

Abstract
Proarrhythmic drugs induce long QT syndrome more frequently in women than men. The present study was designed to determine whether androgens regulate the function and expression of the human ether-á-go-go-related gene (HERG) encoded K+ channel, which is largely responsible for determining the QT interval. In a concentration-dependent manner (10−9 to 10−6 m for 24 h), 5α-dihydrotestosterone (5α-DHT) increased HERG protein abundance in HEK293 cells stably expressing HERG in the presence of coexpressed cardiac androgen receptor (AR) variant [N-terminal truncated isoform of AR (AR45)]. The elevation of HERG protein was seen in endoplasmic reticulum, Golgi, and plasma membrane without clear preferential colocalization. Coexpression of the more common form of the AR did not confer 5α-DHT augmentation of HERG protein. Proteasome inhibitors, N-acetyl-L-leucyl-L-leucyl-L-norleucinal and MG132 prevented the 5α-DHT- dependent enhancement of HERG, as did the lysosome inhibitor, bafilomycin A1. Consistently, the cycloheximide-based protein chase study showed that 5α-DHT prolonged HERG protein half-life. 5α-DHT/AR45 signaling induced phosphorylation of ERK1/2. Blockade of ERK1/2 with PD98059 and U0126 prevented the effect of androgen on HERG protein abundance. Functional studies showed that 5α-DHT treatment for 24 h increased HERG K+ current density in Chinese hamster ovary cells cotransfected with cDNAs of AR45 and HERG channels. Moreover, 5α-DHT also increased ether-á-go-go-related gene-encoded K+ channel protein abundance in isolated rabbit cardiac myocytes. In conclusion, these data provide evidence that stimulation of AR45 receptors by androgens up-regulates HERG K+ channel abundance and activity mainly through stabilizing HERG protein in an ERK1/2 dependent mechanism, and suggest a mechanism to explain the sex difference in the long QT syndrome.
PROARRHYTHMIC drugs induce torsade de pointes (TdP) more frequently in women than men (1). It has long been suspected that the slower rates of cardiac repolarization and the longer QT intervals in women compared with men are responsible for the greater risk in women (2, 3). More recent evidence from clinical and basic studies suggests that androgens may be responsible for the sex difference in the heart rate-corrected QT interval (QTc). In normal populations, as well as in families with inherited long QT syndrome (LQTS), it was found that the QTcs were shorter in postpubescent males (3, 4). Cardiac repolarization periods in castrated men were longer than those in normal men and women with virilization, but similar to those in normal women (5). Moreover, male electrocardiogram patterns of repolarization in the castrated men were restored after testosterone replacement therapy. In animal experiments Liu et al. (6) found that in vivo androgen treatment shortened the QT intervals in orchiectomized male rabbits. These findings indicate that androgens may play major roles in the sex difference in the QTc, which contributes to the higher risk of developing drug-induced cardiac arrhythmias in women (1).
The human ether-á-go-go-related gene (HERG) K+ channel, which encodes the α-subunit of the rapid component of the delayed rectifier K+ current (IKr), is largely responsible for the repolarization of action potential in cardiac myocytes (7, 8, 9). Inherited mutations or drug-induced blockade of the HERG channel can prolong QT intervals and increase the risk of lethal arrhythmia (7, 10, 11). The finding that IKr densities in female rabbit ventricular myocytes are significantly lower than those in the male (12) indicates that the effect of androgen on ether-á-go-go-related gene (ERG)/IKr may contribute to the sex difference in QT intervals. Liu et al. (6) also found that in vivo 5α-dihydrotestosterone (5α-DHT) treatment increased IKr density but not ERG mRNA levels in gonadectomized rabbit hearts, suggesting that a post-translation mechanism may be involved.
The heart has been considered as a target organ of androgens for several decades (13), but the presence of androgen receptor (AR) in the heart had not been clearly demonstrated (14, 15, 16, 17). Recently, Ahrens-Fath et al. (18) found that AR transcript levels in human heart were very low compared with those in the liver or testis; however, the transcript level of the N-terminal truncated isoform of AR (AR45) was high in human heart tissue. These findings suggest that AR45 may play a more important role than AR in the heart. Therefore, in the present study, we investigated the effect of androgens on HERG protein expression and activity in the human embryo kidney 293 (HEK293), and Chinese hamster ovary (CHO) cells transfected with HERG and AR (or AR45) cDNAs.
Materials and Methods
Materials
5β-Dihydrotestosterone (5β-DHT) was purchased from Steraloids, Inc. (Newport, RI). R1881 was from New England Nuclear Life Science Products (Boston, MA). PD98059 and U0126 were from Merck & Co., Inc. (Whitehouse Station, NJ). All other reagents were from Sigma-Aldrich (St. Louis, MO). Testosterone, 5α-DHT, 5β-DHT, progesterone, estradiol, and flutamide were dissolved in ethanol as stock solutions. N-acetyl-L-leucyl-L-leucyl-L-norleucinal (ALLN), cycloheximide, MG132, R1881, and bafilomycin A1 were dissolved in dimethylsulfoxide as stock solutions. The final ethanol and dimethylsulfoxide concentrations were no more than 0.2%.
Cell culture and transfection
HEK293 and CHO cells are AR negative cells (19, 20). HERG-HEK293 and CHO cells were cultured in RPMI 1640 (Invitrogen Corp., Carlsbad, CA) or F-12 (Sigma-Aldrich) cell culture medium, respectively, supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT) and penicillin-streptomycin (Invitrogen) at 37 C in a 5% CO2 atmosphere. We established a HEK293 cell line stably expressing HERG K+ channels, HERG-HEK293. Briefly, HEK293 cells were transfected with HERG-myc plasmid in pCI-Neo vector (Promega Corp., Madison, WI). The stable clones were selected by culture in medium containing G418 (800 μg/ml), and confirmed by Western blot analysis and electrophysiological study. For biochemical analysis, pSG5-AR45, pSG5-AR, or green fluorescence protein (GFP) was transiently transfected into HERG-HEK293 cells with Lipofectamine2000 (Invitrogen). Briefly, HERG-HEK293 cells were cultured in RPMI 1640 medium supplemented with 3% FBS for 1 d, and then were transfected with AR45, AR, or GFP and kept in the aforementioned culture medium overnight. After serum starving for 16 h in RPMI 1640 medium with 0.5% FBS, cells were treated with androgens. For mechanism studies, inhibitors were added to culture medium 1 h before and during androgen treatment. For electrophysiological studies, CHO cells were transiently transfected with cDNAs of HERG, AR45, and GFP by electroporation as described previously (21, 22). Briefly, CHO cells were electroporated (single 160 V pulse for 15 msec) with 4 μg HERG, 4 μg AR45, and 2 μg GFP cDNAs in a 2-mm gap cuvette using a Gene Pulser Xcell (Bio-Rad Laboratories, Inc., Hercules, CA). After electroporation the cells were plated sparsely and grown on sterile glass coverslips in 60-mm tissue culture dishes. Cells were serum starved following the same protocol as mentioned previously in the protocol for biochemical analysis. Forty-eight to 72 h after transfection, cells were used for electrophysiological studies.
