-related BOLD effects in ADHD. This generally concurs with previous fMRI studies in youth with ADHD, which have found that striatal responses during reward anticipation (Paloyelis et al., 2012), as well as striatal responses during more cognitive tasks, that is Go/No-Go paradigms (Durston et al., 2008; Bédard et al., 2010), depend on variations in the DAT1 genotype. A recent study in adults with ADHD failed to extend the effect of the DAT1 genotype on striatal reward responses during reward anticipation, observed in youth (Paloyelis et al., 2012), to adult ADHD (Hoogman et al., 2013). In the current sample with ADHD adults, DAT1 effects on reward-related striatal responses did emerge, but only as a function of cognitive task-related processing. This suggests that, in adults with ADHD, the translation of reward information into (effortful) cognitive processing might be more strongly dependent on variability in the DAT1 gene than reward anticipation itself.
Our study shows that patients with ADHD OFF medication show abnormal BOLD responses in the caudate nucleus during rewarded task-switching, an effect that relied on striatal dopamine signalling as indexed by the DAT1 genotype. In accordance, the caudate nucleus – known to be involved in cognitive flexibility (Cools, 1980; Aarts et al., 2011) – is well positioned to incorporate motivational influences from more ventral regions in the striatum through feed-forward dopaminergic projections (Haber et al., 2000; Grahn et al., 2008; Ikeda et al., 2013). The finding is also remarkably consistent with our previous work using genetic fMRI and PET imaging in healthy volunteers, showing that the effects of reward motivation on cognitive control are altered by dopaminergic transmission in the left caudate nucleus (Aarts et al., 2010, 2014b). In ADHD, Volkow et al. (2009) have shown that dopaminergic transmission in reward-related brain regions is associated with symptoms of inattention, and that connectivity between neural reward and attention networks is impaired (Tomasi and Volkow, 2012). Here, we show that cognitive task-related processing deficits in the striatum (i.e. during task-switching) are modulated by motivation as well as the DAT1 genotype in ADHD. Unlike earlier suggestions (Sonuga-Barke, 2002, 2003; de Zeeuw et al., 2012), ADHD might not be accompanied by isolated deficits in either motivational or cognitive/executive processing pathways, but rather by deficits in the integration between these pathways.
The present finding extends to ADHD our previous work in young healthy volunteers showing that the effects of reward motivation on task-switching and associated striatal signal depend on the DAT1 genotype (Aarts et al., 2010; see also van Holstein et al., 2011). Unlike that previous study, however, the present study did not show any DAT1 genotype effects on rewarded task-switching in healthy controls, in neural or behavioural terms. We are surprised at this lack of effect, but believe that it might reflect a difference in the demographics between the current control group, which was matched to the ADHD group, and the groups in our previous studies that primarily included university students. The most obvious difference is in terms of age, with the current control group being older (mean 38.12 years, SD 10.20) than the healthy volunteers in our previous studies (mean 21.58 years, SD 2.06; and mean 22 years, SD 2.32, for Aarts et al., 2010; van Holstein et al., 2011, respectively). Indeed, studies have consistently observed a reduction in dopamine signalling starting in young adulthood (e.g. Volkow et al., 1996; Reeves et al., 2002). Importantly, the increases in striatal BOLD in the 9R-carrying patients OFF medication were, if anything, accompanied by impaired performance (i.e. increased RT switch cost for high vs. low reward trials, relative to when ON medication). These results contrast with our findings in younger 9R-carrying healthy volunteers who showed increased striatal responses as well as better task-switching performance following high versus low reward cues relative to 10R-homozygotes (Aarts et al., 2010). This suggests that the hyperactivation in the dorsal striatum during rewarded task-switching in the 9R-carrying patients OFF medication is maladaptive for behaviour. The notion of maladaptive striatal hyperactivation in 9R-carrying patients with ADHD is in line with the finding that the 9R allele is the risk allele in adult ADHD (Franke et al., 2010). However, the absence of significant behavioural differences relative to healthy controls precludes statements of normality in terms of performance.
