

Summary
Type 2 diabetes (T2D) is a world-wide epidemic with a medical need for additional targeted therapies. Suppression of hepatic glucose production (HGP) effectively ameliorates diabetes and can be exploited for its treatment. We hypothesized that targeting PGC-1α acetylation in liver, a chemical modification known to inhibit hepatic gluconeogenesis, could be potentially used for treatment of T2D. Thus, we designed a high-throughput chemical screen platform to quantify PGC-1α acetylation in cells and identified small molecules that increase PGC-1α acetylation, suppress gluconeogenic gene expression and reduce glucose production in hepatocytes. Based on the potency and bioavailability, we selected a small molecule, SR-18292, that reduces blood glucose, strongly increases hepatic insulin sensitivity and improves glucose homeostasis in dietary and genetic mouse models of T2D. These studies have important implications for understanding the regulatory mechanisms of glucose metabolism and treatment of T2D.
Graphical abstract
In brief - A small molecule targeting gluconeogenesis improves glucose homeostasis in animals with type 2 diabetes, suggesting a new therapeutic approach for this metabolic disease.
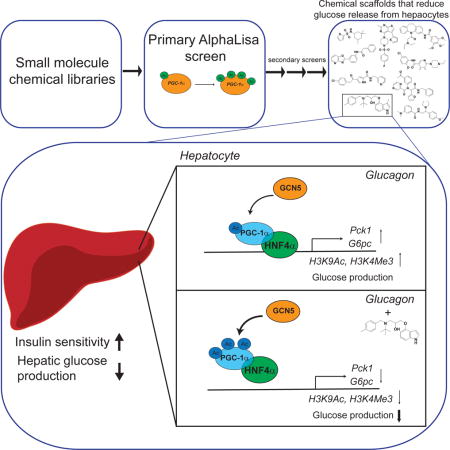
Introduction
T2D has become a worldwide epidemic that affected over 400 million people in 2014 according to the World Health Organization. Uncontrolled increased blood glucose concentration is a hallmark of T2D which can lead to long term complications including micro-and macro-cardiovascular complications, neuropathy, kidney failure and increased risk for developing cancer (Forbes and Cooper, 2013; Richardson and Pollack, 2005). Maintaining blood glucose within a normal range is the foremost objective in the treatment of T2D, as this dramatically reduces the risk of developing diabetes-associated complications (Holman et al., 2008; Kahn et al., 2014). Several drugs are currently being used for treatment of T2D in the clinic; these include primarily metformin, the first line drug used to treat T2D, but also sulfonylureas, thiazolidinediones (TZDs), incretin mimetics and sodium-glucose cotransporter 2 (SGLT2) inhibitors (Bailey, 2013; Kahn et al., 2014). As most patients develop resistance to metformin treatment over time, a strategy of combination therapy that includes treatment with an additional drug or insulin administration is commonly employed (Bailey, 2013). This highlights the need for developing additional drugs to reduce hyperglycemia that can be used either as a monotherapy or as part of a combination therapy.
Whole body glucose homeostasis is achieved through an intricate balance between glucose production, mostly by the liver, and glucose uptake by peripheral tissues (DeFronzo, 2004; Moore et al., 2012). This is regulated primarily through the opposing pancreatic hormones insulin and glucagon. In diabetic states, the liver becomes resistant to the action of insulin and produces an elevated amount of glucose which is a major contributor to the increased blood glucose levels observed in T2D (Rizza, 2010). Hence, reducing HGP is a feasible strategy to manage blood glucose levels in T2D. Importantly, increased HGP in T2D was shown to be primarily a result of dysregulated gluconeogenesis rather than glycogen breakdown (Magnusson et al., 1992), suggesting that targeting components within the gluconeogenic pathway could improve hyperglycemia. Indeed, metformin is believed to exert its effect mainly by reducing hepatic gluconeogenesis (Hundal et al., 2000; Madiraju et al., 2014).
The transcriptional coactivator PGC-1α plays a pivotal role in energy homeostasis by co-activating transcription factors that regulate fat and glucose metabolism (Finck and Kelly, 2006). As part of the fasting response, PGC-1α regulates gluconeogenesis in the liver by binding to the nuclear hormone receptors HNF4α and FoxO1 to control expression of key enzymes in the gluconeogenic pathway (Puigserver et al., 2003; Rhee et al., 2003). PGC-1α is upregulated in several models of diabetes where gluconeogenesis is elevated, highlighting its importance in controlling blood glucose levels (Yoon et al., 2001). While PGC-1α whole body knockout mice are able to maintain normal blood glucose levels by activating PGC-1α-independent pathways (Lin et al., 2004), a partial reduction in PGC-1α levels in the liver, using heterozygous knockout mice, leads to modest but significant defects in gluconeogenesis (Estall et al., 2009). Moreover, isolated livers from PGC-1α−/− mice produce less glucose compared to control mice due to reduced gluconeogenic flux from phosphoenolpyruvate (Burgess et al., 2006), indicating that selective gluconeogenic inhibition of PGC-1α in the liver can potentially result in reduced HGP and ameliorate diabetes.
Post-translational modifications (PTMs) of PGC-1α control its transcriptional activity and metabolic function (Fernandez-Marcos and Auwerx, 2011). Specifically, acetylation of multiple lysines on PGC-1α determines its ability to co-activate transcription factors involved in HGP. As part of the fasting response, SIRT1 deacetylates PGC-1α, resulting in enhancement of its activity and increased glucose production (Rodgers et al., 2005). The acetyl transferase GCN5, on the other hand, catalyzes acetylation of PGC-1α, reducing expression of gluconeogenic genes and glucose output (Dominy et al., 2012; Lerin et al., 2006). In addition, as part of the response to feeding and insulin, the Cyclin D1/Cdk4 complex phosphorylates GCN5 to induce its activity and inhibit gluconeogenic gene expression (Lee et al., 2014). Another node of the insulin response that regulates the activity of PGC-1α during the re-feeding response involves Akt, S6K and Clk2, which phosphorylate PGC-1α on its SR domain to inhibit its activity and reduce HGP (Li et al., 2007; Lustig et al., 2011; Rodgers et al., 2010). Hence, specific manipulation of PGC-1α PTMs can be a potential way to control its gluconeogenic activity to improve hyperglycemia.
Here, we designed and developed a cell-based high-throughput chemical screen using an AlphaLisa assay aimed at identifying chemical scaffolds that induce PGC-1α lysine acetylation. Subsequent secondary assays identified a subset of previously uncharacterized small molecules that were able to reduce glucose production in primary hepatocytes. As a proof of concept of the potential use of these compounds as anti-diabetic drugs, a single hit from our screen reduced fasting blood glucose, significantly increased hepatic insulin sensitivity and improved glucose homeostasis ameliorating diabetes in dietary and genetic mouse models.
Results
An AlphaLisa cell-based high-throughput chemical screening platform identifies small molecules that increase PGC-1α acetylation and suppress HGP
To identify novel anti-diabetic chemical probes that increase PGC-1α acetylation and subsequently inhibit its gluconeogenic activity, we designed a cell-based high-throughput chemical screening platform. We screened the MLP (NIH Molecular Library Probes) and DOS (Diversity-Oriented Synthesis) chemical libraries, totaling ~350,000 compounds, using the U-2 OS cell line transiently co-expressing PGC-1α and GCN5. We developed an AlphaLisa assay (Yasgar et al., 2016) to quantify changes in PGC-1α acetylation upon single dose treatment with each compound from these libraries (Fig 1A). Using this platform, we could reliably measure changes in PGC-1α acetylation and identified 741 active compounds which increased the lysine acetylation signal over 50% compared to vehicle control (Fig 1B). These compounds were considered positive hits from the primary screen. To further winnow our hit list, we re-tested these compounds at dose using the same assay. Based on the potency to increase PGC-1α acetylation (AC50 of <10μM) (Fig. 1C, Fig. S1A and S1B), 381 compounds were selected. Next, a cytotoxicity assay was applied to triage compounds that were toxic to HEK293, HepG2 and A549 cells. As a result, 252 non-toxic compounds were chosen for secondary screening assays.