Isolation of rabbit cardiac myocytes
Myocytes were isolated from rabbit heart via enzymatic digestion as described previously (21, 22). The heart was quickly excised from anesthetized male rabbits (1 month), and perfused with calcium free Tyrode’s solution (at 37 C) containing (in mm) 137 NaCl, 5.4 KCl, 1 MgCl2, 10 HEPES, and 10 glucose. The perfusion solution was then changed to the Tyrode’s solution containing 1 mg/ml collagenase (Worthington type II) and 0.28 mg/ml protease (type XIV; Sigma-Aldrich), and perfused for 20–25 min. The ventricular tissue was then cut into small pieces filtered through a 250-μm mesh screen. The cells were washed, and the calcium concentration of Tyrode’s solution was adjusted to 1.25 mm gradually. Isolated myocytes were resuspended in M199 medium and plated into laminin (Sigma-Aldrich)-coated petri dishes. Cells were treated with androgens with the same protocols as described previously.
Western blotting analysis
Cells were washed with ice-cold PBS and then ice cold lysis buffer [150 mm NaCl, 25 mm Tris-HCl (pH 7.5), 5 mm EDTA, 1% Nonidet P-40, 0.4% deoxycholic acid, and EDTA-free protease inhibitor cocktail tablets (Hoffmann-La Roche Inc., Nutley, NJ)] was added. Lysates were rocked for 1 h at 4 C and then centrifuged at 14,000 × g for 10 min at 4 C. The supernatants were assayed for total protein content (Bio-Rad Laboratories Protein Assay), and equal amounts (50–100 μg) of cell lysate protein were subjected to SDS-PAGE analysis. Protein samples were combined with 4× SDS-PAGE sample buffer [4% (wt/vol) sodium dodecyl sulfate, 40% glycerol, 20% (vol/vol) β-mercaptoethanol, 0.004% (wt/vol) bromphenol blue, and 125 mm Tris buffer (pH 6.8)] incubated for 5 min at room temperature, separated on a 8–10% SDS-PAGE, and electrophoretically transferred onto 0.2 μm nitrocellulose membrane (Bio-Rad Laboratories). Membranes were blocked in 10% nonfat dry milk and 0.1% Tween 20 in Tris-buffered saline (TBS) for 1 h at room temperature, and incubated with appropriate primary antibodies [1:250 to 1:1000 dilutions in 5% BSA (Sigma-Aldrich)] and 0.1% Tween 20 in TBS overnight at 4 C. The membranes were then washed with 0.1% Tween 20 in TBS and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (1:10,000 to 1:50,000 dilution) for 1 h at room temperature. After three washes as described previously, horseradish peroxidase-bound protein was detected by chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Pierce, Rockford, IL). Anti-myc mouse monoclonal 9E10 and antitubulin rabbit monoclonal antibodies were from Sigma-Aldrich. Antiphosphorylated ERK1/2 antibody was from Cell Signaling Technologies (Beverly, MA). Anti-Kv11.1 (HERG)-extracellular antibody was from Alomone Labs, Ltd. (Jerusalem, Israel). Other antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All gels illustrated in the figures are representative examples from four to eight independent experiments. β-Tubulin was applied as an internal control to normalize protein loading. The intensity of bands was quantified using LabWorks Image Analysis software (UVP, LLC, Upland, CA).
Cycloheximide-based protein chase experiment
After posttransfection for 12 h, the medium was replaced with fresh RPMI 1640 plus 0.5% FBS with/without 5α-DHT. Eight hours later, cycloheximide (40 μm) was added into the medium to stop protein synthesis. Cells were then harvested at different time points (0, 6, 12, and 24 h) after cycloheximide treatment. Total cell lysates were analyzed by Western blotting as described previously. The amount of HERG remaining at each time point was normalized with the original HERG amount at time zero. The protein degradation rate is expressed as half-life (t1/2), the time for degradation of 50% of the protein.
RT-PCR
Cells were rinsed twice in cold sterile PBS, and total RNA was extracted by RNeasy Kits (QIAGEN, Inc., Valencia, CA) following the manufacturer’s instruction. Total RNA was quantified by spectrophotometric analysis, and checked for purity and quality by running a small aliquot on a 1% agarose gel. RT-PCR was performed using the QIAGEN one-step RT-PCR kit (QIAGEN). The following primers were used to examine the mRNA expression of HERG: forward, 1999 5′-TCCAGCGGCTGTACTCGGGC-3′; reverse, 2573 3′-GGACCAGAAGTGGTCGGAGAACTC-5′. As a quality control, PCR amplification of the messenger relative to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was performed using the following primer pair: forward, 1644 5′-TGATGACATCAAGAAGGTGGTGAAG-3′; reverse, 1883 5′-TCCTTGGAGGCCATGTAGGCCAT-3′. Reverse transcription was performed at 50 C for 30 min. PCR protocols consisted in a 15-min initial activation step at 95 C, followed by 31 cycles of amplification. Each cycle included 1 min at 94 C, 1 min at 55 C, and 1 min extension at 72 C (30 sec each step for GAPDH). The reactions were terminated by an elongation step at 72 C for 10 min. PCR products were analyzed with 1% agarose gel electrophoresis and imaged by the Multigenius Bioimaging system (Syngene, UK).
Confocal microscopy
HERG-HEK293 cells seeded onto glass coverslips were subject to formaldehyde fixation before indirect immunofluorescent staining. Primary antibodies were detected with Alexa Fluor 568-conjugated antigoat Ig and Alexa Fluor 488-conjugated antirabbit Ig (WGA; Invitrogen). After staining, coverslips were mounted with Prolong Gold antifade reagent (Invitrogen), and confocal images were acquired using a Leica TCS SP5 confocal system (Leica Microsystems GmbH, Wetzlar, Germany). Alexa-488 images were acquired with excitation at 488 nm using an argon laser. Alexa-568 images were acquired with excitation at 543 nm using a helium neon laser. Dual-color analysis was done with appropriate single-color controls with individual channels recorded sequentially. Instrument settings were adjusted to obtain minimal saturated pixels, and where semiquantitative comparisons were done, the settings were kept constant. Negative controls include slides probed with either primary antibody or secondary antibody alone.