The aberrant striatal responses during rewarded task-switching in patients with ADHD (specifically 9R carriers) relative to controls were absent when patients were ON medication. This suggests that methylphenidate normalized striatal responses, although we only obtained trend effects (i.e. at P<0.001 uncorrected for multiple comparisons) when directly comparing patients ON versus OFF methylphenidate. Our findings suggest that the effects of methylphenidate on cognitive task-related processing are accompanied by modulation of the striatum. This generally concurs with previous work showing that methylphenidate can normalize striatal responses during cognitive processes such as response inhibition (Vaidya et al., 1998; Shafritz et al., 2004; Epstein et al., 2007; Rubia et al., 2009, 2011). Here, we show that such normalization of task-related dorsal striatal responses and performance by methylphenidate depends both on the DAT1 genotype and on reward motivation. This suggests that reward motivational factors interact with the effects of the DAT1 genotype to bias the cognitive response to methylphenidate. Future work should address the obvious next question, that is, whether the discrepancy in the extant literature on the effects of the DAT1 genotype on the clinical response to medication (Kambeitz et al., 2014) also reflects variability in the patient’s reward motivational state. Cognitive neuroimaging measures of task-related (motivational) processing might be particularly sensitive to detecting DAT1-dependent effects of methylphenidate in ADHD.
It might be noted that the effects in the OFF state could reflect rebound effects because of short-term medication withdrawal. Future studies, with a longitudinal design or comparing medication-naive patients with medicated patients, will need to determine whether the current findings reflect rebound or withdrawal effects rather than an unmedicated ADHD state.
Our findings were obtained with a sample of 23 patients with ADHD and 26 healthy controls. This limited sample size calls for caution when generalizing to the population (Munafò and Gage, 2013) and precludes definitive conclusions. The findings should therefore be considered preliminary and in need of replication, as was recently also explicitly highlighted (Button et al., 2013). Nevertheless, we believe that our findings are robust, given extensive convergent evidence. Indeed, we have previously observed effects of the DAT1 genotype on the BOLD signal during rewarded task-switching in the same striatal region (i.e. left caudate nucleus) as we report here (Aarts et al., 2010). Moreover, we have observed previously that striatal dopamine synthesis capacity in the (left) caudate nucleus predicted the effects of reward on cognitive performance during a focused attention task (Aarts et al., 2014b). It is unlikely that our whole-brain corrected results represent a false-positive effect as our power calculation based on an independent dataset (Aarts et al., 2010; Button et al., 2013) confirmed that our sample should be large enough to obtain significantly meaningful effects (see the Methods section). Replication of the effect in independent larger samples in future studies will further increase confidence in the reliability of the effect.
Previously, we have obtained similar results in a PET study in healthy volunteers, showing that dopaminergic transmission in the left caudate nucleus altered the effects of reward motivation on cognitive control (Aarts et al., 2014b). In that study, we used a Stroop interference paradigm instead of a task-switching paradigm, suggesting that our present results can be extended to other domains of cognitive control. However, future work should confirm whether our findings in ADHD can be generalized to domains other than task-switching. Moreover, future studies should also examine variation in other dopaminergic genes, such as COMT (Bilder et al., 2004), to investigate whether the current findings are limited to striatal dopamine processing.
Conclusion
Our data suggest a dysfunctional influence of reward motivation on cognitive processing (i.e. task-switching) in the dorsal striatum of adult patients with ADHD, who carry the 9R ADHD risk allele. This deficit is remediated when patients are tested ON methylphenidate. These findings indicate an important role for both reward motivation and interindividual trait differences in striatal dopamine transmission in cognitive processing deficits in adult ADHD.
Acknowledgements
The authors thank Paul Gaalman for assistance during the acquisition of the MRI data. This work was supported by a James McDonnell scholar award to R.C.