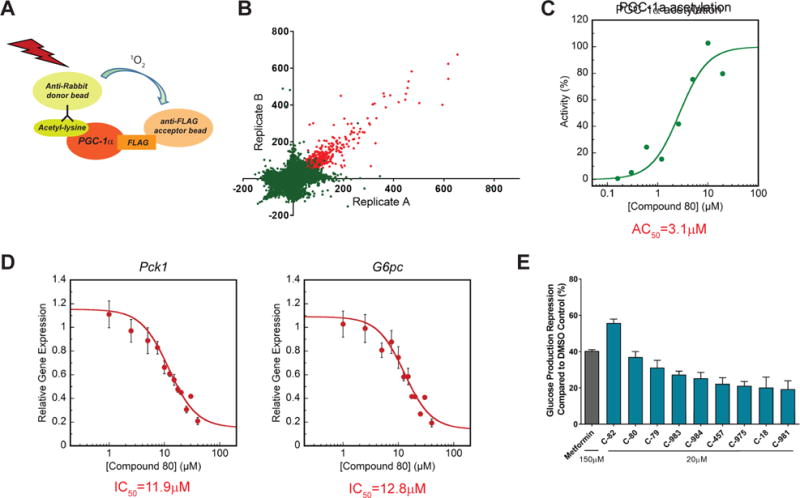
Figure 1. AlphaLisa based high-throughput screen identifies small molecules with inhibitory effect on glucose production in isolated hepatocytes.
See also Fig. S1.
(A) AlphaLisa assay was used to detect acetylation status of PGC-1α in lysates obtained from U-2 OS cells over expressing PGC-1α and GCN5.
(B) Scatter plot of primary screen results. A total of 741 compounds that induced acetylation signal by ≥50% compared to control were identified and considered active (red dots). Axes represent activity score (percentage change in acetylation relative to vehicle control).
(C) The effect of the primary screen hits on PGC-1α acetylation was re-tested in a dose response manner. Dose response curve of lead compound (C-80) is presented as an example.
(D) Isolated mouse primary hepatocytes were used to determine the effect of remaining hits on Pck1 and G6pc gene expression (see also figure S1C). Forskolin was used to induce Pck1 and G6pc expression (considered as 100% activation) and cells were treated with each compound at dose. Data was fit to the Hill equation to calculate estimated AC50. A more accurate dose response curve of lead compound (C-80) was re-generated in-house. For further details, please refer to STAR methods.
(E) Repression of glucose production from hepatocytes following treatment with screen hits. Hepatocytes were infected with Ad-PGC-1α and Ad-GCN5 to induce glucose production. Cells were treated overnight with an overall of 19 compounds, that were identified in the qPCR assay. Pyruvate/lactate were used as substrates for glucose production. Compounds that significantly reduced glucose levels in the medium compared to DMSO control are presented.
PGC-1α activates hepatic gluconeogenic gene expression, and an increase in its acetylation status reduces expression of both Pck1 and G6pc genes (Rodgers et al., 2005; Yoon et al., 2001). We predicted that a set of the selected 252 positive small molecule hits would suppress gluconeogenic gene expression in primary hepatocytes. Thus, we performed a high-throughput screen with these 252 compounds at different doses. Hepatocytes were treated with forskolin for 1.5 hours to induce the expression of both Pck1 and G6pc genes, which mimics the fasting and PGC-1α-mediated gluconeogenic response. The dose response curves were used to calculate an inhibitory activity of the compounds by determining the difference in Ct values between compound treatment and forskolin control at each dose, and provided an estimated potency of the compounds in suppressing Pck1 and G6pc (Fig. 1D, Fig. S1C). As a result, 42 compounds were selected with an estimated inhibitory AC50 of <10μM for either Pck1 or G6pc. These compounds are inhibitors of forskolin-induced gluconeogenic gene expression and predicted to reduce glucose production. To specifically test the inhibitory activity of the remaining small molecule positive hits on PGC-1α-mediated glucose production, we used adenoviruses to ectopically express PGC-1α in primary hepatocytes. Similar to forskolin, PGC-1α overexpression induces gluconeogenic gene expression and promotes glucose release (Herzig et al., 2001; Yoon et al., 2001). Primary hepatocytes were treated with the chosen positive hit compounds that could suppress gluconeogenic gene expression, and the concentration of glucose released to the culture medium was measured. In this small assay screen, 9 compounds significantly suppressed glucose release compared to control (Fig. 1E and Fig. S1D). Interestingly, metformin is only able to suppress glucose production at a higher concentration (100–250μM) (He and Wondisford, 2015), compared to our identified compound collection (20μM), highlighting the potency of these chemical scaffolds to inhibit glucose output. Taken together, using a high-throughput chemical screen platform, we have identified chemical scaffolds that induce acetylation of PGC-1α, suppress expression of gluconeogenic genes and reduce glucose production. Due to their ability to inhibit glucose release from hepatocytes, these compounds can potentially be used as anti-diabetic drugs to reduce blood glucose through suppression of HGP. These scaffolds provide chemical tools that can be used, not only to develop new anti-diabetic drugs, but also to identify molecular and metabolic pathways that regulate HGP.
SR-18292 suppresses hepatic gluconeogenic gene expression and glucose production in primary hepatocytes
To test anti-diabetic effects of the positive hits that scored in all the assays described above, we selected the most potent compounds that decreased glucose production. Compound 82 (C-82) was the top small molecule hit suppressing glucose production (Fig. 1E). However, C-82 showed poor bioavailability and solubility (data not shown) and would require extensive structure-activity relationship (SAR) studies to test this compound in vivo. We therefore selected the second most potent small molecule, compound 80 (C-80), for further validation and characterization. C-80 is part of the NIH Molecular Libraries Compound Collection and has been screened against 545 bioassays that represent diverse targets and a range of readouts. C-80 was found active in only 14 bioassays (2.5%), two of these bioassays are part of our high-throughput screens, highlighting the lack of promiscuity of C-80. Moreover, C-80 showed the highest potency in our confirmatory screen (https://pubchem.ncbi.nlm.nih.gov/assay/bioactivity.html?cid=24747703).
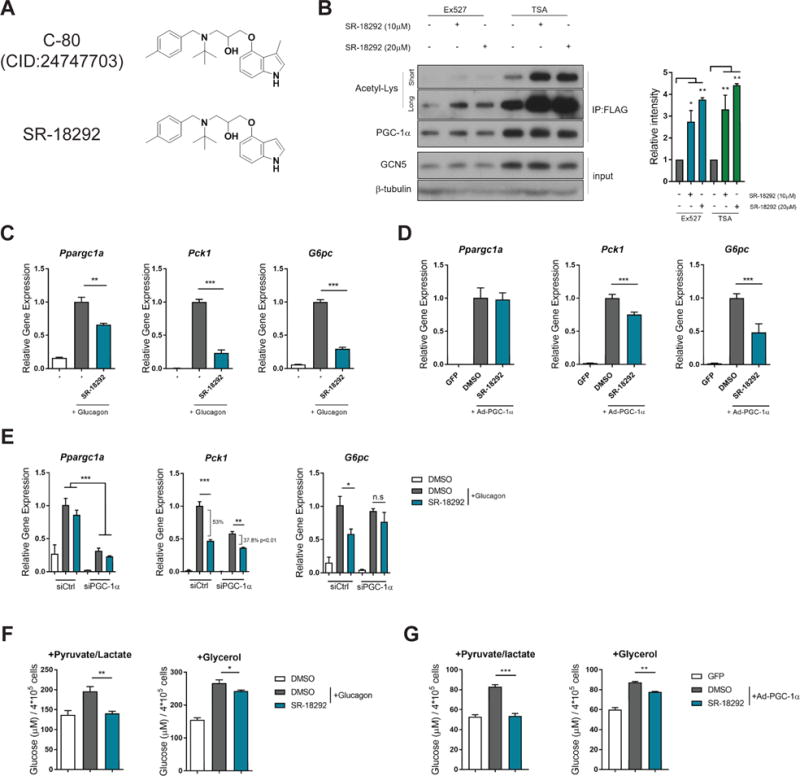
Due to limited commercial availability, we synthesized an analog of C-80 that we named SR-18292. This analog is identical to C-80, except the methyl group on the 3′ position on the indole group that is lacking in SR-18292 (Fig. 2A). To confirm that the effects of SR-18292 were similar to C-80, primary hepatocytes were treated with SR-18292, which increased PGC-1α acetylation (Fig. 2B). The acetylation status of PGC-1α is determined by its dynamic interaction with deacetylases and acetyl transferases, primarily SIRT1 and GCN5 (Lerin et al., 2006; Rodgers et al., 2005). Interestingly, SR-18292 increased acetylation of PGC-1α even in the presence of Ex-527, a selective SIRT1 inhibitor, and Trichostatin A (TSA), a pan-histone deacetylase (HDAC) inhibitor. This result suggests that the increased acetylation of PGC-1α, induced by SR-18292, is mediated by a mechanism that does not involve inhibition of either SIRT1 or other HDACs.