Patch clamp recording
Cells on coverslips were taken directly from the cell culture incubator and placed in an acrylic/polystyrene perfusion chamber (Warner Instruments, Hamden, CT) for electrophysiological measurements. Patch pipettes were pulled and polished to obtain a tip resistance of 2–3 mΩ in the patch clamp solutions. For whole cell voltage clamp, the pipette solution consisted of (in mm) KCl 126, MgSO4 2, CaCl2 0.5, EGTA 5, Mg-ATP 4, and HEPES 25 [(pH 7.2) osmolality: 280 ± 10 mOsm]. External bath solution consisted of (in mm) NaCl 150, CaCl2 1.8, KCl 4, MgCl2 1, glucose 5, and HEPES 10 [(pH 7.4) osmolality: 320 ± 10 mOsm]. All experiments were performed at room temperature (20–22 C). Cells were studied on an inverted microscope (Nikon Corp., Tokyo, Japan) equipped with electronic patch-pipette micromanipulators (Burleigh; EXFO Life Sciences & Industrial Division, Mississauga, Ontario, Canada) and epifluorescence optics for GFP transfected cells. An Axopatch-200B patch clamp amplifier (Axon Instruments, Dowingtown, PA) was used for voltage-clamp measurements. The series resistances were about 9–10 mΩ. Voltage-clamp protocols were controlled using pClamp9.2 acquisition and analysis software. To elicit HERG K+ currents, depolarizing voltage pulses were applied to various levels from a holding potential of −70 mV for 4 sec, followed by stepwise repolarization to −70 mV to measure outward tail currents. Signals were analog filtered at 2000 Hz and sampled at 5000 Hz. Voltage-dependent activation data were fitted to a Boltzmann equation: I = 1/(1 + exp [(Vh-V)/k]), where I is the relative tail current amplitude, V is the applied membrane voltage, Vh is the voltage at half-maximal activation, and k is the slope factor.
Statistics
Values presented are means ± se. ANOVA was used for statistical analysis of the data, and P values less than 0.05 were considered significant.
Results
Androgen up-regulates expression of HERG protein via AR45
As shown in Fig. 1A, 5α-DHT, a potent androgen, significantly increased both mature (∼155 kDa) and immature (∼135 kDa) forms of HERG protein in HERG-HEK293 cells transfected with AR45 but failed to affect HERG protein expression in HERG-HEK293 cells transfected with AR or GFP cDNAs. Similar results were also observed when HERG cDNAs were transiently cotransfected together with AR45, AR, or GFP cDNAs into HEK293 cells (data not shown). These data suggest that 5α-DHT may up-regulate HERG protein abundance via stimulation of AR45, but not AR. Interestingly, transfection with AR alone, but not AR45, was able to increase HERG protein in a hormone-independent manner. This may be due to the intrinsic hormone-independent activity of AR (23).
Fig. 1.
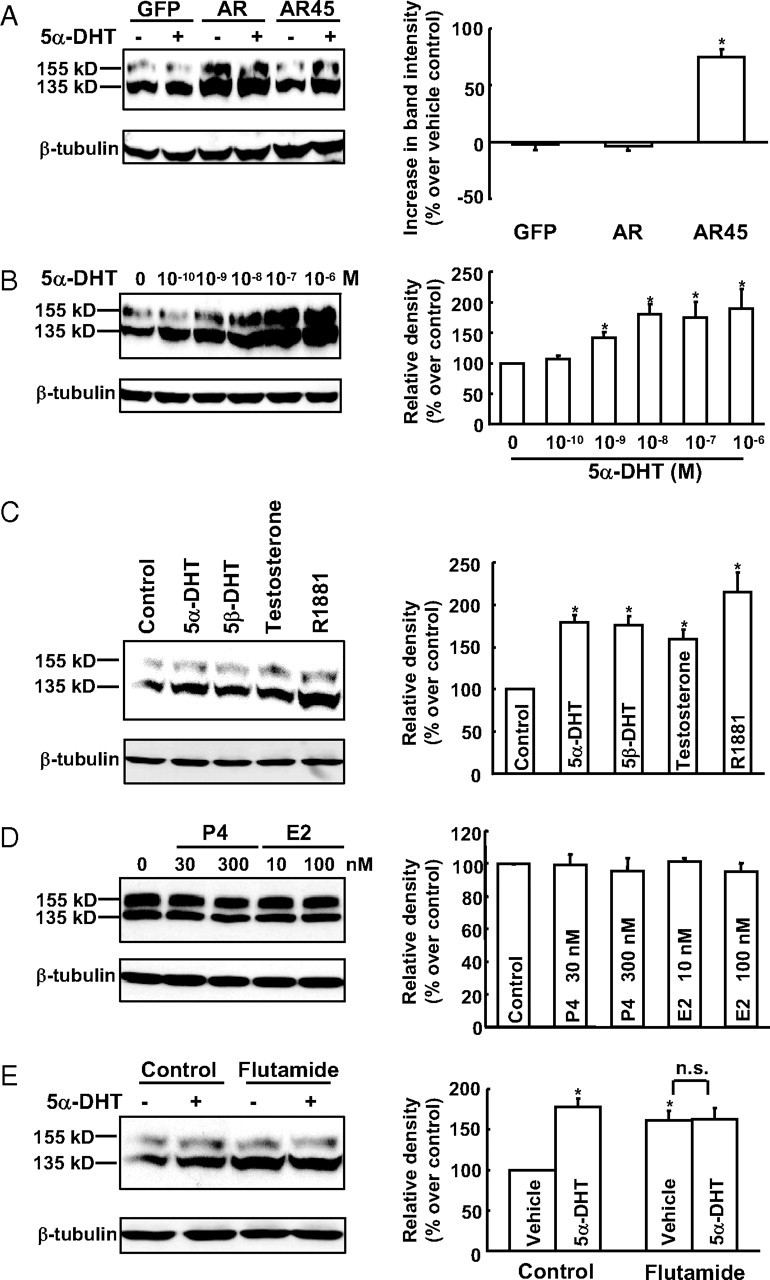
Effect of androgens on HERG protein levels in HERG-HEK293 cells transfected with GFP, AR, or AR45. A, Treatment with 10 nm 5α-DHT for 24 h up-regulated HERG protein expression in HERG-HEK293 cells transfected with AR45, but not in the cells transfected with GFP or AR (n = 6–8 per group). B, Concentration-dependent effect of 5α-DHT in HERG-HEK293 cells transfected with AR45. Western blot analysis indicated that treatment with 5α-DHT (10−10 to 10−6 m) for 24 h increased HERG protein level in a concentration-dependent manner (n = 6–7 per group). C, Treatment with 5α-DHT (10 nm), 5β-DHT (10 nm), testosterone (100 nm), or R1881 (10 nm) for 24 h significantly increased HERG protein abundance (n = 4–8 per group). D, Treatment with progesterone (P4) (30 and 300 nm) or estradiol (E2) (10 and 100 nm) for 24 h failed to affect HERG protein abundance (n = 6 per group). E, The effect of androgen (10 nm 5α-DHT) and antiandrogen (10 μm flutamide) on HERG protein levels (n = 8 per group). Representative Western blots are shown in the left panels. The percent increment in protein density on treatment with 5α-DHT is shown in the right panels. HERG protein abundance was normalized to β-tubulin. Data are means ± se. *, P < 0.05. n.s., Not significant.
