

Abstract
Pseudomyxoma peritonei (PMP) is a subtype of mucinous adenocarcinoma mainly restricted to the peritoneal cavity and most commonly originating from the appendix. The genetic background of PMP is poorly understood and no targeted treatments are currently available for this fatal disease. While RAS signaling pathway is affected in most if not all PMP cases and over half of them also have a mutation in the GNAS gene, other genetic alterations and affected pathways are, to a large degree, poorly known. In this study, we sequenced whole coding genome of nine PMP tumors and paired normal tissues in order to identify additional, commonly mutated genes and signaling pathways affected in PMP. These exome sequencing results were validated with an ultra-deep amplicon sequencing method, leading to 14 validated variants. The validated results contain seven genes that contribute to the protein kinase A (PKA) pathway. PKA pathway, which also contains GNAS, is a major player of overproduction of mucin, which is the characteristic feature of PMP. In addition to PKA pathway, we identified mutations in six genes that belong to the transforming growth factor beta (TGF-β) pathway, which is a key regulator of cell proliferation. Since either GNAS mutation or an alternative mutation in the PKA pathway was identified in 8/9 patients, inhibition of the PKA pathway might reduce mucin production in most of the PMP patients and potentially suppress disease progression.
Introduction
Pseudomyxoma peritonei (PMP) is a rare subtype of intestinal-derived mucinous adenocarcinoma, with prevalence of approximately 1–2 persons per million per year [1]. PMP originates most frequently from the appendix, and through its rupture the tumor cells spread into the peritoneal cavity. A characteristic feature of PMP is the ability of tumor cells to produce large amounts of extracellular mucus, which leads to bowel obstruction, breathing difficulties, malnutrition, and eventually death of the patient. According to the World Health Organization (WHO) classification PMP is categorized into low-grade (LG) and high-grade (HG) disease [2]. LG PMP is a relatively slowly progressing disease with 5-year survival of 63%, while the HG type of the disease has the ability to invade and metastasize, leading to 5-year survival of only 23% [3]. Current standard treatment of PMP is aggressive cytoreductive surgery combined with hyperthermic intraperitoneal chemotherapy [4]. This combinatory treatment, however, causes morbidity and mortality, and is only amenable for 60–70% of the patients [5]. Furthermore, as a significant proportion of PMPs usually relapse, additional targeted treatments are needed.
The genetic background of PMP is poorly understood due to its low incidence and challenges related to collection of representative tissue material of this remarkably mucinous tumor type. The published next-generation sequencing (NGS) studies [6–11] have mostly used targeted sequencing of mutational hotspot areas of known cancer related genes. These studies have discovered the characteristic KRAS and GNAS alterations in PMP, together with a lower frequency of e.g. SMAD4, SMAD2, SMAD3, TP53, PIK3CA, and AKT1 mutations. Exome sequencing enables identification of variants in the whole coding region of the human genome and allows to expand the analysis of the mutations to pathway level.
In our previous study covering 212 somatic mutation hotspots in 48 cancer related genes in 19 patients [9], we found that all sequenced PMP tumors had an activating KRAS mutation and 12/19 (63%) of the patients harbored a mutation in the GNAS gene. Here, we have sequenced the whole coding genome of nine of these PMP tumors and paired normal tissues in order to identify common mutated genes and pathways outside the scope of the targeted hotspot panels. Mutations in the protein kinase A (PKA) pathway were under special focus, since PKA pathway activation is involved in the overproduction of mucin [7], and 4/9 tumors in our sample set lacked the typical GNAS mutation, which led to hypothesis that mutations in other genes in PKA pathway may lead to similar phenotype. Exome sequencing results were validated with an independent measurement method that is based on ultra-deep amplicon sequencing.
Materials and methods
Patients
Nine PMP tumor samples (6 LG and 3 HG) with histologically confirmed appendiceal origin were analyzed with exome sequencing. The tumor cell content of the samples was between 10% and 60% as estimated from hematoxylin and eosin stained (HE) slides. Nine matching control samples (from skin or gallbladder) were included into the analysis. Tissue samples were processed at Division of Pathology and Genetics, HUSLAB, at the Helsinki University Central Hospital between 2006 and 2013. Grading of the tumor samples was done according to the WHO 2010 classification [2]. This study was approved by the Ethics Committee of the Helsinki University Central Hospital (code 408/13/03/03/2009) and the patients gave written informed consent.
DNA extraction
From the formalin-fixed paraffin-embedded (FFPE) tissue specimens, 10 μm flakes were cut with a Leica SM2000R microtome (Leica Microsystems GmbH, Wetzlar, Germany). Macrodissection was used when applicable to increase the yield of tumor DNA. Two to fifteen tissue flakes were deparaffinized and genomic DNA was extracted using QIAamp DNA Mini Kit (Qiagen, Venlo, the Netherlands) as previously reported [9]. DNA quality was inspected with NanoDrop spectrophotometer (Thermo Fischer Scientific, Waltham, MA) and agarose gel electrophoresis (with Midori Green Advanced DNA Stain; Nippon Genetics EUROPE GmbH, Dueren, Germany), and DNA concentration was determined using Qubit dsDNA HS Assay Kit and Qubit 2.0 Fluorometer (Molecular Probes/Life Technologies, Paisley, UK).