Figure 2. Effects of SR-18292 on PGC-1.
α acetylation and gluconeogenic genes in isolated primary hepatocytes. See also Fig. S2.
(A) SR-18292 is identical to C-80 except the methyl group on the 3′ position on the indole group.
(B) Western blot analysis of PGC-1α acetylation following treatment with SR-18292 in the presence of the SIRT1 inhibitor, Ex527 (5μM) and the pan HDACs inhibitor TSA (1μM). Hepatocytes were infected with Ad-PGC-1α and Ad-GCN5 and treated with SR-18292 for 18 h. IP, immunoprecipitation.
(C) – (D) qPCR analysis of Ppargc1a, Pck1 and G6pc mRNA expression levels following treatment with SR-18292 (20μM) in isolated primary hepatocytes.
(E) qPCR analysis of Ppargc1a, Pck1 and G6pc mRNA expression levels following SR-18292 (20μM) treatment and siRNA to reduce to expression of Ppargc1a. Difference in % repression of Pck1 was calculated using two-way ANOVA with Sidak posttest.
(F) – (G) Glucose production by primary hepatocytes following treatment with SR-18292 (20μM). Hepatocytes were treated with either glucagon (200nM) or infected with Ad-PGC-1α to induce glucose production.
All data presented as mean +/− S.E.M. n=3, one-way ANOVA with Tuckey posttest. Representative of at least 2 independent experiments. *P<0.05, **P<0.01, ***P<0.001. n.s=not significant
Upon fasting, increased plasma glucagon levels induce gluconeogenic genes in the liver, and PGC-1α is a component of this response (Altarejos and Montminy, 2011; Herzig et al., 2001; Rodgers et al., 2008). In agreement with SR-18292 inhibiting PGC-1α gluconeogenic function, treatment of hepatocytes with SR-18292 significantly reduced the ability of glucagon to stimulate Pck1 and G6pc gene expression (Fig. 2C). This was achieved without disrupting the canonical glucagon-induced increases in phosphorylation of CREB and dephosphorylation of CRTC2 (Fig. S2A). In addition, cell autonomous enhancement of insulin signaling was not observed (Fig. S2B), suggesting that the effect of SR-18292 on gene expression at the cellular level is probably downstream of the signaling cascade that results in activation of CREB and Akt. Importantly, SR-18292 did not cause hepatocyte toxicity as measured by cell viability or total protein levels (Fig. S2C–E).
As a major regulator of Pck1 and G6pc transcription, overexpression of PGC-1α in hepatocytes is sufficient to induce expression of these genes (Herzig et al., 2001; Yoon et al., 2001). Similar to its effects with glucagon, SR-18292 significantly reduced gluconeogenic gene and protein expression induced by ectopic expression of PGC-1α (Fig. 2D Fig. S2F). Since increased gluconeogenic gene expression is driven solely by PGC-1α in these experiments, the suppression of these genes suggests a direct effect of SR-18292 on the PGC-1α transcriptional complex that inhibits expression of Pck1 and G6pc genes. Reducing the expression of PGC-1α by siRNA significantly impairs the ability of SR-18292 to suppress Pck1 and G6pc, suggesting that its effect is at least partially mediated by PGC-1α (Fig. 2E). Consistent with the effects on suppression of gluconeogenic genes, SR-18292 significantly reduced glucagon- and PGC-1α-mediated glucose production in isolated hepatocytes (Fig. 2F and G). Interestingly, SR-18292 decreases glucose production when pyruvate and lactate are used as substrates for gluconeogenesis as well as when glycerol is used as a substrate (Fig. 2F and G). The reduction in glucose production correlated with suppression of Pck1 and G6pc since short treatment with SR-18292 (3 hr) had no effect on gene expression or glucose release (Fig. S2G and H). Together, these results show that SR-18292 is a potent inhibitor of the gluconeogenic gene expression and glucose production in hepatocytes.
SR-18292 increases the interaction of PGC-1α with GCN5 and reduces co-activation of HNF4α by PGC-1α
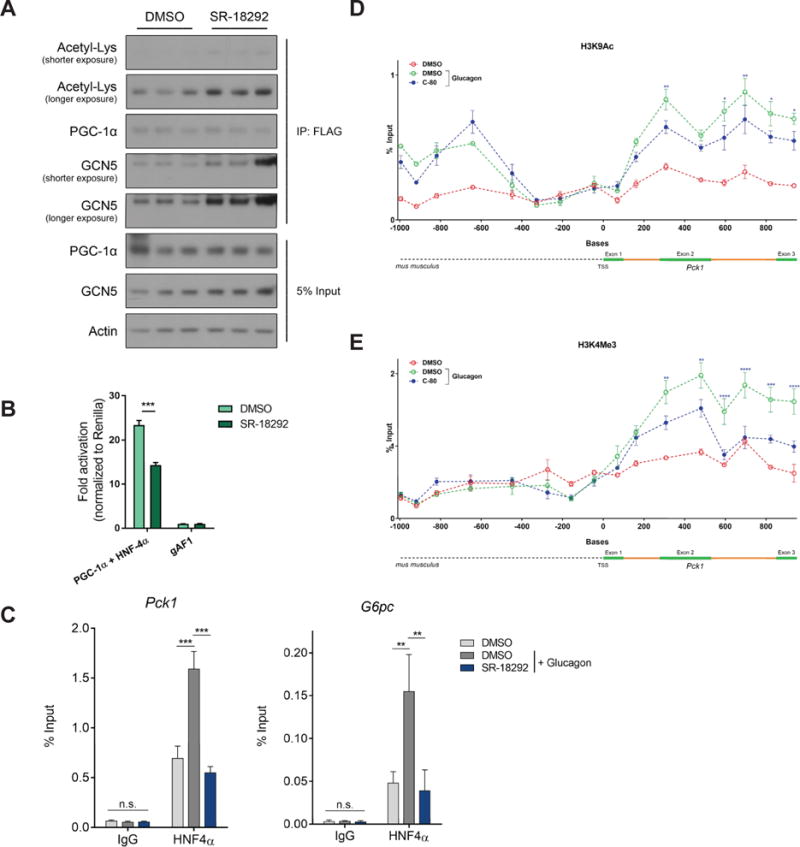
The PGC-1α acetylation data from primary hepatocytes (Fig. 2B) implies that SR-18292 induces acetylation by a mechanism that does not involve inhibition of HDACs. We therefore tested the possibility that the SR-18292-mediated increase in PGC-1α acetylation is modulated by GCN5, an acetyl transferase known to acetylate PGC-1α (Lerin et al., 2006). First, we tested if the catalytic activity of GCN5 is increased as a result of SR-18292 treatment. To this end, GCN5 was overexpressed and subsequently immunoprecipitated (IP) from U-2 OS cells treated with SR-18292. We found that the catalytic activity was not changed compared to GCN5 IPed from control cells (Fig. S3A). In addition, SR-18292 does not directly activate GCN5 since in vitro treatment of IPed GCN5 did not result in increased activity (Fig. S3B). We then speculated that while the catalytic activity of GCN5 is not changed by SR-18292, its physical interaction with PGC-1α might be modulated by this treatment. Ectopic expression and co-IP of PGC-1α from U-2 OS cells revealed that the interaction of endogenous GCN5, as well as ectopically expressed GCN5, with PGC-1α was increased in cells that were treated with SR-18292 (Fig. 3A and S3C). To identify the PGC-1α lysine residues that are affected by treatment with SR-18292, we performed mass spectrometry analysis and identified 7 lysine residues whose acetylation was increased (Fig. S3D). Two of these sites (K277 and K346) were previously described, and the increased acetylation of these sites is associated with reduced gluconeogenic activity of PGC-1α (Rodgers et al., 2005). Together, these results suggest that by increasing the interaction of GCN5 with PGC-1α, SR-18292 increases the acetylation of specific PGC-1α lysine residues that might subsequently decrease its gluconeogenic activity.