Figure 1B shows that 5α-DHT increased HERG protein levels in a concentration-dependent manner. The EC50 for the effect was 1.04 ± 0.54 nm. The serum 5α-DHT level is 1.5 ± 0.8 nm in men (24) but only approximately 0.02 nm in women (25). Thus, the EC50 of 5α-DHT was within the physiological concentration range in men. The maximum response was observed when the concentration of 5α-DHT was at 10 nm. For this reason, this concentration (10 nm) was chosen for the following experiments.
We also tested whether other sex hormones can also produce a similar effect. We first examined the effect of different androgens. As shown in Fig. 1C, three other androgens (testosterone, 5β-DHT, and R1881) likewise significantly increased HERG abundance.
However, other sex hormones, like progesterone (30 and 300 nm) and estradiol (10 and 100 nm), failed to significantly affect HERG protein levels (Fig. 1D). These data suggest that androgens selectively up-regulate HERG protein expression via stimulation of AR45.
To test whether the effect of 5α-DHT is via AR/AR45 receptors, flutamide, an antiandrogen, was used. As shown in Fig. 1E, flutamide (10 μm) attenuated the effect of 5α-DHT. However, it is interesting to note that flutamide itself also up-regulated HERG protein expression. This could be caused by the nongenomic androgen-like effects of the drug (26, 27).
The effect of 5α-DHT on HERG protein expression was mediated via a posttranslational mechanism
This series of experiments was designed to exclude the possibility that the effect of androgen is a genomic effect. RT-PCR was first performed to examine the mRNA level of HERG upon androgen treatment. Figure 2A shows that 5α-DHT treatment did not change the HERG mRNA level. We tested with another plasmid that contains the same promoter and kir2.1 gene. The activation of AR45 by 5α-DHT did not change the kir2.1 protein level (Fig. 2B). To test whether up-regulation in HERG protein expression was due to enhanced protein synthesis, cycloheximide, a protein translation inhibitor, was used. As shown in Fig. 2C, cycloheximide, which alone decreased HERG protein levels, did not abolish the stimulatory effect of 5α-DHT. These data suggest that the effect of 5α-DHT was via a posttranslational mechanism.
Fig. 2.

Posttranslational effect of 5α-DHT on HERG protein levels. A, RT-PCR for the measurement of HERG mRNA levels with 5α-DHT treatment; the HERG mRNA levels were normalized with GAPDH (n = 4). B, Immunoblot analysis showed that 5α-DHT (10 nm) had no effect on kir2.1 protein level (n = 4). C, Immunoblot analysis showed that 5α-DHT (10 nm) increased HERG protein levels in the presence or absence of cycloheximide (40 μm). Cycloheximide was applied 1 h before administration of 5α-DHT (n = 5 per group). Data are means ± se. *, P < 0.05.
5α-DHT augments HERG protein abundance at cell surface and in the intracellular compartments
To examine the effect of 5α-DHT on HERG channel trafficking, we performed double-staining immunofluorescence of control and 5α-DHT-treated cells with GM130 as a marker of Golgi complex or calnexin as a marker for endoplasmic reticulum (ER) (Fig. 3). After 24 h 5α-DHT treatment, there was no clear preferential colocalization of HERG, indicating that a general increase in HERG occurred in all compartments. This suggests that the 5α-DHT effects on HERG are not due to altered trafficking.
Fig. 3.
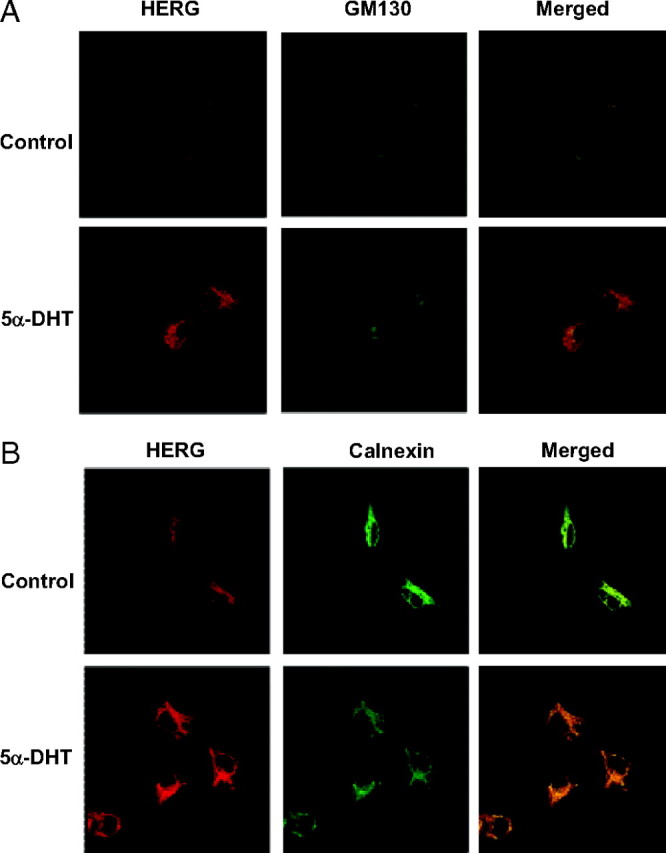
Effect of 5α-DHT on subcellular localization of HERG. Immunofluorescence assays with double stating for HERG-myc (red channel) and either the Golgi marker GM130 (A) or the ER marker calnexin (B) (green channel). 5α-DHT treatment (10 nm for 24 h) resulted in globally increased HERG signal in both compartments as well as on the surface.