Exome sequencing
Target enrichment for exome sequencing was done with the Agilent SureSelect 51M Capture Kit (Santa Clara, CA). Paired-end sequencing with 90 base pair read length was performed with Illumina HiSeq 2000 (San Diego, CA). Alignment of the reads was done by BWA [12] with parameters bwa aln -o 1 -e 50 -m 100000 -t 4 -i 15 -q 10 -l, and the human genome build 37 (hg19) was used as the reference. Duplicate reads were removed with Picard Tools (http://picard.sourceforge.net).
Data analyses
Sequence data were analyzed with the Anduril workflow framework [13] (Fig 1). Variants were called by VarScan somatic (v.2.3.6) [14] and MuTect (v1.1.4) [15] in tumor-normal pairs. The pileup files for VarScan somatic were created using SAMTools mpileup (v0.1.19) [16] with default parameters except for -q 1 -Q 10. We estimated tumor cell percentage for each sample both visually from the HE stained slides and from the variant allele frequencies (VAF) of KRAS and GNAS variants obtained from our previous targeted deep sequencing of the same samples [9] (Table 1). The average read coverage of the targeted sequencing was 3,580X, making the VAFs and, thus, tumor cell percentages, more reliable than using VAFs from exome sequencing (average coverage 120X). Tumor purity estimated from the targeted sequencing, ranging from 6% to 28%, was then used as the purity parameter for each tumor sample in the VarScan variant caller algorithm. Other default parameters for VarScan were used except—min-avg-qual 10. VarScan derived somatic single nucleotide variation (SNV) and short insertion and deletion (indel) calls were filtered with Fisher test p-value < 0.05 to obtain statistically significant calls. VarScan results were further filtered to contain cancer related genes from the Cancer Gene Census [17] in addition with kinome genes [18] to prioritize functionally relevant calls. SNV calls from MuTect were not filtered, since the method itself contains stringent filtering, and allowed thus calls from the whole exonic regions. In order not to miss true positives we preferred sensitivity over specificity and combined the calls from VarScan and MuTect. The increase in false positives in the discovery set is overcome by the use of independent validation method. Synonymous changes were disregarded. Resulting variants were annotated with Annovar [19] and CADD (v1.2) functional scores [20]. Scaled CADD score > 10 was used to gain the set of most pathogenic variants. All resulting variants were inspected visually by unpublished genome analysis tool Rikurator (R. Katainen, V. Mäkinen, L.A. Aaltonen, E. Pitkänen, manuscript under preparation). Variants with mis-alignment or having reads only in single direction were regarded as unreliable and removed. Variant genes were annotated with Gene Ontology terms [21] and pathway analyses were done with DAVID Bioinformatics Resources 6.7 [22].
Fig 1. Workflow used for exome sequencing of nine pseudomyxoma peritonei specimens and their matching control samples.

Table 1. Tumor cell content of pseudomyxoma peritonei FFPE samples estimated from HE stainings, and variant allele frequencies (VAF) of KRAS and GNAS from targeted sequencing.
| VAF from targeted seq* | |||||
|---|---|---|---|---|---|
| Patient | Grade | HE T% | KRAS | GNAS | Seq T%** |
| 2 | LG | 40 | 0.179 | 0.077 | 16 |
| 201 | LG | 20 | 0.083 | 0.062 | 12 |
| 202 | HG | 10 | 0.054 | 10 | |
| 206 | HG | 60 | 0.103 | 20 | |
| 210 | LG | 20 | 0.034 | 6 | |
| 214 | LG | 25 | 0.041 | 0.034 | 6 |
| 28 | HG | 50 | 0.078 | 0.079 | 16 |
| 29 | LG | 30 | 0.144 | 0.278 | 28 |
| 31 | LG | 20 | 0.081 | 16 | |
LG: low-grade; HG: high-grade; HE: hematoxylin and eosin; T%: tumor cell percentage; VAF: variant allele frequency; seq: sequencing
* Ref. [9]
**Tumor cell percentage calculated by multiplying the heterozygote VAF (lower value of either KRAS or GNAS) by factor of two.
Amplicon sequencing
Ultra-deep amplicon sequencing was used to validate exome sequencing results. Ninety-four most relevant variants detected with exome sequencing were validated using ultra-deep amplicon sequencing (S1 Table). We selected variants that belong to the protein kinase A (PKA) or transforming growth factor beta (TGF-β) pathway, and sites in genes which were mutated in at least two patients. Variants on PKA pathway, which is shown to play a key role in PMP pathogenesis [7], were selected since 4/9 PMP tumors lacked GNAS mutation and we hypothesized that in these cases mutations in other genes in the PKA pathway may lead to similar phenotype. Variants on TGF-β pathway were selected since it showed high enrichment in pathway analysis (Bonferroni corrected p-value 0.001; S2 Table). MAPK and ErbB pathways showed the highest enrichment of signaling pathways (Bonferroni corrected p-values 1.53x10-6 and 3.89x10-5, respectively), but these pathways were not validated due to the fact that all PMP tumor samples had an activating mutation of the KRAS oncogene [9], which is sufficient to activate these pathways [23]. Sample 29 could not be validated due to lack of DNA. PCR primers (Sigma-Aldrich, St. Louis, MO; S3 Table) were designed by Primer3 [24] with amplicon size of 150–200 bp, GC percentage 30–70%, primer length 18–35 bp, and melting temperature 60–62°C. PCR reactions were done in 20 μl reaction volume containing 20 ng gDNA, 0.4 μl dNTP mix (100 mM), 2 μl of each primers (10 μM), 2.5 U Expand High FidelityPLUS Enzyme Blend and 1X Expand High FidelityPLUS Reaction buffer with final concentration of 1.5 mM MgCl2 (Roche, Basel, Switzerland). The cycling conditions were as previously reported [25] except that denaturation time in each cycle was 30 seconds, annealing temperature was 60°C, and the number of PCR cycles was 32–35. Half of the PCR reaction was then analyzed with gel electrophoresis and the other half was used for amplicon purification. Each separate amplicon was purified with Beckman Coulter AMPure XP Beads (Pasadena, CA) according to the manual with bead:DNA ratio of 2X (20 μl of nuclease free water and 60 μl of AMpureXP Beads were added to 10 μl of sample). Purified amplicons were quantified using Qubit and all the amplicons of each patient sample were pooled in equimolar basis. The concentrations of all the amplicon pools were then adjusted to 0,54 ng/μl and 10 μl of these pools were used for library preparation. Library preparation was done using ThruPLEX DNA-seq Kit (Rubicon Genomics, Ann Arbor, MI) according to the instruction manual and libraries were checked with Agilent 2100 Bioanalyzer. Finally, the samples were sequenced with MiSeq v2 (Illumina) using paired-end sequencing with 150 base pair read length. Reads were aligned by BWA mem with default parameters and human genome build 37 (hg19) as the reference. Variants were calculated by SAMtools mpileup (v0.1.19) and VarScan (v.2.3.6) readcounts commands. Variants with coverage greater than 1,000X and frequency at least one third of the corresponding KRAS VAF in the same sample were regarded as confident.