Conflicts of interest
E.A. is supported by a VENI grant of the Netherlands Organisation for Scientific Research (NWO) (016.135.023) and an AXA Research Fund fellowship. M.v.H. is supported by a Donders Institute Top Talent grant. B.F.’s research is supported by grants by NWO Brain and Cognition (443-09-223) and VICI (016.130.669), the EU FP7 grants Aggressotype (602805) and IMAGEMEND (602450), and the Hersenstichting Nederland. J.B. is supported by NWO Large Investment Grant (1750102007010), NWO Brain and Cognition grants (433-09-242 and 056-13-015), and the EU FP7 grants TACTICS (278948), Aggressotype (602805) and IMAGEMEND (602450) and J.B. has been a consultant in the past 3 years to/member of advisory board of and/or speaker for Janssen Cilag BV, Eli Lilly and Servier, but he is not an employee or a stock shareholder. R.C. was supported by a VIDI grant of NWO (016095340), and currently holds a James McDonnell Scholar award. R.C. is a consultant to Abbott Laboratories and Pfizer, but she is not an employee or a stock shareholder. For the remaining authors there are no conflicts of interest.
Footnotes
Esther Aarts and Mieke van Holstein contributed equally to the writing of this article.
References
- Aarts E, Roelofs A, Franke B, Rijpkema M, Fernández G, Helmich RC, Cools R. (2010). Striatal dopamine mediates the interface between motivational and cognitive control in humans: evidence from genetic imaging. Neuropsychopharmacology 35:1943–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarts E, van Holstein M, Cools R. (2011). Striatal dopamine and the interface between motivation and cognition. Front Psychol 2:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarts E, Helmich RC, Janssen MJ, Oyen WJ, Bloem BR, Cools R. (2012). Aberrant reward processing in Parkinson's disease is associated with dopamine cell loss. Neuroimage 59:3339–3346. [DOI] [PubMed] [Google Scholar]
- Aarts E, Nusselein AA, Smittenaar P, Helmich RC, Bloem BR, Cools R. (2014a). Greater striatal responses to medication in Parkinson’s disease are associated with better task-switching but worse reward performance. Neuropsychologia 62:390–397. [DOI] [PubMed] [Google Scholar]
- Aarts E, Wallace DL, Dang LC, Jagust WJ, Cools R, D'Esposito M. (2014b). Dopamine and the cognitive downside of a promised bonus. Psychol Sci 25:1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. (1994). Diagnostic and Statistical Manual of Mental Disorders 4th ed.Washington, DC: American Psychiatric Publishing. [Google Scholar]
- American Psychiatric Association. (2013). Diagnostic and Statistical Manual of Mental Disorders 5th ed.Arlington, VA: American Psychiatric Publishing. [Google Scholar]
- Arnsten AF, Li BM. (2005). Neurobiology of executive functions: catecholamine influences on prefrontal cortical functions. Biol Psychiatry 57:1377–1384. [DOI] [PubMed] [Google Scholar]
- Aron AR, Dowson JH, Sahakian BJ, Robbins TW. (2003). Methylphenidate improves response inhibition in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry 54:1465–1468. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. (2005). Unified segmentation. Neuroimage 26:839–851. [DOI] [PubMed] [Google Scholar]
- Barkley RA. (1997). Behavioral inhibition, sustained attention, and executive functions: constructing a unifying theory of ADHD. Psychol Bull 121:65–94. [DOI] [PubMed] [Google Scholar]
- Bédard AC, Schulz KP, Cook EH, Jr, Fan J, Clerkin SM, Ivanov I, et al. (2010). Dopamine transporter gene variation modulates activation of striatum in youth with ADHD. Neuroimage 53:935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Devilbiss DM, Andrzejewski ME, Arnsten AF, Kelley AE, Schmeichel B, et al. (2006). Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry 60:1111–1120. [DOI] [PubMed] [Google Scholar]