The effect of 5α-DHT on HERG protein in the presence of protein degradation inhibitors
There are two documented pathways for HERG degradation: lysosomal and proteasomal degradation (28, 29). To determine the involvement of these pathways in the androgen effect, a lysosomal protease inhibitor, bafilomycin A1, and two proteasome inhibitors, MG132 and ALLN, were used. As shown in Fig. 4, all these inhibitors alone significantly increased HERG protein abundance, confirming that both pathways are involved in HERG protein degradation. Treatment with these inhibitors significantly attenuated the effect of 5α-DHT (Fig. 4). These data suggest that the effect of androgen may be via inhibition of protein degradation.
Fig. 4.

Effect of 5α-DHT on HERG protein levels in the presence and absence of proteasome and lysosomal protease inhibitors. Immunoblot analysis shows that the up-regulation of HERG protein abundance was abolished by two proteasome inhibitors, ALLN (50 μm) and MG132 (50 μm), and a lysosome inhibitor, bafilomycin A1 (100 nm). Inhibitors were applied 1 h before administration of 5α-DHT. Data are means ± se (n = 5–8 per group). *, P < 0.05. n.s., Not significant.
5α-DHT prolongs the half-life of HERG protein degradation
To confirm the aforementioned findings, we examined the effect of androgen on HERG protein turnover rates with the cycloheximide chase assay. As shown in Fig. 5, 5α-DHT treatment significantly delayed protein degradation at different time points. The half-life time (t1/2) of HERG (12.4 ± 1.5 h without 5α-DHT treatment) was prolonged to 16.3 ± 1.2 h when treated with 5α-DHT. These data suggest that 5α-DHT may increase HERG protein stability.
Fig. 5.

Effect of 5α-DHT on HERG protein stability. HERG protein stability was examined with cycloheximide-based chase assay. Immunoblot analysis shows that treatment with 5α-DHT (10 nm) prolonged HERG protein half-life. Data are means ± se (n = 5 per group). *, P < 0.05.
The effect of 5α-DHT on HERG protein expression in the presence and absence of ERK1/2 inhibitors
Recent studies have shown that androgens exert nongenomic effects by activating the MAPK signaling pathway (30, 31, 32, 33, 34). For this reason we examined whether MAPK mediates the effect of androgens on HERG protein abundance via AR45. In HERG-HEK293 cells transfected with AR45, 5α-DHT induced phosphorylation of ERK1/2 (a marker for ERK activation) in a concentration-dependent manner (Fig. 6A). In HERG-HEK293 cells transfected with GFP cDNA, 5α-DHT did not induce significant phosphorylation of ERK1/2. Two ERK1/2 inhibitors (PD98059 and U0126), neither of which alone changed HERG protein abundance, abolished the effect of 5α-DHT on HERG protein levels (Fig. 6B). These data suggest that the effect of AR45 stimulation is at least partly mediated by ERK1/2.
Fig. 6.

Effect of 5α-DHT on HERG protein levels was via activation of ERK1/2. A, Immunoblot analysis showing that treatment with 5α-DHT (10−10 to 10−7 m) for 30 min concentration dependently induced phosphorylation of ERK1/2 in cells transfected with AR45 (n = 4), but not in cells transfected with GFP (n = 6). B, Immunoblot analysis showing that two ERK1/2 inhibitors, PD98059 (20 μm) and U0126 (20 μm), abolished the effect of 5α-DHT on HERG protein levels (n = 4–7 per group). Inhibitors were applied 1 h before administration of 5α-DHT. Data are means ± se. *, P < 0.05. n.s., Not significant.
Effect of 5α-DHT on HERG currents in CHO cells transfected with both HERG K+ channel and AR45 cDNAs
We further investigated whether 5α-DHT-induced up-regulation of HERG protein abundance may also result in a commensurate increase in HERG current. Because of bearing little endogenous potassium currents, CHO cells were chosen for the following electrophysiological study. Using the whole cell configuration, we found that treatment with 5α-DHT for 24 h increased HERG current densities (Fig. 7, A and B). As shown in Fig. 7B, the tail current densities of cells were significantly increased when test voltages were between −20 and 40 mV, whereas the current kinetic properties were not changed ([half-activation voltage (V1/2:) Control: −20.4 ± 3.4 mV; 5α-DHT: −21.3 ± 2.2 mV; n = 8, P > 0.05]). These data suggest that 5α-DHT may not have a direct effect on HERG channel current kinetics and that the increased current density was caused only by the elevated expression of HERG protein.
Fig. 7.

Effect of 5α-DHT on HERG K+ channel activity in CHO cells transfected with HERG K+ channels and AR45. A, Representative current tracings showing the effect of 5α-DHT (10 nm, 24 h). Currents were elicited by 4.5-sec depolarizing steps to various levels (between 40 and −80 mV), followed by repolarizing steps to −70 mV. Scale bars, 20 pA/pF and 1 sec. B (left), Current-voltage (I-V) relation curve plotted from current measured at the end of the depolarizing test pulses. Inset, Data were normalized to unity with and without 5α-DHT. B (right), Voltage-dependent activation curves plotted from peak tail currents during a repolarizing step to −70 mV after depolarizing to various voltages. Inset, The same data normalized to the maximum current in the same curve. Data are means ± se (n = 8 per group). *, P < 0.05. ▴, Without 5α-DHT; ▪,with 5α-DHT treatment.
Effect of 5α-DHT on ERG protein level in isolated rabbit cardiac myocytes
To determine whether sustained stimulation of AR could also increase native ERG protein abundance, isolated rabbit cardiac myocytes were treated with or without 5α-DHT for 24 h. As shown in Fig. 8, treatment with 5α-DHT for 24 h also significantly increased ERG protein abundance, which is consistent with the previous study that in vivo 5α-DHT replacement increased ERG/IKr density in rabbits (6).
Fig. 8.

Effect of 5α-DHT on ERG protein levels in isolated rabbit cardiac myocytes. Immunoblot analysis shows that treatment with 5α-DHT (10 nm) for 24 h up-regulated ERG protein expression in rabbit cardiac myocytes. Data are means ± se (n = 4 per group). *, P < 0.05.