Comparison of KRAS and GNAS mutations detected with targeted sequencing, exome sequencing, and validation sequencing
Average coverage of the exome sequencing was 120X, and on average 95.9% of the target was covered with at least 20X. With this coverage, the two applied variant caller algorithms were not able to detect all the somatic mutations that we previously detected in the same samples by the targeted sequencing with the average coverage of 3,580X [9]. For KRAS we detected 5/9, and for GNAS 5/5 of the mutations (Table 2). To validate the exome sequencing results we used ultra-deep amplicon sequencing with average coverage ranging from 60,000X to 400,000X per sample. With this validation sequencing we were able to detect the KRAS and GNAS variants with similar VAF as previously with the targeted sequencing (Table 2).
Table 2. Comparison of KRAS and GNAS mutations detected with targeted sequencing, exome sequencing and validation sequencing.
| KRAS | ||||||||||
| Targeted sequencing* | Exome sequencing | Validation sequencing | ||||||||
| Patient | Position | Coverage | Call | Frequency | Coverage | Call | Frequency | Coverage | Call | Frequency |
| 2 | 12:25398284 | 2346 | C/T | 0.179 | 29 | C/T | 0.207 | 56166 | C/T | 0.163 |
| 201 | 12:25398284 | 3775 | C/T | 0.083 | 54 | ND | ND | 310144 | C/T | 0.050 |
| 202 | 12:25398284 | 4159 | C/T | 0.054 | 55 | C/T | 0.073 | 415544 | C/T | 0.052 |
| 206 | 12:25398284 | 3640 | C/T | 0.103 | 48 | ND | ND | 84351 | C/T | 0.096 |
| 210 | 12:25398284 | 8797 | C/T | 0.034 | 48 | C/T | 0.083 | 160119 | C/T | 0.033 |
| 214 | 12:25398284 | 2844 | C/T | 0.041 | 56 | ND | ND | 165996 | C/T | 0.035 |
| 28 | 12:25398284 | 3495 | C/T | 0.078 | 49 | C/T | 0.061 | 259844 | C/T | 0.080 |
| 29 | 12:25398281 | 2765 | C/T | 0.144 | 47 | C/T | 0.085 | NA | NA | NA |
| 31 | 12:25398284 | 1611 | C/T | 0.081 | 28 | ND | ND | 25378 | C/T | 0.085 |
| GNAS | ||||||||||
| Targeted sequencing* | Exome sequencing | Validation sequencing | ||||||||
| Patient | Position | Coverage | Call | Frequency | Coverage | Call | Frequency | Coverage | Call | Frequency |
| 2 | 20:57484420 | 2811 | C/T | 0.077 | 116 | C/T | 0.060 | 61926 | C/T | 0.094 |
| 201 | 20:57484421 | 3405 | G/A | 0.062 | 117 | G/A | 0.103 | 368512 | G/A | 0.048 |
| 202 | ||||||||||
| 206 | ||||||||||
| 210 | ||||||||||
| 214 | 20:57484420 | 2509 | G/A | 0.034 | 153 | G/A | 0.033 | 265484 | G/A | 0.031 |
| 28 | 20:57484420 | 2726 | G/A | 0.079 | 139 | G/A | 0.094 | 164773 | G/A | 0.082 |
| 29 | 20:57484420 | 1763 | G/A | 0.278 | 134 | G/A | 0.164 | NA | NA | NA |
| 31 | ||||||||||
* [9]; ND: not detected; NA: not available.
Results
Somatic exome-wide SNVs and short indels in PMP tumors
Results of exome-wide analysis of the filtered somatic SNVs and indels are summarized in Fig 2. After all the filtering steps, we retained an average 67 high confidence, protein chancing somatic SNVs and indels per tumor (range 15–180) (S2 Table). In total, we found 535 transitions (Ts; A↔G or C↔T) and 59 transversions (Tv; A:G↔C:T), giving an overall Ts to Tv ratio 9.1. Ts to Tv ratios for each tumor ranged from 2.8 to 28.3. High rate of Ts is a common feature in FFPE samples caused by PCR induced errors, promoted by DNA modifications [26, 27].
Fig 2. Exome-wide statistics of filtered somatic SNV and indel calls.