- Bilder RM, Volavka J, Lachman HM, Grace AA. (2004). The catechol-O-methyltransferase polymorphism: relations to the tonic-phasic dopamine hypothesis and neuropsychiatric phenotypes. Neuropsychopharmacology 29:1943–1961. [DOI] [PubMed] [Google Scholar]
- Bond AJ, Lader MH. (1974). The use of analogue scales in rating subjective feelings. Br J Med Psychol 47:211–218. [Google Scholar]
- Brookes KJ, Xu X, Anney R, Franke B, Zhou K, Chen W, et al. (2008). Association of ADHD with genetic variants in the 5′-region of the dopamine transporter gene: evidence for allelic heterogeneity. Am J Med Genet B Neuropsychiatr Genet 147B:1519–1523. [DOI] [PubMed] [Google Scholar]
- Bush G, Frazier JA, Rauch SL, Seidman LJ, Whalen PJ, Jenike MA, et al. (1999). Anterior cingulate cortex dysfunction in attention-deficit/hyperactivity disorder revealed by fMRI and the Counting Stroop. Biol Psychiatry 45:1542–1552. [DOI] [PubMed] [Google Scholar]
- Button KS, Ioannidis JP, Mokrysz C, Nosek BA, Flint J, Robinson ES, Munafò MR. (2013). Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci 14:365–376. [DOI] [PubMed] [Google Scholar]
- Carmona S, Hoekzema E, Ramos-Quiroga JA, Richarte V, Canals C, Bosch R, et al. (2012). Response inhibition and reward anticipation in medication-naïve adults with attention-deficit/hyperactivity disorder: a within-subject case-control neuroimaging study. Hum Brain Mapp 33:2350–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliax BJ, Drash GW, Staley JK, Haber S, Mobley CJ, Miller GW, et al. (1999). Immunocytochemical localization of the dopamine transporter in human brain. J Comp Neurol 409:38–56. [DOI] [PubMed] [Google Scholar]
- Clatworthy PL, Lewis SJ, Brichard L, Hong YT, Izquierdo D, Clark L, et al. (2009). Dopamine release in dissociable striatal subregions predicts the different effects of oral methylphenidate on reversal learning and spatial working memory. J Neurosci 29:4690–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colzato LS, van den Wildenberg WP, Van der Does AJ, Hommel B. (2010). Genetic markers of striatal dopamine predict individual differences in dysfunctional, but not functional impulsivity. Neuroscience 170:782–788. [DOI] [PubMed] [Google Scholar]
- Cools AR. (1980). Role of the neostriatal dopaminergic activity in sequencing and selecting behavioural strategies: facilitation of processes involved in selecting the best strategy in a stressful situation. Behav Brain Res 1:361–378. [DOI] [PubMed] [Google Scholar]
- Cools R, D'Esposito M. (2011). Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry 69:e113–e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools AR, van den Bercken JH, Horstink MW, van Spaendonck KP, Berger HJ. (1984). Cognitive and motor shifting aptitude disorder in Parkinson's disease. J Neurol Neurosurg Psychiatry 47:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese S, Kelly C, Chabernaud C, Proal E, Di Martino A, Milham MP, Castellanos FX. (2012). Toward systems neuroscience of ADHD: a meta-analysis of 55 fMRI studies. Am J Psychiatry 169:1038–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Riedel M, Müller U, Möller HJ, Ettinger U. (2011). Relationship between SLC6A3 genotype and striatal dopamine transporter availability: a meta-analysis of human single photon emission computed tomography studies. Synapse 65:998–1005. [DOI] [PubMed] [Google Scholar]
- Cubillo A, Halari R, Ecker C, Giampietro V, Taylor E, Rubia K. (2010). Reduced activation and inter-regional functional connectivity of fronto-striatal networks in adults with childhood Attention-Deficit Hyperactivity Disorder (ADHD) and persisting symptoms during tasks of motor inhibition and cognitive switching. J Psychiatr Res 44:629–639. [DOI] [PubMed] [Google Scholar]
- Desman C, Petermann F, Hampel P. (2008). Deficit in response inhibition in children with attention deficit/hyperactivity disorder (ADHD): impact of motivation? Child Neuropsychol 14:483–503. [DOI] [PubMed] [Google Scholar]