Discussion
Female gender is a risk factor for drug-induced cardiac arrhythmias, with an approximate 2:1 preponderance of women to men developing TdP arrhythmias in response to similar concentrations of a wide variety of drugs (1). We found in the present study that androgen up-regulated HERG protein expression and activity via a nongenomic mechanism. Activation of AR45 stabilized HERG protein through stimulation of ERK1/2. Therefore, the findings in the present study reveal the underlying mechanism for the gender differences in drug-induced arrhythmias.
AR45 is a naturally occurring variant of the human AR (18). It lacks the entire region encoded by exon 1 of the AR gene and is composed of the AR DNA-binding domain, hinge region, and ligand-binding domain, preceded by a novel seven amino acid long N-terminal extension. A survey of human tissues revealed that AR45 is expressed mainly in heart and skeletal muscle (18). In the present study, we found that stimulation of AR45, but not AR, up-regulated HERG protein abundance. Therefore, we further investigated the signaling and molecular mechanisms for the effect of AR45.
The nongenomic effect of AR45 stimulation on HERG protein abundance was first demonstrated in the present study. We observed the effect of androgen on HERG abundance in the presence of cycloheximide, a protein synthesis inhibitor. We found that cycloheximide alone decreased HERG protein abundance, but failed to abolish the effect of 5α-DHT. Using RT-PCR analysis, we also found that 5α-DHT had no effect on the mRNA level of HERG. Moreover, flutamide, a pure antiandrogen that binds to ARs and produces nongenomic effects (27), also up-regulated HERG protein expression. Together, these data strongly suggest that the up-regulation of HERG protein levels is not via de novo protein synthesis. This is also supported by the previous finding that AR45, which lacks the N-terminal activation function-1 (AF-1) domain, has very weak or no transactivation efficiency (18).
Gong et al. (29) reported that HERG proteins, especially the immature form, undergo proteasomal degradation, whereas Chapman et al. (28) demonstrated that HERG proteins could also be degraded via the lysosomal degradation pathway. We found in the present study that inhibition of either pathway significantly increased HERG protein abundance and attenuated the effect of androgen. These data indicate that both proteasomal and lysosomal degradation pathways are involved in the stimulatory effect of androgen on HERG abundance. To confirm the effect of androgen via inhibiting HERG channel protein degradation, we measured HERG protein stability with cycloheximide chase assay. We found that 5α-DHT significantly prolonged HERG half-life, confirming that androgens can stabilize HERG protein.
Recent studies showed that ERK1/2 activation regulates protein stability via modulating protein degradation both directly and indirectly (35, 36, 37). It was reported that ERK1/2 may directly phosphorylate MAPK phosphatase-1 and, therefore, reduce the degradation of this protein (35). ERK1/2 can also regulate protein degradation indirectly. For instance, activation of ERK1/2 inhibited GATA3 degradation via the ubiquitylation-dependent proteasome pathway by stimulating E3 ligase Mdm2 (37) and promoted p21 protein stability in the ubiquitylation-independent proteasome pathway by enhancing cyclin D1-imposed block (36). Therefore, we examined whether the inhibitory effect of androgens on HERG protein degradation is also mediated by ERK1/2. In the present study, we found that AR45 stimulation induced ERK1/2 phosphorylation within the same hormone concentration range as the effect on HERG. Blockade of ERK1/2 with two specific inhibitors, PD98059 and U0126, abolished the effect of 5α-DHT on HERG protein degradation. These data clearly suggest that activation of AR45 in the heart may modulate HERG protein stability via the ERK1/2 pathway.
The N terminus of AR not only determines the ability of AR to regulate gene transcription but also affects the binding affinity of signaling molecules (i.e. p85α of PI3K) to the receptor (18, 38). Interestingly, a similar isoform of ERα, ERα46, which like AR45 lacks AF-1, more efficiently modulates membrane initiated estrogen actions than ERα (39), and less efficiently regulates transcriptional activation evoked by estrogen (40). These findings and the data from our study imply that AF-1 in the N terminus of sex hormone receptors is critical for the genomic effects of sex hormones. AR and ERα, both of which have AF-1, may contribute more to the genomic effects, whereas AR45 and ERα46 may play a more important role in nongenomic functions within a restricted subset of tissues. The ratios of ERα46 to ERα (41, 42) and AR45 to AR (18) vary in different types of cells and at various developmental stages, which suggests that differential expression of alternatively spliced ER or AR variants may contribute to tissue-specific actions of sex hormones.
Our results also show that long-term 5α-DHT treatment only increased HERG current density but failed to alter HERG current kinetics or voltage dependence. However, Liu et al. (6) found that in vivo 5α-DHT replacement on orchiectomized male rabbits shifted the V1/2 of the IKr to a more negative membrane potential. The discrepancy between our and Liu et al.’s data could occur from the effect of 5α-DHT on other isoforms of ERG, β-subunits, or other impinging signaling pathways not present in our heterologous system. The channel contribution to the cardiac IKr is comprised not only of ERG, but also of ERG1b (N-terminal truncated transcriptional variant of ERG) and β-subunits such as MiRP1 (43, 44, 45). To understand better the modulation on IKr by androgens, further studies are warranted to investigate the effect of androgen on ERG1b and β-subunit expression and function.
In this study, we also examined the effect of other sex hormones on HERG protein expression. We found that progesterone and estradiol at the physiological concentration range failed to alter HERG protein abundance. These data suggest that androgens may selectively up-regulate HERG channel protein via stimulation of AR45.
We also compared the effect of different androgens on HERG protein expression. Testosterone is the predominant androgen present in the peripheral circulation. It can be converted to 5α-DHT and 5β-DHT. Compared with 5α-DHT, testosterone is at a 10- to 12-fold greater concentration in the blood. 5β-DHT has low affinity for the intracellular AR (46). R1881 is a widely used synthetic androgen that has high affinity for the AR (27). In this study we found that all of these androgens increased HERG protein abundance. Because 5α-DHT binds to the AR with higher affinity and specificity, it was chosen to test the effect of androgens on HERG protein expression in all other experiments. Flutamide binds the AR with relatively low affinity but does not possess any androgenic activity (27, 46). It has been widely accepted that antiandrogens can antagonize the genomic effect of androgens. In the present study, we found that flutamide mimics the effect of 5α-DHT, implying that the effect of androgen on HERG abundance was not a genomic effect. Recent studies showed that similar to androgen, antiandrogen can also activate ERK1/2 (30, 47). This may explain why flutamide, as an antiandrogen, also up-regulated HERG protein expression. The general cardiovascular safety of clinical uses of flutamide may be due, in part, to its lack of inhibition of the nongenomic functions of AR45.