(A) Number of SNVs and indels. (B) Total number of somatic nucleotide base changes. (C) Ts to Tv ratio of each tumor. SNV: single nucleotide variation; indel: insertion/deletion; Ts: transition; Tv: transversion.
Validation of the mutations
We performed ultra-deep sequencing validation for altogether 94 potentially relevant variants in 53 genes detected in the exome sequencing experiment. As internal controls, we included amplicons for the KRAS and GNAS hotspot areas. Since all the sequenced samples had a KRAS mutation, we filtered our validation results using the corresponding KRAS variant allele frequencies (S1 Table). Altogether 14 mutations in 13 genes passed all the quality filtering steps, resulting in validation rate of 14.9%. All validated 14 variants were considered to affect protein function as ten of them are missense mutations, three are nonsense mutations, and one is a canonical splice site mutation. All of these mutations were predicted to be potentially functional by the in silico prediction tool CADD and six of them exist in the COSMIC database [28] (Table 3; the gene, transcript, and chromosomal positions are from Ensembl build 37 [29]). Evaluation of the reliability of these variants, considering the bias in FFPE samples, was accomplished by applying six separate criteria specified in the Table 3.
Table 3. Variants detected on pseudomyxoma peritonei by exome sequencing and validated by ultra-deep amplicon sequencing.
| Patient | Gene | Ensembl gene | Ensembl transcript | Chromosomal position | Nucleotide (cDNA) | Amino acid (protein) | CADD score | Evidence* | Criteria for annotating variant as a potential artefact** |
|---|---|---|---|---|---|---|---|---|---|
| 2 | OXTR | ENSG00000180914 | ENST00000316793 | 3:8809260 | c.C614G | p.T205R | 19.8 | 2, 3, 4 | |
| 201 | LTBP1 | ENSG00000049323 | ENST00000354476 | 2:33335707 | c.G922A | p.V308M | 11.3 | Cosmic | 1, 2, 3, 5 |
| 202 | THBS1 | ENSG00000137801 | ENST00000260356 | 15:39885810 | c.A3208G | p.T1070A | 24.3 | 3, 4 | |
| 206 | DUSP22 | ENSG00000112679 | ENST00000344450 | 6:348195 | c.G356A | p.R119H | 34 | 1, 2, 3, 4 | |
| 206 | EDNRA | ENSG00000151617 | ENST00000324300 | 4:148441016 | c.G434A | p.R145H | 23.8 | 1, 3, 4 | |
| 206 | PRKAR1A | ENSG00000108946 | ENST00000589228 | 17:66518896 | c.178-1G>C | splice site variant | 24.7 | 1, 3, 4, 6 | |
| 206 | RYR2 | ENSG00000198626 | ENST00000366574 | 1:237693756 | c.G2852T | p.G951V | 23 | 3, 4 | |
| 210 | TGFBR2 | ENSG00000163513 | ENST00000295754 | 3:30729968 | c.C1489T | p.R497X | 50 | Cosmic | |
| 210 | PPP2R1B | ENSG00000137713 | ENST00000527614 | 11:111626081 | c.C781T | p.R261X | 38 | 1, 3, 4 | |
| 210 | ACVR1B | ENSG00000135503 | ENST00000257963 | 12:52374813 | c.G641T | p.G214V | 31 | Cosmic | 3 |
| 210 | GUSB | ENSG00000169919 | ENST00000304895 | 7:65439369 | c.G1304A | p.R435H | 26.9 | 1, 3, 4 | |
| 214 | TGFBR2 | ENSG00000163513 | ENST00000295754 | 3:30732957 | c.G1570A | p.D524N | 35 | Cosmic | 1, 6 |
| 31 | GRIA2 | ENSG00000120251 | ENST00000296526 | 4:158233974 | c.C613T | p.R205W | 28 | Cosmic | 1, 3, 6 |
| 31 | CNGB1 | ENSG00000070729 | ENST00000251102 | 16:57950065 | c.C2185T | p.R729X | 17.9 | Cosmic | 1, 3 |
X: stop-gain. CADD score reflects the deleteriousness of the mutation.
*Cosmic: Occurence of similar mutation in the COSMIC database [28].
- C→T/G→A reported only by one variant caller
- VAF < average in a sample
- found only in a single sample
- not reported as recurrently mutated in COSMIC
- CADD score < 15
- coverage < 50% of sample average
Validation confirmed mutations in the PKA and TGF-β pathways
From the selected 21 variants in 19 genes that belong to the PKA pathway, the validation confirmed seven variants in seven genes occurring in 4/9 patients (OXTR, EDNRA, PRKAR1A, RYR2, GUSB, GRIA2, and CNGB1) (Table 3 and Fig 3). From the selected 19 variants in 14 genes that belong to the TGF-β pathway, validation confirmed six variants in five genes occurring in 4/9 patients (LTBP1, THBS1, TGFBR2, PPP2R1B, and ACVR1B) (Table 3 and Fig 3). Each of these genes was mutated exclusively in one patient except TGFBR2 which was found to be mutated in two cases.
Fig 3. Gene mutations identified in pseudomyxoma peritonei (PMP).