- De Zeeuw P, Weusten J, van Dijk S, van Belle J, Durston S. (2012). Deficits in cognitive control, timing and reward sensitivity appear to be dissociable in ADHD. PLoS One 7:e51416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibbets P, Evers EA, Hurks PP, Bakker K, Jolles J. (2010). Differential brain activation patterns in adult attention-deficit hyperactivity disorder (ADHD) associated with task switching. Neuropsychology 24:413–423. [DOI] [PubMed] [Google Scholar]
- Dickstein SG, Bannon K, Castellanos FX, Milham MP. (2006). The neural correlates of attention deficit hyperactivity disorder: an ALE meta-analysis. J Child Psychol Psychiatry 47:1051–1062. [DOI] [PubMed] [Google Scholar]
- Dodds CM, Müller U, Clark L, van Loon A, Cools R, Robbins TW. (2008). Methylphenidate has differential effects on blood oxygenation level-dependent signal related to cognitive subprocesses of reversal learning. J Neurosci 28:5976–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher JC, Kohn P, Kolachana B, Weinberger DR, Berman KF. (2009). Variation in dopamine genes influences responsivity of the human reward system. Proc Natl Acad Sci U S A 106:617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durston S, Fossella JA, Mulder MJ, Casey BJ, Ziermans TB, Vessaz MN, Van Engeland H. (2008). Dopamine transporter genotype conveys familial risk of attention-deficit/hyperactivity disorder through striatal activation. J Am Acad Child Adolesc Psychiatry 47:61–67. [DOI] [PubMed] [Google Scholar]
- Durston S, de Zeeuw P, Staal WG. (2009). Imaging genetics in ADHD: a focus on cognitive control. Neurosci Biobehav Rev 33:674–689. [DOI] [PubMed] [Google Scholar]
- Durston S, van Belle J, de Zeeuw P. (2011). Differentiating frontostriatal and fronto-cerebellar circuits in attention-deficit/hyperactivity disorder. Biol Psychiatry 69:1178–1184. [DOI] [PubMed] [Google Scholar]
- Epstein JN, Casey BJ, Tonev ST, Davidson MC, Reiss AL, Garrett A, et al. (2007). ADHD- and medication-related brain activation effects in concordantly affected parent-child dyads with ADHD. J Child Psychol Psychiatry 48:899–913. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA, Sklar P. (2005). Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry 57:1313–1323. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Spencer TJ, Madras BK, Zhang-James Y, Biederman J. (2014). Functional effects of dopamine transporter gene genotypes on in vivo dopamine transporter functioning: a meta-analysis. Mol Psychiatry 19:880–889. [DOI] [PubMed] [Google Scholar]
- Forbes EE, Brown SM, Kimak M, Ferrell RE, Manuck SB, Hariri AR. (2009). Genetic variation in components of dopamine neurotransmission impacts ventral striatal reactivity associated with impulsivity. Mol Psychiatry 14:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke B, Hoogman M, Arias Vasquez A, Heister JG, Savelkoul PJ, Naber M, et al. (2008). Association of the dopamine transporter (SLC6A3/DAT1) gene 9-6 haplotype with adult ADHD. Am J Med Genet B Neuropsychiatr Genet 147B:1576–1579. [DOI] [PubMed] [Google Scholar]
- Franke B, Vasquez AA, Johansson S, Hoogman M, Romanos J, Boreatti-Hümmer A, et al. (2010). Multicenter analysis of the SLC6A3/DAT1 VNTR haplotype in persistent ADHD suggests differential involvement of the gene in childhood and persistent ADHD. Neuropsychopharmacology 35:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa E, Bado P, Tripp G, Mattos P, Wickens JR, Bramati IE, et al. (2014). Abnormal striatal BOLD responses to reward anticipation and reward delivery in ADHD. PLoS One 9:e89129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Jones SR, Caron MG. (1999). Functional hyperdopaminergia in dopamine transporter knock-out mice. Biol Psychiatry 46:303–311. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379:606–612. [DOI] [PubMed] [Google Scholar]