Knowing that the androgen is capable of regulation of protein expression or channel function in a hetero-expression system is not enough. We need to return to the native tissue to identify its physiological significance. In the present study, we found that 5α-DHT also significantly increased ERG protein expression in rabbit cardiac myocytes, confirming the existence of such regulation by androgens on endogenous ERG in cardiac tissue.
In conclusion, our study demonstrates that prolonged stimulation of AR45, but not AR, up-regulated HERG K+ channels mainly through stabilizing HERG protein via stimulation of ERK1/2. These findings may provide a molecular mechanism for the sex differences in QT intervals and drug-induced arrhythmias.
Acknowledgments
We thank Miss Neo Kay Li and Miss Ester Khin Sandar Win for technical support and Dr. Gavin Dawe for critical readings.
Footnotes
This work is supported by National Medical Research Council research Grant NMRC0881/2004 and Biomedical Research Council research Grant BMRC 04/1/21/19/335.
Disclosure Statement: The authors have nothing to disclose.
First Published Online July 3, 2008
Abbreviations: AF-1, Activation function-1; ALLN, N-acetyl-L-leucyl-L-leucyl-L-norleucinal; AR, androgen receptor; CHO, Chinese hamster ovary; 5α-DHT, 5α-dihydrotestosterone; 5β-DHT, 5β-dihydrotestosterone; ER, endoplasmic reticulum; ERG, ether-á-go-go-related gene; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescence protein; HEK, human embryo kidney; HERG, human ether-á-go-go-related gene; IKr, rapid component of the delayed rectifier K+ current; LQTS, long QT syndrome; AR45, N-terminal truncated isoform of androgen receptor; QTc, heart rate-corrected QT interval; TBS, Tris-buffered saline; TdP, torsade de pointes.
References
- 1.Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH 1993. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA 270:2590–2597 [DOI] [PubMed] [Google Scholar]
- 2.Bazett H 1920. An analysis of the time relationship of electrocardiograms. Heart 7:353–370 [Google Scholar]
- 3.Rautaharju PM, Zhou SH, Wong S, Calhoun HP, Berenson GS, Prineas R, Davignon A 1992. Sex differences in the evolution of the electrocardiographic QT interval with age. Can J Cardiol 8:690–695 [PubMed] [Google Scholar]
- 4.Lehmann MH, Timothy KW, Frankovich D, Fromm BS, Keating M, Locati EH, Taggart RT, Towbin JA, Moss AJ, Schwartz PJ, Vincent GM 1997. Age-gender influence on the rate-corrected QT interval and the QT-heart rate relation in families with genotypically characterized long QT syndrome. J Am Coll Cardiol 29:93–99 [DOI] [PubMed] [Google Scholar]
- 5.Bidoggia H, Maciel JP, Capalozza N, Mosca S, Blaksley EJ, Valverde E, Bertran G, Arini P, Biagetti MO, Quinteiro RA 2000. Sex differences on the electrocardiographic pattern of cardiac repolarization: possible role of testosterone. Am Heart J 140:678–683 [DOI] [PubMed] [Google Scholar]
- 6.Liu XK, Katchman A, Whitfield BH, Wan G, Janowski EM, Woosley RL, Ebert SN 2003. In vivo androgen treatment shortens the QT interval and increases the densities of inward and delayed rectifier potassium currents in orchiectomized male rabbits. Cardiovasc Res 57:28–36 [DOI] [PubMed] [Google Scholar]
- 7.Sanguinetti MC, Jiang C, Curran ME, Keating MT 1995. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81:299–307 [DOI] [PubMed] [Google Scholar]
- 8.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA 1995. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269:92–95 [DOI] [PubMed] [Google Scholar]
- 9.Scheve BK, Benedict AT, Petrecca K, Van Wagoner DR, Shrier A, Nerbonne JM 2000. Expression of distinct ERG proteins, in rat, mouse, and human heart. J Biol Chem 275:5997–6006 [DOI] [PubMed] [Google Scholar]
- 10.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT 1995. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80:795–803 [DOI] [PubMed] [Google Scholar]
- 11.Sanguinetti MC, Curran ME, Spector PS, Keating MT 1996. Spectrum of HERG K+ channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci USA [Erratum (1996) 93:8796] 93:2208–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu XK, Katchman A, Drici MD, Ebert SN, Ducic I, Morad M, Woosley RL 1998. Gender difference in the cycle length-dependent QT and potassium currents in rabbits. J Pharmacol Exp Ther 285:672–679 [PubMed] [Google Scholar]
- 13.McGill Jr HC, Anselmo VC, Buchanan JM, Sheridan PJ 1980. The heart is a target organ for androgen. Science 207:775–777 [DOI] [PubMed] [Google Scholar]
- 14.Takeda H, Chodak G, Mutchnik S, Nakamoto T, Chang C 1990. Immunohistochemical localization of androgen receptors with mono- and polyclonal antibodies to androgen receptor. J Endocrinol 126:17–25 [DOI] [PubMed] [Google Scholar]
- 15.Kimura N, Mizokami A, Oonuma T, Sasano H, Nagura H 1993. Immunocytochemical localization of androgen receptor with polyclonal antibody in paraffin-embedded human tissues. J Histochem Cytochem 41:671–678 [DOI] [PubMed] [Google Scholar]
- 16.Doyu M, Sobue G, Kimata K, Yamamoto K, Mitsuma T 1994. Androgen receptor mRNA with increased size of tandem CAG repeat is widely expressed in the neural and nonneural tissues of X-linked recessive bulbospinal neuronopathy. J Neurol Sci 127:43–47 [DOI] [PubMed] [Google Scholar]
- 17.Knowlton AA, Sun L 2001. Heat-shock factor-1, steroid hormones, and regulation of heat-shock protein expression in the heart. Am J Physiol Heart Circ Physiol 280:H455–H464 [DOI] [PubMed]
- 18.Ahrens-Fath I, Politz O, Geserick C, Haendler B 2005. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J 272:74–84 [DOI] [PubMed] [Google Scholar]
- 19.Pandini G, Mineo R, Frasca F, Roberts Jr CT, Marcelli M, Vigneri R, Belfiore A 2005. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res 65:1849–1857 [DOI] [PubMed] [Google Scholar]