Figure 3. SR-18292 increases interaction of GCN5 with PGC-1 α and reduces transcriptional activity of HNF4α.
See also Fig. S3.
(A) Co-IP of FLAG-PGC-1α following treatment with SR-18292 (10μM). IP, immunoprecipitation.
(B) U-2 OS cells were transfected with the indicated plasmids and luciferase reporter levels were measured after 24 hours. n=3, two-way ANOVA with Sidak posttest. Representative of at least 2 independent experiments.
(C) The Pck1 and G6pc promoters’ occupancy by HNF4α was determined following chromatin IP (ChIP) using HNF4α specific antibody. Glucagon (200nM) was used to promote HNF4α binding to these promoter regions. n=3, one-way ANOVA with Tuckey posttest. Representative of at least 2 independent experiments.
(D) and (E) H3K9Ac and H3K4me3 marks on Pck1 gene are reduced upon treatment. n=3, two-way ANOVA with Tuckey posttest.
*P<0.05, **P<0.01, ***P<0.001.
We previously shown that by interacting with PGC-1α, the GCN5 acetyl transferase complex decreases PGC-1α gluconeogenic transcriptional activation (Lerin et al., 2006). This is achieved at least in part by reducing the ability of PGC-1α to co-activate the nuclear hormone receptor HNF4α. Co-activation of HNF4α by PGC-1α is required to promote transcription of the gluconeogenic genes Pck1 and G6pc (Rhee et al., 2003). To test whether SR-18292 reduces the transcriptional activity of HNF4α that is induced by PGC-1α, we used a luciferase reporter construct driven by a fragment of the Pck1 promoter containing an HNF4α binding site (Sugiyama et al., 2000). In agreement with the enhanced interaction between PGC-1α and GCN5, the transcriptional activity of HNF4α was significantly reduced in cells treated with SR-18292 (Fig. 3B). To further characterize the SR-18292-mediated inhibition of HNF4α transcriptional activity, we performed chromatin immunoprecipitation (ChIP) assays to examine the promoter occupancy of HNF4α on Pck1 and G6pc promoters. Primary hepatocytes were treated with glucagon to induce gluconeogenic gene transcripts, and antibodies against HNF4α were used to IP chromatin fragments bound to this protein. In accordance with the inhibitory effect of SR-18292 on Pck1 and G6pc expression, the glucagon-induced Pck1 and G6pc promoter occupancy by HNF4α was significantly reduced in cells treated with SR-18292 (Fig. 3C). The FoxO1 promoter occupancy was not altered (Fig. S3E), suggesting that this effect is specific to HNF4α. Epigenetic changes accompany the response to glucagon in the fasted state, including an increase in activating H3K9 acetylation (H3K9Ac) and H3K4 trimethylation (H3K4Me3) of the Pck1 and G6pc genes (Ravnskjaer et al., 2013). Interestingly, compound-treated hepatocytes had reduced H3K9Ac and H3K4Me3 marks on the Pck1 gene (Fig. 3D and E ), further supporting our findings that upon treatment, transcription of this gene is less active. These results suggest that by modulating the interaction between GCN5 and PGC-1α, SR-18292 inhibits the gluconeogenic activity of PGC-1α and reduces co-activation of HNF4α. In addition, SR-18292 treatment decreases occupancy of HNF4α on Pck1 and G6pc promoters, leading to reduced expression of gluconeogenic genes.
Identification of SR-18292 small molecule analogs lacking gluconeogenic inhibitory effects
To gain further insights into the mode of action of SR-18292, we performed SAR studies to search for chemical modifications that impair its activity, as this might reveal the active site of the compound. We generated a panel of chemical analogs and tested their ability to suppress gluconeogenic gene expression induced by either glucagon or PGC-1α overexpression. Interestingly, we found that replacement of the indole group with phenyl (Fig. S4A, SR-19138) reduced the inhibitory effect of the new scaffold on glucagon or PGC-1α-increased Pck1 and G6pc expression (Fig. S4B and C). Other analogs of the compound had little or no effect on gluconeogenic activity (Fig. S4B and C). This suggests that hydrogen bond interaction in this part of the molecule might be important for activity. The reduced effect of the new analog on suppression of Pck1 and G6pc correlated with a reduced ability to induce acetylation of PGC-1α compared to the parent compound, SR-18292 (Fig. S4D). These results indicate that the indole group is involved in the bioactivity of SR-18292.
SR-18292 reduces fasting blood glucose, increases hepatic insulin sensitivity and improves glucose homeostasis in diabetic mice
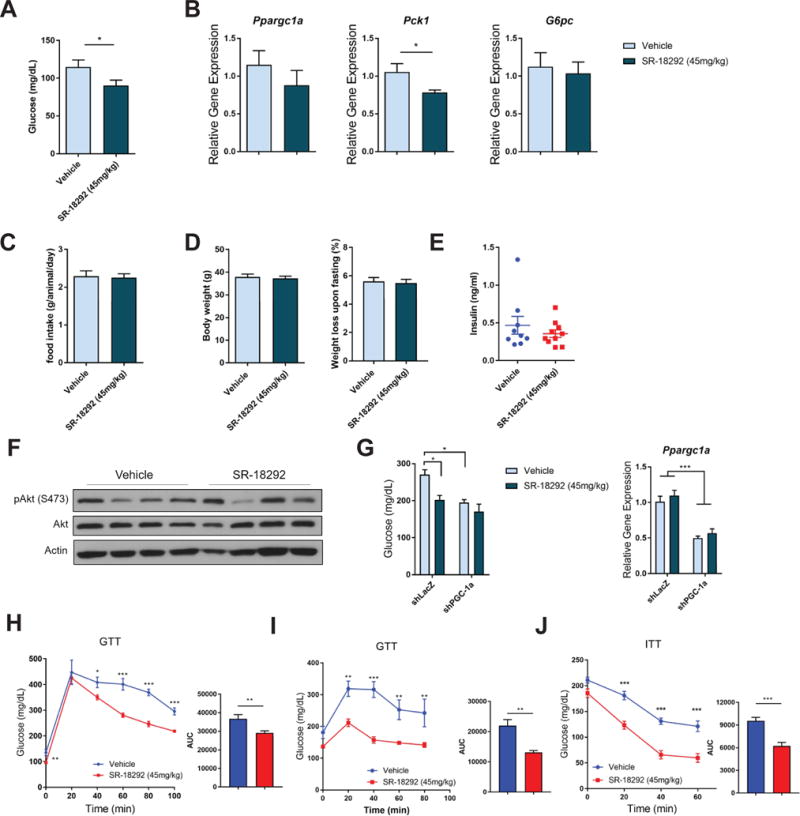
Based on the potent gluconeogenic suppressor effects of SR-18292 in primary hepatocytes, we tested its potential use as an anti-diabetic drug. In the fasted state, the liver contributes to regulating blood glucose levels through the release of glucose into the circulation (Cherrington, 1999; Moore et al., 2012). Therefore, we focused on measuring blood glucose in the fasted state during which the liver plays a major role maintaining glycemia. Preliminary pharmacokinetics (PK) studies with the original C-80 revealed that the compound concentration in the liver, 2 h following a single dose (30 mg/kg) administration via intraperitoneal (I.P.) injection, was 2.3μM. We projected that multiple injections of a higher dose might be sufficient to reach active concentration in the liver. We used high fat diet (HFD) fed mice, a dietary model of obesity and T2D, which were treated with SR-18292 (45mg/kg) via I.P. injection for 3 consecutive days and again on day 4 before measuring fasting blood glucose. Strikingly, mice that were treated with SR-18292 had significantly lower levels of fasting blood glucose concentrations compared to matched vehicle-treated control mice (Fig. 4A). The induction of gluconeogenic gene expression is a regulatory component of the response to fasting. Importantly, gluconeogenic gene expression, specifically that of Pck1, is inhibited in livers isolated from mice treated with SR-18292 (Fig. 4B), suggesting that SR-18292 targets similar hepatic glucose metabolic pathways both in vitro and in vivo.