- Gizer IR, Ficks C, Waldman ID. (2009). Candidate gene studies of ADHD: a meta-analytic review. Hum Genet 126:51–90. [DOI] [PubMed] [Google Scholar]
- Glow PH, Glow RA. (1979). Hyperkinetic impulse disorder: a developmental defect of motivation. Genet Psychol Monogr 100:159–231. [PubMed] [Google Scholar]
- Grahn JA, Parkinson JA, Owen AM. (2008). The cognitive functions of the caudate nucleus. Prog Neurobiol 86:141–155. [DOI] [PubMed] [Google Scholar]
- Haber SN. (2003). The primate basal ganglia: parallel and integrative networks. J Chem Neuroanat 26:317–330. [DOI] [PubMed] [Google Scholar]
- Haber SN, Fudge JL, McFarland NR. (2000). Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci 20:2369–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogman M, Aarts E, Zwiers M, Slaats-Willemse D, Naber M, Onnink M, et al. (2011). Nitric oxide synthase genotype modulation of impulsivity and ventral striatal activity in adult ADHD patients and healthy comparison subjects. Am J Psychiatry 168:1099–1106. [DOI] [PubMed] [Google Scholar]
- Hoogman M, Onnink M, Cools R, Aarts E, Kan C, Arias Vasquez A, et al. (2013). The dopamine transporter haplotype and reward-related striatal responses in adult ADHD. Eur Neuropsychopharmacol 23:469–478. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Saigusa T, Kamei J, Koshikawa N, Cools AR. (2013). Spiraling dopaminergic circuitry from the ventral striatum to dorsal striatum is an effective feed-forward loop. Neuroscience 241:126–134. [DOI] [PubMed] [Google Scholar]
- Kambeitz J, Romanos M, Ettinger U. (2014). Meta-analysis of the association between dopamine transporter genotype and response to methylphenidate treatment in ADHD. Pharmacogenomics J 14:77–84. [DOI] [PubMed] [Google Scholar]
- Karalunas SL, Huang-Pollock CL. (2011). Examining relationships between executive functioning and delay aversion in attention deficit hyperactivity disorder. J Clin Child Adolesc Psychol 40:837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Adler L, Barkley R, Biederman J, Conners CK, Demler O, et al. (2006). The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry 163:716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad K, Gauggel S, Manz A, Schöll M. (2000). Lack of inhibition: a motivational deficit in children with attention deficit/hyperactivity disorder and children with traumatic brain injury. Child Neuropsychol 6:286–296. [DOI] [PubMed] [Google Scholar]
- Kooij JJ, Francken MH.2007Diagnostisch Interview Voor ADHD (DIVA) bij volwassenen. Available at: http://www.psyq.nl/Programma/Kenniscentrum-ADHD-bij-volwassenen/diagnostiek-en-protocollen-adhd [Accessed 12 November 2014].
- Kooij JJ, Buitelaar JK, van den Oord EJ, Furer JW, Rijnders CA, Hodiamont PP. (2005). Internal and external validity of attention-deficit hyperactivity disorder in a population-based sample of adults. Psychol Med 35:817–827. [DOI] [PubMed] [Google Scholar]
- Lewis R, Kupke T. (1977). The Lafayette clinic repeatable neuropsychological test battery. Its development and research applications Hollywood, FL: Southeastern Psychological Association. [Google Scholar]
- Majdandzić J, Grol MJ, van Schie HT, Verhagen L, Toni I, Bekkering H. (2007). The role of immediate and final goals in action planning: an fMRI study. Neuroimage 37:589–598. [DOI] [PubMed] [Google Scholar]
- McCarthy H, Skokauskas N, Frodl T. (2014). Identifying a consistent pattern of neural function in attention deficit hyperactivity disorder: a meta-analysis. Psychol Med 44:869–880. [DOI] [PubMed] [Google Scholar]
- Munafò MR, Gage SH. (2013). Improving the reliability and reporting of genetic association studies. Drug Alcohol Depend 132:411–413. [DOI] [PubMed] [Google Scholar]
- Onnink AM, Zwiers MP, Hoogman M, Mostert JC, Kan CC, Buitelaar J, Franke B. (2014). Brain alterations in adult ADHD: effects of gender, treatment and comorbid depression. Eur Neuropsychopharmacol 24:397–409. [DOI] [PubMed] [Google Scholar]