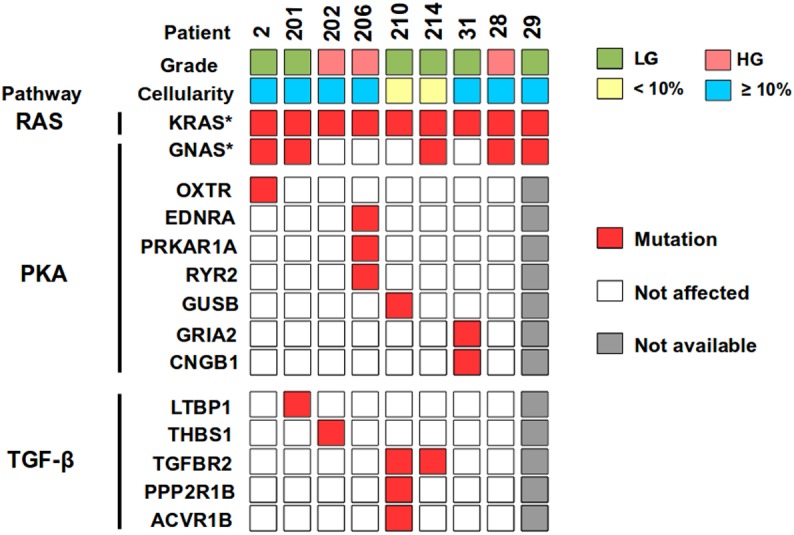
Nine appendix-derived PMP samples were analyzed for grade and cellularity, using HE slides and NGS, respectively. Somatic mutations were identified using exome sequencing and validated by ultra-deep amplicon sequencing. HG: high-grade; LG: low-grade. * [9].
Other validated mutations
Finally, we selected 25 genes that were mutated in at least two PMP patients in the exome data. This led to validation sequencing of 52 variants in 25 genes. TGFBR2 was the only mutated gene that was confirmed in multiple PMP patients (2/9). Interestingly, a mutation in DUSP22 was confirmed in one HG PMP patient (Table 3). DUSP22 activates the JNK pathway [30] and is involved in apoptosis. Thus, mis-regulation in apoptosis may contribute to the high-cellularity of this PMP tumor with 20% tumor cell percentage.
Discussion
The genetic background of PMP is incompletely characterized. It is known that the RAS signaling pathway activation is frequently if not always present in PMP and most often facilitated by a mutation of the KRAS oncogene in codons 12 or 13 (S4 Table) [6–11,31–34]. In addition to KRAS, one NRAS mutation has been reported in a patient lacking KRAS mutation [10], emphasizing the crucial role of RAS pathway activation in PMP. These mutant RAS proteins are constitutively active promoting cell proliferation and suppressing apoptosis [35]. KRAS and NRAS mutated colorectal cancers (CRC) are known to be resistant to epithelial growth factor receptor (EGFR)-targeted therapy [36], suggesting that this type of treatment may also be ineffective in PMP. In addition, BRAF mutations are rare events in PMP [7–11] and thus anti-BRAF therapy may also not be applicable. In addition to RAS pathway activating mutations, GNAS codon 201 is mutated with high frequency in PMP [6–11,34]. GNAS encodes the α-subunit of the stimulatory G protein that transduces signals from seven-transmembrane receptors. A mutation at codon 201 leads to constitutive activation of adenylyl cyclase leading to elevated levels of cyclic AMP (cAMP) and activation of the PKA pathway. In a CRC cell line, activated GNAS has been shown to induce expression of MUC2 and MUC5AC [34], which belong along with MUC5B to the most prominent mucins produced by PMP tumors [37,38]. Further, activated PKA has been shown to stimulate secretion of mucins in human colonic epithelial cells [39]. Thus, GNAS or PKA inhibition might reduce mucin secretion in PMP patients, relieving obstruction related symptoms and disease progression.
PKA has been reported to be strongly phosphorylated and activated in PMP tumors even in the absence of GNAS mutation [7], which lead to the hypothesis that in addition to GNAS, also other members of PKA pathway may be affected. As four out of nine of our exome sequenced samples lacked the GNAS hotspot mutation (Fig 3), we targeted our search to identify other mutations of the PKA pathway (Fig 4A). One of our HG PMP patients harbored three mutated genes of this pathway: PRKAR1A, EDNRA, and RYR2. PRKAR1A encodes regulatory subunit of PKA and had a canonical splice site mutation (c.178-1G>C) in the dimerization/docking domain, suggesting a loss of function. Similarly, Alakus et al. [7] found a mutation in another subunit of PKA (PRKACA) in a PMP patient lacking GNAS mutation. Interestingly, patients of Carney complex, which is an autosomal dominant disease caused by inactivation of the PRKAR1A gene, have a high prevalence of pancreatic tumors, including intraductal papillary mucinous neoplasms (IPMN) [41]. This also supports functionality of PRKAR1A in PMP, since mutational profiles of IPMN and PMP are highly similar [7,9,34]. Importantly, inactivation of PRKAR1A gene in Carney complex cells has been found to constitutively activate PKA [42,43]. In our series, mutations in the EDNRA (p.R145H) and RYR2 (p.G951V) genes were missense, altering the amino acid structure of seven-transmembrane receptor and RyR domain, respectively. One LG PMP patient had a missense mutation in the GUSB (p.R435H) gene, which is known to interact with the PRKACA subunit of PKA [44]. Furthermore, germline mutations of GUSB gene cause autosomal recessive disorder, mucopolysaccharidoses, characterized with production of excessive mucus. Another LG PMP patient had two mutated genes of the PKA pathway, CNGB1 (p.R729X) and GRIA2 (p.R205W), harboring a nonsense and a missense mutations, respectively. In summary, we were able to detect mutations in PKA pathway in 3/4 patients lacking GNAS mutation. Altogether, 8/9 patients seemed to have affected PKA pathway, making it a potentially interesting molecular target in PMP.