Figure 4. SR-18292 reduces fasting blood glucose and improves overall glucose homeostasis.
See also Fig. S5.
(A) Fasting blood glucose levels of mice fed HFD for 13 weeks. n=10, two-tailed student’s t-test.
(B) qPCR analysis of Ppargc1a, Pck1 and G6pc from liver tissue isolated from fasted mice treated with SR-18292. n=10, two-tailed student’s t-test.
(C) Food intake was measured during the first 48 hours of treatment with SR-18292. Pulled data from 4 different experiments is presented. In each experiment n=7–9.
(D) Body weight of mice following treatment with SR-18292. n=10. Weight loss data upon overnight fasting is pulled from 3 different experiments.
(E) Fasting insulin levels of mice fed HFD for 13 weeks and treated with SR-18292.
(F) Western blot analysis of pAkt levels in liver tissues from fasted mice treated with SR-18292.
(G) Lep°b/°b mice were administered adenoviruses expressing shLacZ or shPGC-1α via tail vein injection. Blood glucose was measured on day 8 post infection. Livers were collected to verify knockdown efficiency.
(H) GTT in mice fed HFD for 14 weeks. n=7/vehicle, n=8/SR-18292, Two-way ANOVA with Sidak posttest.
(I) GTT in Lep°b/°b mice. 9 weeks old mice were used. n=6, Two-way ANOVA with Sidak posttest.
(J) ITT in mice fed HFD for 3 weeks. n=6, Two-way ANOVA with Sidak posttest.
*P<0.05, **P<0.01, ***P<0.001. AUC, area under the curve.
The reduction in fasting blood glucose concentration is not likely to be a result of reduced calorie consumption as neither food intake nor body weight were changed as a result of treatment with SR-18292 (Fig. 4C and D). In addition, liver toxicity, as measured by serum levels of alanine transaminase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH) and bilirubin, was not observed in mice treated with SR-18292 (Table S1). Basal blood insulin levels, while trending lower, were not significantly changed as a consequence of SR-18292 treatment as indicated by similar fasting blood insulin levels in both the SR-18292-treated and the vehicle control groups (Fig. 4E). In addition, phosphorylation of Akt was also not changed (Fig. 4F), suggesting that SR-18292 does not exert its blood glucose-lowering effects by affecting insulin secretion or canonical insulin signaling. To assess the contribution of PGC-1α in mediating the in vivo effects of SR-18292, we depleted hepatic PGC-1α in Lep°b/°b mice using shRNA and observed a significant reduction in basal blood glucose concentrations compared to control. Importantly, reduced levels of PGC-1α in the liver blunted SR-18292-mediated reductions in blood glucose concentrations, indicating that PGC-1α contributes to the SR-18292 metabolic action (Fig. 4G). To fully assess the effects of SR-18292 on whole body glucose homeostasis and insulin resistance, we performed a glucose tolerance test (GTT) and insulin tolerance test (ITT) following the same protocol used to determine fasting blood glucose. Interestingly, SR-18292 treatment significantly enhanced glucose tolerance both in DIO mice and Lep°b/°b mice (Fig. 4H and I). As expected, insulin levels after GTT were lower in the treated group (Fig. S5A). In addition, the response to insulin was also enhanced in the drug treated group (Fig. 4J), suggesting an improved response to insulin at the whole body level. To test if SR-18292 chronic treatment will cause similar anti-diabetic effects, we treated mice for a period of 14 days. The ability of SR-18292 to reduce blood glucose was sustained during this period without resulting in changes in body weight (Fig S5G and H). Importantly, this longer treatment did not cause any signs of toxicity as measured by serum ALT and AST levels, H&E staining of liver slices and the expression of proinflammatory genes (Fig. S5I–K).
SR-18292 suppresses HGP and increases liver insulin sensitivity in vivo
The strong effects of SR-18292 in primary hepatocytes prompted us to specifically test the ability of the liver to produce glucose upon treatment with SR-18292. To this end, we performed a pyruvate tolerance test (PTT), in which pyruvate is injected to mice to increase HGP. In accordance with the effects in isolated hepatocytes, glucose levels after pyruvate injection were significantly reduced in the SR-18292 treated group, suggesting that HGP from pyruvate is impaired as a result of the compound treatment (Fig. 5A and S5B). Interestingly, the defects in glucose production were comparable to that mediated by metformin treatment. To further analyze the cause of changes in blood glucose we carried out hyperinsulinemic-euglycemic clamp studies in HFD-fed mice treated with SR-18292. The glucose infusion rate in the SR-18292-treated mice was substantially higher compared to vehicle-treated mice (Fig. 5B), reflecting the improved insulin sensitivity. Importantly, this was not due to improved peripheral glucose uptake (Fig. 5C) but rather by reduced endogenous glucose production both under basal and clamp conditions (Fig. 5D), further corroborating that SR-18292 improves hyperglycemia via reduction in HGP and strongly increases selective hepatic insulin sensitivity. Inhibition of HGP can result in a side effect in which carbons are diverted toward lipid synthesis, leading ultimately to accumulation of triglycerides (TG) and cholesterol in the liver causing hepatic steatosis. The treatment with SR-18292 did not cause this side effect, as liver TG and cholesterol levels were not changed even after prolonged treatment (Fig. 5E and S5L). Another concern associated with inhibition of gluconeogenesis is accumulation of lactate or ketone bodies in the blood that can lead to lactic- or keto-acidosis, respectively, a major concern in diabetes management. Treatment with SR-18292 is not likely to induce such an effect since both serum lactate and ketone bodies were not changed (Fig. S5C and M). The transcriptional lipid metabolic profile (Fig. S5D), serum levels of TG, free fatty acids and cholesterol (Fig. S5E) were not significantly altered with SR-18292 treatment. In addition, the expression level of mitochondrial genes, also targeted by PGC-1α, was not changed, further showing the selectivity of SR-18292 toward inhibition of gluconeogenesis (Fig. S5F). Collectively, these data define SR-18292 as a chemical scaffold that significantly improves overall glucose homeostasis and support the potential use of this compound as a drug treatment to control blood glucose levels in diabetic states. It also shows that selective chemical inhibition of PGC-1α gluconeogenic activity in hepatocytes is a feasible strategy to reduce fasting blood glucose and improve hyperglycemia in pre-clinical mouse models of T2D.
Figure 5. SR-18292 suppresses HGP in vivo.
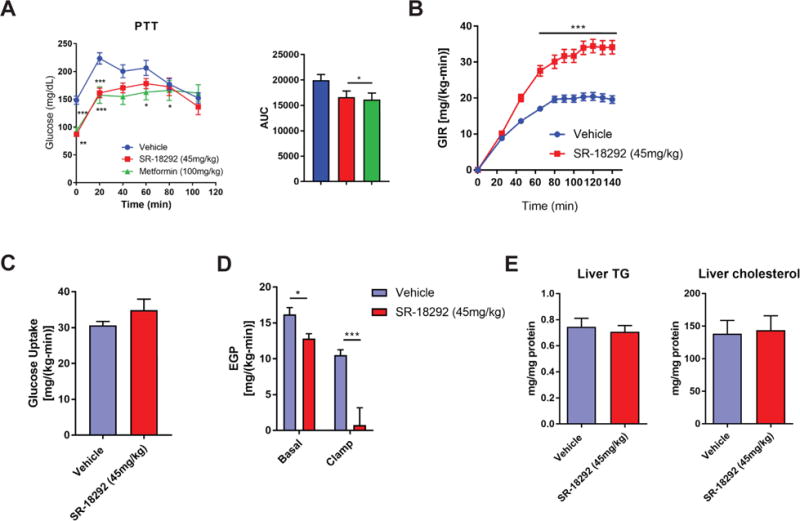
(A) PTT following treatment with SR-18292 and metformin in mice fed HFD for 17 weeks. n=10/vehicle, n=10/SR-18292, n=7/Metformin. Two-way ANOVA with Sidak posttest.