- Oosterlaan J, Sergeant JA. (1998). Effects of reward and response cost on response inhibition in AD/HD, disruptive, anxious, and normal children. J Abnorm Child Psychol 26:161–174. [DOI] [PubMed] [Google Scholar]
- Paloyelis Y, Mehta MA, Faraone SV, Asherson P, Kuntsi J. (2012). Striatal sensitivity during reward processing in attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 51:722–732e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton JH, Stanford MS, Barratt ES. (1995). Factor structure of the Barratt impulsiveness scale. J Clin Psychol 51:768–774. [DOI] [PubMed] [Google Scholar]
- Plichta MM, Scheres A. (2014). Ventral-striatal responsiveness during reward anticipation in ADHD and its relation to trait impulsivity in the healthy population: a meta-analytic review of the fMRI literature. Neurosci Biobehav Rev 38:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plichta MM, Vasic N, Wolf RC, Lesch KP, Brummer D, Jacob C, et al. (2009). Neural hyporesponsiveness and hyperresponsiveness during immediate and delayed reward processing in adult attention-deficit/hyperactivity disorder. Biol Psychiatry 65:7–14. [DOI] [PubMed] [Google Scholar]
- Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. (2007). The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry 164:942–948. [DOI] [PubMed] [Google Scholar]
- Reeves S, Bench C, Howard R. (2002). Ageing and the nigrostriatal dopaminergic system. Int J Geriatr Psychiatry 17:359–370. [DOI] [PubMed] [Google Scholar]
- Rokem A, Landau AN, Prinzmetal W, Wallace DL, Silver MA, D'Esposito M. (2012). Modulation of inhibition of return by the dopamine D2 receptor agonist bromocriptine depends on individual DAT1 genotype. Cereb Cortex 22:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubia K, Halari R, Cubillo A, Mohammad AM, Brammer M, Taylor E. (2009). Methylphenidate normalises activation and functional connectivity deficits in attention and motivation networks in medication-naïve children with ADHD during a rewarded continuous performance task. Neuropharmacology 57:640–652. [DOI] [PubMed] [Google Scholar]
- Rubia K, Halari R, Cubillo A, Smith AB, Mohammad AM, Brammer M, Taylor E. (2011). Methylphenidate normalizes fronto-striatal underactivation during interference inhibition in medication-naïve boys with attention-deficit hyperactivity disorder. Neuropsychopharmacology 36:1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salthouse TA. (1996). The processing-speed theory of adult age differences in cognition. Psychol Rev 103:403–428. [DOI] [PubMed] [Google Scholar]
- Scheres A, Milham MP, Knutson B, Castellanos FX. (2007). Ventral striatal hyporesponsiveness during reward anticipation in attention-deficit/hyperactivity disorder. Biol Psychiatry 61:720–724. [DOI] [PubMed] [Google Scholar]
- Schmeichel BE, Zemlan FP, Berridge CW. (2013). A selective dopamine reuptake inhibitor improves prefrontal cortex-dependent cognitive function: potential relevance to attention deficit hyperactivity disorder. Neuropharmacology 64:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant JA, Geurts H, Huijbregts S, Scheres A, Oosterlaan J. (2003). The top and the bottom of ADHD: a neuropsychological perspective. Neurosci Biobehav Rev 27:583–592. [DOI] [PubMed] [Google Scholar]
- Shafritz KM, Marchione KE, Gore JC, Shaywitz SE, Shaywitz BA. The effects of methylphenidate on neural systems of attention in attention deficit hyperactivity disorder. (2004). Am J Psychiatry 161:1990–1997. [DOI] [PubMed] [Google Scholar]
- Shanahan MA, Pennington BF, Willcutt EW. (2008). Do motivational incentives reduce the inhibition deficit in ADHD? Dev Neuropsychol 33:137–159. [DOI] [PubMed] [Google Scholar]
- Sheskin DJ. (2004). Handbook of parametric and non-parametric statistical procedures 3rd ed.Boca Raton, Florida: Chapman and Hall/CRC Press. [Google Scholar]