Fig 4. Gene mutations related to PKA and TGF-β pathways in pseudomyxoma peritonei.

(A) Variants of PKA pathway. (B) Variants of TGF-β pathway. Pathways have been modified from KEGG [40]. PKA: protein kinase A; TGF-β: transforming growth factor beta.
TGF-β pathway is dysregulated in many cancers, including PMP (S4 Table) [7,9–11]. Thus far, SMAD4, SMAD2, SMAD3, TGFBR1, and TGFBR2 mutations have been the most often detected ones in PMP. Similarly to Alakus et al. [7], we detected TGFBR2 mutations (p.R497X and p.D524N) in two PMP patients, both affecting kinase domains. Since TGFBR2-R497X is a nonsense mutation and mutations in the same residue as TGFBR2-D524N have been detected in inherited diseases [45], it indicates loss of function for both cases. TGFBR2 has been found mutated also in CRC leading to inactivation of the TGF-β pathway [46]. Interestingly, only the combination of KRAS activation and TGFBR2 inactivation in mice have been found to induce formation of intestinal neoplasms [47]. In addition, we detected other mutations in the TGF-β pathway: LTBP1 (p.V308M), THBS1 (p.T1070A), PPP2R1B (p.R261X), and ACVR1B (p.G214V) genes, which may contribute to the development and progression of PMP (Fig 4B). Both LTBP1 and THBS1 have been shown to control the activity of TGFB1 [48,49] and they were mutated in different patients. PPP2R1B encodes the beta isoform of the A subunit of the serine/threonine protein phosphatase 2A (PP2A), which has been found to be mutated also in colon and lung cancers [50]. ACVR1B mutation in the catalytic domain was found in a patient who also had mutated TGFBR2. ACVR1B encodes activin A type IB receptor and is frequently mutated in CRC [46]. Interestingly, loss of ACVR1B was recently reported to accelerate development of IPMN in mice with activated KRAS [51]. Altogether, 4/9 PMP patients were identified to have TGF-β pathway alterations, which further highlight the importance of the TGF-β pathway in PMP pathogenesis.
Most likely also other dysregulated pathways than RAS, PKA and TGF-β have a role in the development and progression of PMP, and further investigation of these pathways is needed (S2 and S4 Tables). For example, TP53 has been shown to be mutated in HG PMPs [7–11]. The PI3K-AKT pathway is affected through PIK3CA and AKT1 mutations [9–11], and MCL1 amplification [6], and the Wnt pathway through CTNNB1 mutation [7,11] and amplification of JUN [6]. Furthermore, mechanisms, such as somatic structural rearrangements, intronic variants, microRNA expression changes, and epigenetic events, may also influence the development of PMP but have not yet been examined.
Due to the low proportion of neoplastic cells in the PMP samples, sensitivity and functional prioritizing was emphasized in the exome sequencing data analysis. Ultra-deep targeted sequencing was then used to eliminate false positives resulting from the reduced specificity. Transition substitutions, C→T or G→A, are the most abundant mutations in PMP [7], which was evident also in our results (Fig 2B), suggesting similar mechanisms of DNA mutagenesis as in CRC [52]. It is, however, important to note that hydrolytic deamination of cytosines leads to overrepresentation of C to T (C→T/G→A), and therefore variant artefacts are common in next-generation amplicon sequencing of formalin-fixed tumor samples [27]. Prevalence of C to T artefacts is indeed highest in low concentration samples in VAFs < 10%. Although such variants are rarely of high functional relevance and do not usually locate in mutational hotspots [53], some clinically actionable mutations have been shown to be false positives [54,55]. To minimize the number of formalin-fixation caused false positive alterations in the final results, we applied the filtering steps represented in the Fig 1. In total, 14 variants (Table 3) survived after filtering and included 64% of C→T/G→A transitions (9 out of 14 substitutions), which is a higher proportion compared to the average of 40.8% in the human genome [53]. It is, however, extremely unlikely that identical artifacts emerge and pass all our filtering steps in two independent experiments utilizing distinct targeting methods, i.e. Agilent’s whole-exome hybrid capture assay for mutation identification and custom-designed PCR amplicons for ultra-deep validation sequencing. However, mutations which resemble typical artefacts in formalin-fixed tumor sequences must always be treated with a caution (Table 3).
In conclusion, we have used FFPE samples with deep sequencing to characterize the mutational landscape in PMP. FFPE samples are more abundant and easier to access than fresh samples but their computational analysis is challenging. Thus, the successful use of the freely available computational pipeline developed herein paves way to use FFPE samples in future PMP studies. Our major finding include novel mutated genes in the PKA pathway possibly contributing to the characteristic mucin production of the PMP disease. Inhibition of the PKA pathway could potentially be a universal target in reducing mucin production in the PMP patients, which could prolong survival of these patients. Our results also highlight the importance of TGF-β pathway in the development and progression of PMP and therapies targeting this pathway could also suppress progression of the disease.
Conclusions
Genetic background of pseudomyxoma peritonei (PMP) is not well characterized and there are no targeted therapies for the treatment of this disease. In this study, we used exome sequencing to expand the mutational analysis of PMP tumors beyond the mutational hotspot areas of known cancer genes and validated the results with an ultra-deep amplicon sequencing method. Validated results indicate that in addition to the fundamental KRAS and GNAS mutations, somatic mutations in the PKA and TGF-β pathways possibly account for the increased mucus secretion and cell proliferation, respectively.