(B) – (D) hyperinsulinemic euglycemic clamp studies from mice fed HFD for 16 weeks. Glucose infusion rate (GIR), glucose uptake and endogenous glucose production (EGP) are presented. n=9/vehicle, n=7/SR-18292.
(E) Liver triglycerides and cholesterol levels from livers isolated from mice fed HFD for 17 weeks and treated with SR-18292. n=10.
*P<0.05, **P<0.01, ***P<0.001. AUC, area under the curve.
Discussion
The dramatic increase in the prevalence of T2D as part of the metabolic syndrome is believed to be primarily a result of modern life style, which creates an imbalance between energy expenditure and energy consumption. There are currently several drug classes that are mainly being used in the clinic to treat T2D. The first class is the biguanides, which includes metformin, and is the most widely used class and considered the first choice as a mean to reduce HbA1c index. Other classes include primarily the sulfonylureas, GLP-1 receptor agonists and DPP-4 inhibitors, which ultimately stimulate insulin secretion, and the TZDs, which increase insulin sensitivity by activation of the nuclear receptor PPARγ. When desired HbA1c index is not achieved using metformin as a monotherapy, a two-drug combination, that includes metformin and an additional drug, is the recommended strategy (Bailey, 2013). Administration of insulin is also considered as an approach to reduce HbA1c levels, but the risk of developing hypoglycemia is higher compared to other treatments. Despite the low cost and high efficiency of metformin treatment, the common need for a two-drug combination approach highlights the importance of developing new drug classes that could be used in the clinic either as a monotherapy or as part of a combination approach. HGP can be suppressed by targeting PGC-1α leading to reduced hyperglycemia (Herzig et al., 2001; Lee et al., 2014; Lerin et al., 2006; Yoon et al., 2001). These studies suggest that drugs that selectively inhibit hepatic PGC-1α gluconeogenic function might constitute a new class of anti-diabetic drugs. Here, we describe the identification of such a class of compounds that reduce glucose release from isolated hepatocytes and select one of these small molecules hits to show that it ameliorates diabetes.
Based on the fact that increased PGC-1α acetylation reduces gluconeogenic activity (Rodgers et al., 2005), we designed and developed a cell-based high-throughput chemical screening platform using PGC-1α acetylation as a read out. Hepatic PGC-1α depletion blunted the effects of SR-18292 reducing blood glucose levels and while our results correlate PGC-1α acetylation with suppression of hepatic gluconeogenic gene expression, we cannot conclude at this point that the increased acetylation of PGC-1α is necessary and/or sufficient to mediate the effect of these compounds. It is unclear at this point how these compounds mediate increases of PGC-1α acetylation and which sites are specifically linked to suppression of hepatic gluconeogenic gene expression. A possible mechanism suggested by our data with SR-18292 points towards the dissociation of the HNF4α and GCN5-PGC1α complex from the chromatin of these promoters. Displacement of this complex will be consistent with the decreases in H3K9 acetylation/H3K4 trimethylation activation marks observed at the Pck1 promoter and decreases in gene expression after SR-18292 treatment. Regardless of whether the effects of the compounds we have found are mediated solely by mechanisms linked to PGC-1α acetylation or not, it will be of interest to use these compounds as a discovery tool to study how glucose production is regulated in hepatocytes. Since the direct molecular targets of these compounds are currently unknown, further investigation into their targets might shed new light on the components that are important for the regulation of glucose output and hepatic insulin action. These molecular targets can also constitute potential new therapeutic strategies to treat T2D.
Several studies have demonstrated the importance of GCN5 and its close homolog PCAF in regulating the gluconeogenic activity of PGC-1α by catalyzing its acetylation (Dominy et al., 2011; Sun et al., 2014). This study highlights the potential targeting of the GCN5/PGC-1α axis by small molecules for diabetes treatment, and is in agreement with our previous studies that found that inhibition of the Cdk4 kinase complex reduces GCN5 activity and can result in less acetylated PGC-1α and elevated blood glucose levels (Lee et al., 2014). The increased binding between PGC-1α and GCN5, mediated by SR-18292, leads to reduced HNF4α transcriptional activity and to its removal from Pck1 and G6pc promoter regions. GCN5 is normally associated with increased histone acetylation and more active chromatin (Bodo et al., 1977), suggesting its effect on Pck1 and G6pc expression through PGC-1α acetylation overcomes its effect on histone modification. A negative effect of GCN5 on transcription was also documented by its acetylation-mediated regulation of the SWI/SNF complex, which leads to dissociation of SWI/SNF from chromatin (Kim et al., 2010). SR-18292 promotes the displacement of HNF4α from the promoter regions, and probably of the protein complex, supporting a model in which GCN5 acetylates PGC-1α, which in turn results in a remodeled transcription complex that dissociates from the Pck1 and G6pc promoters. The reduction in H3K9Ac levels may be a result of the removal of GCN5 from the Pck1 promoter. The magnitude of HNF4α removal from the Pck1 promoter exceeds the reduction of H3K9Ac and H3K4Me3 marks and probably forms part of a sequence of chromatin events at gluconeogenic promoters caused by SR-18292.
At the cellular level, it is not clear at this point what protein(s) or enzyme(s) are directly targeted by SR-18292. It is possible that the target of SR-18292 modifies GCN5, increasing its binding to PGC-1α., Along these lines, we have previously shown that the Cyclin D1/Cdk4 complex can activate GCN5 in response to insulin and amino acids through phosphorylation of T272 and S372 (Lee et al., 2014), and that SIRT6 increases GCN5 activity through deacetylation of K549 (Dominy et al., 2012), demonstrating the existence of such effectors that regulate GCN5 activity by PTMs. Although these sites were not modulated by SR-18292 (data not shown), suggesting that Cyclin D1/Cdk4 and SIRT6 do not mediate the effect of SR-18292, modification of other sites might be responsible to promote the interaction between GCN5 and PGC-1α. Alternatively, since GCN5 and PGC-1α are part of larger complexes that regulate transcription, SR-18292 could directly target upstream regulators or a component of these complexes that enhance the interaction between PGC-1α and GCN5. The identification of this target would further define the anti-diabetic effects of this compound and expand the therapeutic possibilities.
It is generally agreed upon that the elevated HGP in T2D is a result of increased gluconeogenesis (Hundal et al., 2000; Magnusson et al., 1992). This probably reflects the chronic insulin resistance in T2D that impairs the ability of insulin to suppress gluconeogenesis. Hence, targeting components within the gluconeogenic pathway, like PEPCK and G6Pase, can potentially improve hyperglycemia. However, gluconeogenic fluxes can be controlled independently of changes in gluconeogenic enzymes levels, such as PEPCK (Burgess et al., 2007; Madiraju et al., 2014; Perry et al., 2015; Perry et al., 2014; Ramnanan et al., 2010; Samuel et al., 2009) raising the question whether inhibiting gluconeogenesis via manipulation of these enzymes can indeed reduce HGP in diabetic states. While it is clear that acute insulin-mediated suppression of HGP (Ramnanan et al., 2010) and gluconeogenesis (Perry et al., 2015) can occur independent of changes in gluconeogenic gene expression, the tight regulation of gluconeogenic genes by the feeding status is an important feature of insulin action on HGP (Pilkis and Granner, 1992). Thus, it is possible that in diabetic conditions, where gluconeogenesis is chronically elevated, modulation of expression and activity of gluconeogenic enzymes affect the flux of this pathway and control HGP and insulin sensitivity. In fact, early studies show that inhibition of gluconeogenesis by 3-mercaptopicolinic acid, a preferential PEPCK inhibitor, results in hypoglycemia, supporting PEPCK’s importance in glucose homeostasis (DiTullio et al., 1974). Our findings support that non-genetic inhibition of gluconeogenic gene expression reduces HGP and improves hyperglycemia. The mechanism of action of SR-18292 is to decrease expression of gluconeogenic genes, mainly PEPCK in vivo, however, it is conceivable that additional mechanisms contribute to the potent effects of this compound on HGP and insulin sensitivity. While we cannot exclude at this point that SR-18292 mediates its effect by targeting other tissues other than the liver, based on the potent and selective effects on glucose production and hepatic insulin sensitivity, our results suggest that the liver is a major target of SR-18292 and probably accounts for a significant part of its anti-diabetic effects.