- Simon V, Czobor P, Bálint S, Mészáros A, Bitter I. (2009). Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry 194:204–211. [DOI] [PubMed] [Google Scholar]
- Slusarek M, Velling S, Bunk D, Eggers C. (2001). Motivational effects on inhibitory control in children with ADHD. J Am Acad Child Adolesc Psychiatry 40:355–363. [DOI] [PubMed] [Google Scholar]
- Sonuga-Barke EJ. (2002). Psychological heterogeneity in AD/HD – a dual pathway model of behaviour and cognition. Behav Brain Res 130:29–36. [DOI] [PubMed] [Google Scholar]
- Sonuga-Barke EJ. (2003). The dual pathway model of AD/HD: an elaboration of neuro-developmental characteristics. Neurosci Biobehav Rev 27:593–604. [DOI] [PubMed] [Google Scholar]
- Sonuga-Barke EJ. (2005). Causal models of attention-deficit/hyperactivity disorder: from common simple deficits to multiple developmental pathways. Biol Psychiatry 57:1231–1238. [DOI] [PubMed] [Google Scholar]
- Spreen FO, Benton AL. (1977). Manual of Instructions for the Neurosensory Center Comprehensive Examination for Aphasia VIC, British Columbia, Canada: University of Victoria. [Google Scholar]
- Ströhle A, Stoy M, Wrase J, Schwarzer S, Schlagenhauf F, Huss M, et al. (2008). Reward anticipation and outcomes in adult males with attention-deficit/hyperactivity disorder. Neuroimage 39:966–972. [DOI] [PubMed] [Google Scholar]
- Tomasi D, Volkow ND. (2012). Abnormal functional connectivity in children with attention-deficit/hyperactivity disorder. Biol Psychiatry 71:443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebel H, Albrecht B, Asherson P, Börger NA, Butler L, Chen W, et al. (2010). Performance variability, impulsivity errors and the impact of incentives as gender-independent endophenotypes for ADHD. J Child Psychol Psychiatry 51:210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya CJ, Austin G, Kirkorian G, Ridlehuber HW, Desmond JE, Glover GH, Gabrieli JD. (1998). Selective effects of methylphenidate in attention deficit hyperactivity disorder: a functional magnetic resonance study. Proc Natl Acad Sci U S A 95:14494–14499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Holstein M, Aarts E, van der Schaaf ME, Geurts DE, Verkes RJ, Franke B, et al. (2011). Human cognitive flexibility depends on dopamine D2 receptor signaling. Psychopharmacology (Berl) 218:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen L, Dijkerman HC, Grol MJ, Toni I. (2008). Perceptuo-motor interactions during prehension movements. J Neurosci 28:4726–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij MM, Cools AR. (2008). Twenty years of dopamine research: individual differences in the response of accumbal dopamine to environmental and pharmacological challenges. Eur J Pharmacol 585:228–244. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley SJ, et al. (1995). A new PET ligand for the dopamine transporter: studies in the human brain. J Nucl Med 36:2162–2168. [PubMed] [Google Scholar]
- Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley SJ, et al. (1996). Dopamine transporters decrease with age. J Nucl Med 37:554–559. [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Gatley SJ, Logan J, Ding YS, et al. (1998). Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry 155:1325–1331. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang G, Ding Y, Gatley SJ. (2002). Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord 6 Suppl 1:S31–S43. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Kollins SH, Wigal TL, Newcorn JH, Telang F, et al. (2009). Evaluating dopamine reward pathway in ADHD: clinical implications. JAMA 302:1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Tomasi D, Kollins SH, Wigal TL, Newcorn JH, et al. (2012). Methylphenidate-elicited dopamine increases in ventral striatum are associated with long-term symptom improvement in adults with attention deficit hyperactivity disorder. J Neurosci 32:841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]