Supporting information
(XLSX)
(XLSX)
(DOCX)
(XLSX)
Acknowledgments
We thank Carita Liikanen, Merja Haukka, and Biomedicum Functional Genomics Unit (FuGU) (University of Helsinki, Helsinki, Finland) for excellent technical assistance, and Katherine Icay for proof-reading.
Data Availability
All relevant data are within the paper and its Supporting Information files. The exome sequencing data is available from the European Genome-phenome Archive database (accession number EGAS00001002418).
Funding Statement
This study was supported by University of Helsinki, Sigrid Jusélius Foundation, the Finnish Cancer Organizations, Helsinki University Central Hospital Research Funds, the Academy of Finland (Center of Excellence in Cancer Genetics Research), Ida Montin Foundation (LS), Cancer Society of Finland (LS), Orion Research Foundation (LS), and K. Albin Johansson Foundation (LS). The funding sources had no involvement in planning, execution, or reporting of this research.
References
- 1.Smeenk RM, van Velthuysen MLF, Verwaal VJ, Zoetmulder FAN. Appendiceal neoplasms and pseudomyxoma peritonei: a population based study. Eur J Surg Oncol. 2008;34:196–201. [DOI] [PubMed] [Google Scholar]
- 2.Carr NJ, Sobin LH. Adenocarcinoma of the appendix In Bosman FT, Carneiro F, Hruban RH, Theise ND, editors. WHO classification of tumours of the digestive system, 4th edition Lyon: IARC; 2010. p. 122–5. [Google Scholar]
- 3.Carr NJ, Finch J, Ilesley IC, Chandrakumaran K, Mohamed F, Mirnezami A, et al. Pathology and prognosis in pseudomyxoma peritonei: a review of 274 cases. J Clin Pathol. 2012;65:919–23. [DOI] [PubMed] [Google Scholar]
- 4.Sugarbaker PH, Kern K, Lack E. Malignant pseudomyxoma peritonei of colonic origin. Natural history and presentation of a curative approach to treatment. Dis Colon Rectum. 1987;30:772–9. [DOI] [PubMed] [Google Scholar]
- 5.Järvinen P, Ristimäki A, Kantonen J, Lepistö A. Feasibility of radical cytoreductive surgery and hyperthermic intraperitoneal chemotherapy for pseudomyxoma peritonei of appendiceal origin. Scand J Surg. 2013;102:145–51. 10.1177/1457496913490463 [DOI] [PubMed] [Google Scholar]
- 6.Sio TT, Mansfield AS, Grotz TE, Graham RP, Molina JR, Que FG, et al. Concurrent MCL1 and JUN amplification in pseudomyxoma peritonei: a comprehensive genetic profiling and survival analysis. J Hum Genet. 2013;59:124–8. 10.1038/jhg.2013.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alakus H, Babicky ML, Ghosh P, Yost S, Jepsen K, Dai Y, et al. Genome-wide mutational landscape of mucinous carcinomatosis peritonei of appendiceal origin. Genome Med. 2014;6:43 10.1186/gm559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, Mody K, de Abreu FB, Pipas JM, Peterson JD, Gallagher TL, et al. Molecular profiling of appendiceal epithelial tumors using massively parallel sequencing to identify somatic mutations. Clin Chem. 2014;60:1004–11. 10.1373/clinchem.2014.225565 [DOI] [PubMed] [Google Scholar]
- 9.Nummela P, Saarinen L, Thiel A, Järvinen P, Lehtonen R, Lepistö A, et al. Genomic profile of pseudomyxoma peritonei analyzed using next-generation sequencing and immunohistochemistry. Int J Cancer. 2015;136:E282–9. 10.1002/ijc.29245 [DOI] [PubMed] [Google Scholar]
- 10.Noguchi R, Yano H, Gohda Y, Suda R, Igari T, Ohta Y, et al. Molecular profiles of high-grade and low-grade pseudomyxoma peritonei. Cancer Med. 2015;4:1809–16. 10.1002/cam4.542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pietrantonio F, Perrone F, Mennitto A, Gleeson EM, Milione M, Tamborini E, et al. Toward the molecular dissection of peritoneal pseudomyxoma. Ann Oncol. 2016;27:2097–2103. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ovaska K, Laakso M, Haapa-Paananen S, Louhimo R, Chen P, Aittomäki V, et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010;2:65 10.1186/gm186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9. 10.1038/nbt.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–83. 10.1038/nrc1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. 10.1126/science.1075762 [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66. 10.1038/nprot.2015.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 23.Thomas RK, Baker AC, DeBiasi RM, Winckler W, Laframboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–51. 10.1038/ng1975 [DOI] [PubMed] [Google Scholar]
- 24.Untergrasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiel A, Moza M, Kytölä S, Orpana A, Jahkola T, Hernberg M, et al. Prospective immunohistochemical analysis of BRAF V600E mutation in melanoma. Hum Pathol. 2015;46:169–75. 10.1016/j.humpath.2014.08.018 [DOI] [PubMed] [Google Scholar]
- 26.Brodin J, Mild M, Hedskog C, Sherwood E, Leitner T, Andersson B, et al. PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS One. 2013;8:e70388 10.1371/journal.pone.0070388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong SQ, Li J, Tan AY-C, Vedururu R, Pang J-MB, Do H, et al. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics. 2014;7:23 10.1186/1755-8794-7-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2014;43:D805–11. 10.1093/nar/gku1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–9. 10.1093/nar/gku1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen Y, Luche R, Wei B, Gordon ML, Diltz CD, Tonks NK. Activation of the Jnk signaling pathway by a dual-specificity phosphatase, JSP-1. Proc Natl Acad Sci USA. 2001;98:13613–8. 10.1073/pnas.231499098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szych C, Staebler A, Connolly DC, Wu R, Cho KR, Ronnett BM. Molecular genetic evidence supporting the clonality and appendiceal origin of Pseudomyxoma peritonei in women. Am J Pathol. 1999;154:1849–55. 10.1016/S0002-9440(10)65442-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shetty S, Thomas P, Ramanan B, Sharma P, Govindarajan V, Loggie B. Kras mutations and p53 overexpression in pseudomyxoma peritonei: association with phenotype and prognosis. J Surg Res. 2013;180:97–103. 10.1016/j.jss.2012.10.053 [DOI] [PubMed] [Google Scholar]