In summary, the findings presented in this study have implications for the regulatory mechanisms underlying diabetes. We have identified small molecules that can be used as chemical tools to study the regulation of glucose metabolism. These chemical compounds target components within the hepatic PGC-1α gluconeogenic pathway and reduce glucose production. In particular, we have found a chemical scaffold that improves glucose homeostasis in a preclinical mouse model. These small molecules have the potential to be used either as a single agent or in combination with current anti-diabetic drugs.
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pere Puigserver (pere_puigserver@dfci.harvard.edu)
Experimental Model and Subject Details
Animal procedures
All mice were purchased from Jackson laboratories and housed under a 12 h light/12 h dark cycle at 22 °C. Before handling, mice were allowed to acclimate for at least 1 week in our animal facility. For in vivo studies with DIO mice, males 6–8 weeks old were fed HFD for the indicated time. For drug administration, SR-18292 was re-suspended in a 10% DMSO/10% Tween80/80% PBS solution at a final concentration of 6–12 mg/ml. Metformin was re-suspended in the same solution at a 20mg/ml concentration. SR-18292 was injected via I.P. for 3 days between 4–5pm and food was removed on day 3 at 5pm. The following morning (day 4) SR-18292 was injected again (for a total of 4 injections) and blood glucose was measured after 3 hours. Injection volume did not exceed 275μl per mouse. Metformin was administered via oral gavage. Pyruvate tolerance and glucose tolerance tests were performed following the same dosing protocol, but 3 hours after the last injection mice were injected with either pyruvate (2g/Kg) or glucose (2g/kg or 0.5g/kg for Lep°b/°b) and blood glucose was measured every 20 minutes. Insulin tolerance test was performed following the same dosing protocol, but mice were fasted for 4 hours after the forth injection before performing the test. Insulin was injected via I.P. (0.5U/Kg) and blood glucose was measured every 20 minutes. Glycemia was measured by tail bleed using a glucometer (OneTouch). For all experiments, age- and body weight- matched animals were used. For protein, RNA extracts and biochemistry studies, livers were removed following each experiment and snap frozen in liquid nitrogen. For information on number of animals in each experiment please refer to figures 4, 5 and S5 legends. All studies were performed according to protocols approved by Beth Israel Deaconess Medical Center’s Animal Care and Use Committee.
Primary hepatocytes and cell cultures
U-2 OS cell lines were maintained in DMEM containing 10% FBS. Transfections were performed with Polyfect (QIAGEN) with a fixed total quantity of DNA. Cells were collected after 24 h or 48 h of transfection as indicated. Medium was changed every day as well as 3 h before collecting cells.
Primary hepatocytes were isolated from 8- to 12-week-old male C57BL/6 mice by perfusion with liver digest medium (Invitrogen, 17703-034) followed by 70μm mesh filtration. Percoll (Sigma, P7828) gradient centrifugation allowed primary hepatocytes isolation from other cell types and debris. Cells were seeded in plating medium (DMEM with 10% FBS, 2mM sodium pyruvate, 1% penicillin/streptomycin, 1 M dexamethasone, and 100nM insulin). After 4 h of seeding, the medium was changed and incubated overnight in maintenance medium (DMEM, 0.2% BSA, 2mM sodium pyruvate, 1% penicillin/streptomycin, 0.1 M dexamethasone, and 1nM insulin). To infect cells, the indicated adenoviruses were added at MOI of 2.5 to 5 for 4 h. Cells were collected within 48 h after infection and medium was changed every day.
Method details
Primary and secondary AlphaLisa screen to determine single dose and dose response effects on PGC-1α acetylation
U-2 OS cells were thawed and grown in 4 Triple flasks (NUNC) with growth medium. After 48 hours, cells were harvested and seeded in 4 Hyper flasks at a density of 15–20 million cell each. Cells were then grown for 4–5 days until reached confluency. After reaching the desired confluency, medium was changed to assay medium and cells were infected with Ad-PGC-1α and Ad-GCN5 for 4 hours. Following infection, medium was changed to phenol-free medium and cells were incubated overnight. The following day, cells were harvested and seeded on a 384-well plate at 7000 cells/well in 50μl assay media. Cells were allowed to adhere for 4–8 hours after which 100nl of compound was added. After overnight incubation with compound, media was aspirated and cells were incubated with lysis buffer for 1 hour at RT. For detection of acetylated PGC-1α, cells were incubated with 6μl of Acetyl-Lys antibody for 1 hour at RT. Lysates were then incubated with Alpha acceptor beads (6μl, 1 hour RT) and Alpha donor beads (6μl, 1 hour RT). Perkin Elmer EnVision plate reader was used for signal detection using Alpha protocol. For each compound, activity score was calculated based on the mean of the normalized and corrected sample activity replicates. Compounds with activity score ≥50 (50% activity above neutral control) were called as hits. As a positive control, each plate contained wells that were treated with fascaplysin, a CDK4 inhibitor, that reduces the acetylation of PGC-1α. For a compound to be called a hit all replicates had to be considered hits (also, please refer to: https://pubchem.ncbi.nlm.nih.gov/bioassay/651723). Active hits from primary screen were then used in a secondary screen to determine AC50 toward PGC-1α acetylation. Same protocol for detection of PGC-1α acetylation was applied but compounds were added at different doses. For each sample the highest valid tested concentration was determined and set as Maximum concentration. Raw signals of the plate wells were normalized using the ‘Neutral Controls Minus Inhibitors’ method in Genedata Assay Analyzer (v10.0.2). AC values were calculated using the curve fitting strategies in Genedata Screener Condoseo (7.0.3) and up to the active concentration limit (Maximum concentration) described for each sample (also, please refer to: https://pubchem.ncbi.nlm.nih.gov/bioassay/720513).
Highthroughput cytotoxicity panel
To determine potential cytotoxicity of identified compounds 3 different cell lines were used (HEK293, HepG2 and A549). Cells were grown in Triple flask to reach ~95% confluence and then re-suspended for dispensing at 50,000 cells/mL of medium (DMEM, 10% FBS, Penicillin//Strep/L-Glutamine). The following day, cells were plated on 384-well plates in 40μl medium and incubated for 24 hours in standard tissue culture conditions. Compounds (100nl) were then added at different doses into 40μl assay volume and incubated for 72 hours. To determine cell viability, plates were removed from the incubator and allowed to cool down for 15 minutes and viability was determined by addition of 20μl 50% Promega CellTiterGlo (diluted 1:1 with PBS, pH 7.4) and incubation at RT for 5 minutes. PerkinElmer EnVision was used to measure luminescence.