- 20.Roy P, Salminen H, Koskimies P, Simola J, Smeds A, Saukko P, Huhtaniemi IT 2004. Screening of some anti-androgenic endocrine disruptors using a recombinant cell-based in vitro bioassay. J Steroid Biochem Mol Biol 88:157–166 [DOI] [PubMed] [Google Scholar]
- 21.Bian J, Cui J, McDonald TV 2001. HERG K(+) channel activity is regulated by changes in phosphatidyl inositol 4,5-bisphosphate. Circ Res 89:1168–1176 [DOI] [PubMed] [Google Scholar]
- 22.Bian JS, Kagan A, McDonald TV 2004. Molecular analysis of PIP2 regulation of HERG and IKr. Am J Physiol Heart Circ Physiol 287:H2154–H2163 [DOI] [PubMed]
- 23.Huang ZQ, Li J, Wong J 2002. AR possesses an intrinsic hormone-independent transcriptional activity. Mol Endocrinol 16:924–937 [DOI] [PubMed] [Google Scholar]
- 24.Platz EA, Leitzmann MF, Rifai N, Kantoff PW, Chen YC, Stampfer MJ, Willett WC, Giovannucci E 2005. Sex steroid hormones and the androgen receptor gene CAG repeat and subsequent risk of prostate cancer in the prostate-specific antigen era. Cancer Epidemiol Biomarkers Prev 14:1262–1269 [DOI] [PubMed] [Google Scholar]
- 25.Burger HG 2002. Androgen production in women. Fertil Steril 77(Suppl 4):S3–S5 [DOI] [PubMed]
- 26.Shet MS, McPhaul M, Fisher CW, Stallings NR, Estabrook RW 1997. Metabolism of the antiandrogenic drug (Flutamide) by human CYP1A2. Drug Metab Dispos 25:1298–1303 [PubMed] [Google Scholar]
- 27.Gao W, Bohl CE, Dalton JT 2005. Chemistry and structural biology of androgen receptor. Chem Rev 105:3352–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapman H, Ramstrom C, Korhonen L, Laine M, Wann KT, Lindholm D, Pasternack M, Tornquist K 2005. Downregulation of the HERG (KCNH2) K(+) channel by ceramide: evidence for ubiquitin-mediated lysosomal degradation. J Cell Sci 118:5325–5334 [DOI] [PubMed] [Google Scholar]
- 29.Gong Q, Keeney DR, Molinari M, Zhou Z 2005. Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J Biol Chem 280:19419–19425 [DOI] [PubMed] [Google Scholar]
- 30.Peterziel H, Mink S, Schonert A, Becker M, Klocker H, Cato AC 1999. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene 18: 6322–6329 [DOI] [PubMed]
- 31.Estrada M, Espinosa A, Muller M, Jaimovich E 2003. Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology 144: 3586–3597 [DOI] [PubMed]
- 32.Fix C, Jordan C, Cano P, Walker WH 2004. Testosterone activates mitogen-activated protein kinase and the cAMP response element binding protein transcription factor in Sertoli cells. Proc Natl Acad Sci USA 101:10919–10924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen TV, Yao M, Pike CJ 2005. Androgens activate mitogen-activated protein kinase signaling: role in neuroprotection. J Neurochem 94:1639–1651 [DOI] [PubMed] [Google Scholar]
- 34.Cheng J, Watkins SC, Walker WH 2007. Testosterone activates mitogen-activated protein kinase via Src kinase and the epidermal growth factor receptor in Sertoli cells. Endocrinology 148:2066–2074 [DOI] [PubMed] [Google Scholar]
- 35.Brondello JM, Pouyssegur J, McKenzie FR 1999. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 286:2514–2517 [DOI] [PubMed] [Google Scholar]
- 36.Coleman ML, Marshall CJ, Olson MF 2003. Ras promotes p21(Waf1/Cip1) protein stability via a cyclin D1-imposed block in proteasome-mediated degradation. EMBO J 22:2036–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamashita M, Shinnakasu R, Asou H, Kimura M, Hasegawa A, Hashimoto K, Hatano N, Ogata M, Nakayama T 2005. Ras-ERK MAPK cascade regulates GATA3 stability and Th2 differentiation through ubiquitin-proteasome pathway. J Biol Chem 280:29409–29419 [DOI] [PubMed] [Google Scholar]
- 38.Baron S, Manin M, Beaudoin C, Leotoing L, Communal Y, Veyssiere G, Morel L 2004. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J Biol Chem 279:14579–14586 [DOI] [PubMed] [Google Scholar]
- 39.Li L, Haynes MP, Bender JR 2003. Plasma membrane localization and function of the estrogen receptor α variant (ER46) in human endothelial cells. Proc Natl Acad Sci USA 100:4807–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Figtree GA, McDonald D, Watkins H, Channon KM 2003. Truncated estrogen receptor α 46-kDa isoform in human endothelial cells: relationship to acute activation of nitric oxide synthase. Circulation 107:120–126 [DOI] [PubMed] [Google Scholar]
- 41.Flouriot G, Brand H, Denger S, Metivier R, Kos M, Reid G, Sonntag-Buck V, Gannon F 2000. Identification of a new isoform of the human estrogen receptor-α (hER-α) that is encoded by distinct transcripts and that is able to repress hER-α activation function 1. EMBO J 19:4688–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Staub C, Rauch M, Ferriere F, Trepos M, Dorval-Coiffec I, Saunders PT, Cobellis G, Flouriot G, Saligaut C, Jegou B 2005. Expression of estrogen receptor ESR1 and its 46-kDa variant in the gubernaculum testis. Biol Reprod 73:703–712 [DOI] [PubMed] [Google Scholar]
- 43.Lees-Miller JP, Kondo C, Wang L, Duff HJ 1997. Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circ Res 81:719–726 [DOI] [PubMed] [Google Scholar]
- 44.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA 1999. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97:175–187 [DOI] [PubMed] [Google Scholar]
- 45.Jones EM, Roti Roti EC, Wang J, Delfosse SA, Robertson GA 2004. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J Biol Chem 279:44690–44694 [DOI] [PubMed] [Google Scholar]
- 46.Fang H, Tong W, Branham WS, Moland CL, Dial SL, Hong H, Xie Q, Perkins R, Owens W, Sheehan DM 2003. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem Res Toxicol 16:1338–1358 [DOI] [PubMed] [Google Scholar]
- 47.Zhu X, Li H, Liu JP, Funder JW 1999. Androgen stimulates mitogen-activated protein kinase in human breast cancer cells. Mol Cell Endocrinol 152:199–206 [DOI] [PubMed] [Google Scholar]