- 33.Davison JM, Choudry HA, Pingpank JF, Ahrendt SA, Holtzman MP, Zureikat AH, et al. Clinicopathologic and molecular analysis of disseminated appendiceal mucinous neoplasms: identification of factors predicting survival and proposed criteria for a three-tiered assessment of tumor grade. Mod Pathol. 2014;27:1521–39. 10.1038/modpathol.2014.37 [DOI] [PubMed] [Google Scholar]
- 34.Nishikawa G, Sekine S, Ogawa R, Matsubara A, Mori T, Taniguchi H, et al. Frequent GNAS mutations in low-grade appendiceal mucinous neoplasms. Br J Cancer. 2013;108:951–8. 10.1038/bjc.2013.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74. 10.1038/nrc3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission Possible? Nat Rev Drug Discov. 2014;13:828–51. 10.1038/nrd4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Connell JT, Hacker CM, Barsky SH. MUC2 is a molecular marker for pseudomyxoma peritonei. Mod Pathol. 2002;15:958–72. 10.1097/01.MP.0000026617.52466.9F [DOI] [PubMed] [Google Scholar]
- 38.Amini A, Masoumi-Moghaddam S, Ehteda A, Morris DL. Secreted mucins in pseudomyxoma peritonei: pathophysiological significance and potential therapeutic prospects. Orphanet J Rare Dis. 2014;9:71 10.1186/1750-1172-9-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradbury NA. Protein kinase-A-mediated secretion of mucin from human colonic epithelial cells. J Cell Physiol. 2000;185:408–15. [DOI] [PubMed] [Google Scholar]
- 40.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaujoux S, Tissier F, Ragazzon B, Rebours V, Saloustros E, Perlemoine K, et al. Pancreatic ductal and acinar cell neoplasms in Carney complex: a possible new association. J Clin Endocrinol Metab. 2011;96:E1888–95. 10.1210/jc.2011-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson-White A, Hundley TR, Shiferaw M, Bertherat J, Sandrini F, Stratakis CA. Protein kinase-A activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinases ERK1/2. Hum Mol Genet. 2003;12:1475–84. [DOI] [PubMed] [Google Scholar]
- 43.Rhayem Y, Stunff CL, Khalek WA, Auzan C, Bertherat J, Linglart A, et al. Functional characterization of PRKAR1A mutations reveals a unique molecular mechanism causing acrodysostosis but multiple mechanisms causing Carney complex. J Biol Chem. 2015;290:27816 10.1074/jbc.M115.656553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ono M, Taniguchi N, Makita A, Fujita M, Sekiya C, Namiki M. Phosphorylation of beta-glucuronidases from human normal liver and hepatoma by cAMP-dependent protein kinase. J Biol Chem. 1988;263:5884–9. [PubMed] [Google Scholar]
- 45.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–98. 10.1056/NEJMoa055695 [DOI] [PubMed] [Google Scholar]
- 46.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trobridge P, Knoblaugh S, Washington MK, Munoz NM, Tsuchiya KD, Rojas A, et al. TGF-beta receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a beta-catenin-independent pathway. Gastroenterology. 2009;136:1680–8. 10.1053/j.gastro.2009.01.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saharinen J, Keski-Oja J. Specific sequence motif of 8-Cys repeats of TGF-β binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-β. Mol Biol Cell. 2000;11:2691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy-Ullrich JE, Schultz-Cherry S, Höök M. Transforming growth factor-beta complexes with thrombospondin. Mol Biol Cell. 1992;3:181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang SS, Esplin ED, Li JL, Huang L, Gazdar A, Minna J, et al. Alterations of the PPP2R1B gene in human lung and colon cancer. Science. 1998;282:284–7. [DOI] [PubMed] [Google Scholar]
- 51.Qiu W, Tang SM, Lee S, Turk AT, Sireci AN, Qiu A, et al. Loss of activin receptor type 1B accelerates development of intraductal papillary mucinous neoplasms in mice with activated KRAS. Gastroenterology. 2016;150:218–28. 10.1053/j.gastro.2015.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–98. 10.1038/nrg3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA. 2010;107:961–8. 10.1073/pnas.0912629107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yost SE, Smith EN, Schwab RB, Bao L, Jung H, Wang X, et al. Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens. Nucleic Acids Res. 2012;40:e107 10.1093/nar/gks299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fassunke J, Haller F, Hebele S, Moskalev EA, Penzel R, Pfarr N, et al. Utility of different massive parallel sequencing platforms for mutation profiling in clinical samples and identification of pitfalls using FFPE tissue. Int J Mol Med. 2015;36:1233–43. 10.3892/ijmm.2015.2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(DOCX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. The exome sequencing data is available from the European Genome-phenome Archive database (accession number EGAS00001002418).