High throughput qPCR screen
To determine the effects of compounds on expression level of Pck1 and G6pc genes, primary hepatocytes were isolated as described in the Experimental Model and Subject Details section. Following isolation, hepatocytes were seeded in 384-well plates at 4000 cells/well in 40μl plating medium and incubated overnight at 37°C. The next morning, plating media was aspirated and 40μl maintenance medium (DMEM, 5% FBS, 2mM sodium pyruvate, 1% Pen/Strep, 1nM insulin, 0.1μM dexamethasone) was added. Incubation at 37°C was continued until afternoon when compounds w ere added (100nl) at dose using 384 well pin tool on pin table (Walkup Cybi Well). After overnight incubation, maintenance medium was aspirated and low serum medium was added (DMEM, 5% FBS, 2mM sodium pyruvate, 1% Pen/Strep). Compounds were re pinned to cells and plates were returned to incubator for additional 3 hours. After 3 hours forskolin stimulation started for additional 1.5 hours. For cDNA synthesis Cells-to-Ct kit (Ambion, 4391851C) was used. The medium was aspirated and cells were washed twice (100μL with PBS) using the ELX405 Plate washer (Biotek). The assay plates were flipped upside down and centrifuged at 1000 rpm 2 minutes to remove the excess liquid. 10μL of Lysis solution with DNase I (from Cell to CT Lysis kit, Ambion) was added to each well using the MultiDrop Combi/Standard tube dispensing cassette (Thermo Scientific). Each assay plate was then shaken for 2 minutes and incubated for an additional 8 minutes at RT. At the end of the incubation, 1μL of stop solution was added with the Multidrop Combi-nl (Thermo Scientific) and the assay plates were centrifuged (face up) (1000 rpm, 2minutes). The assay plates were incubated for 2 minutes at RT and processed for reverse transcription. For reverse transcription, 8μL of RT master mix (RT buffer, RT Enzyme mix, H2O) was dispensed into each well of a RT assay plate (Axygen, PCR-384 RGD C). 2μL of the lysed cells were transferred into RT assay plate using Vario transfer unit (CyBi Well). The RT assay plates were incubated at 37°C for 1h and the reverse transcript ase is inactivated by incubating the plates for 1 minute at 95°C. qPCR assay was performed by the a ddition of 4μl/well of PCR master mix (Roche) to PCR plate (Roche Light Cycler 480 MultiWell Plate 384, Cat# 04 729 749 001) using the Multidrop Combi-nl (Thermo Scientific). 1μL/well of RT DNA is then transferred to the 4μL/well PCR plate using CyBi Well. The PCR plates were centrifuged (face up) for 2 minutes at 1000 rpm. PCR was performed using ThermoCycler (Roche Light Cycler 480 II) with Macro Probe Protocol (95°C for 10min, 55 cycles of 95°C f or 10 sec, 60°C for 30 sec and 40°C for 30 sec). TaqMan probe sets that were used (Mouse Pck1, Applied Biosystems, 4351370 (Mm01247058_m1), Mouse G6pc, Applied Biosystems, 4351370 (Mm00839363_m1), mouse Gapdh, Applied Biosystems (4352339E)). To calculate inhibitory AC50, dose response curves were generated by determining the difference in Ct values between compound treatment and forskolin control at each dose and were fit to the Hill equation. Curves were forced through 100% inhibitory activity (which was determined to be the DMSO control), so as to generate AC50 values calculated based on these curve, that were comparable between compounds. This was done even if in some cases higher inhibition than the DMSO control was observed. AC values were calculated using the curve fitting strategies in Genedata Screener Condoseo (7.0.3). (also, please refer to: https://pubchem.ncbi.nlm.nih.gov/bioassay/743465 and https://pubchem.ncbi.nlm.nih.gov/bioassay/743463).
Procedures for Chemical Synthesis of SR-18292
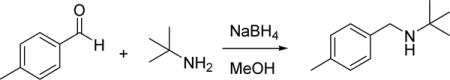
To a room temperature solution of 4-methylbenzaldehyde (2 g, 16.65 mmol) in MeOH (25 mL) was added 2-methylpropan-2-amine (1.217 g, 16.65 mmol). The solution was stirred at room temperature for 1 hour, cooled to 0 °C, and then treated with NaBH4 (1.260 g, 33.3 mmol) in one portion. The reaction mixture was was allowed to warm to room temperature overnight with stirring. The reaction was quenched with saturated aqueous NH4Cl solution and extracted with ethyl acetate. The organic layer was washed with saturated sodium bicarbonate solution, brine, dried (Na2SO4) and concentrated under reduced pressure. The crude residue was purified by flash chromatography on silica gel (EtOAc/hexanes) to give 2-methyl-N-(4-methylbenzyl)propan- 2-amine (2.42 g, 13.65 mmol, 82% yield) as white solid. HRMS (ESI+), m/z: [M + H]+, calcd. for C12H20N+ 178.1590; found 178.1598.
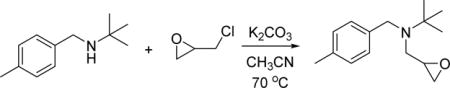
To a solution of 2-methyl-N-(4-methylbenzyl)propan-2-amine (2.42 g, 13.65 mmol) in acetonitrile (25 mL) was added 2-(chloromethyl)oxirane (2.53 g, 27.3 mmol), K2CO3 (3.77 g, 27.3 mmol) and KI (4.53 g, 27.3 mmol). The mixture was heated to 70 °C for 16h, cooled, and filtered, washing with ethyl acetate. The filtrate was concentrated in vacuo and purified by flash chromatography on silica gel (EtOAc/hexanes) to afford 2-methyl-N-(4-methylbenzyl)-N-(oxiran-2-ylmethyl)propan-2-amine (2.1 g, 9.0 mmol, 66% yield) as colorless oil. 1H NMR (400 MHz, CDCl3) δ 1.17 (s, 9H), 2.33 (s, 3H), 3.60 (d, J = 14.8 Hz, 1H), 3.83 (d, J = 14.8 Hz, 1H), 7.09 (t, J = 3.0 Hz, 1H), 7.10 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 7.9 Hz, 2H). HRMS (ESI+), m/z: [M + H]+, calcd. for C15H24NO+ 234.1852; found 234.1862.
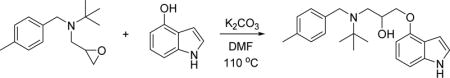
To a solution of 2-methyl-N-(4-methylbenzyl)-N-(oxiran-2-ylmethyl)propan-2-amine (100 mg, 0.43 mmol) in DMF (2 ml) was addeds 1H-indol-4-ol (114 mg, 0.86 mmol) and K2CO3 (119 mg, 0.86 mmol). The mixture was heated to 110oC for 16h and then cooled. The reaction mixture was diluted with ethyl acetate, washed with saturated sodium bicarbonate solution, brine, dried (Na2SO4) and concentrated in vacuo. The residue was purified by reverse-phase preparative HPLC to give 1-((1H-indol-4-yl)oxy)-3-(tert-butyl(4-methylbenzyl)amino)propan-2-ol (TFA salt, 55 mg, 0.11 mmol, 27% yield) as colorless solid. 1H NMR (400 MHz, CDCl3) δ 1.21 (s, 9H), 2.33 (s, 3H), 2.80–2.95 (m, 2H), 3.30 (brs, 1H), 3.57–3.63 (m, 2H), 3.83–3.95 (m, 3H), 6.39 (d, J = 7.5 Hz, 1H), 6.62 (t, J = 3.1 Hz, 1H), 7.00 (d, J = 7.3 Hz, 1H), 7.05 (d, J = 7.4 Hz, 1H), 7.09 (t, J = 3.0 Hz, 1H), 7.13 (d, J = 7.8 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 8.19 (brs, 1H). HRMS (ESI+), m/z: [M + H]+, calcd. for C23H31N2O2+2 367.2380; found 367.2398. See also Data S1 for HRMS.
Drug treatment of primary hepatocytes for measurement of protein modification, gene expression and glucose output
For measurement of acetylation, gene expression and glucose production SR-18292 (20μM) was added to cells overnight and fresh compound was added the following day for additional 3 h before collecting the cells (for total of ~18 h treatment). Where insulin and glucagon stimulation were performed, the medium was changed to starvation medium (DMEM, 0.2% BSA, 2mM sodium pyruvate, and 1% penicillin/streptomycin) with fresh compound for the last 3 h and cells were stimulated with either 100nM insulin or 200nM glucagon for an extra 1.5 h. For glucose measurements following PGC-1α over expression cells were incubated for the last 3 h in glucose-free medium (phenol-red/glucose free DMEM, 0.2% BSA, 2mM sodium pyruvate and 20mM sodium lactate or 10mM glycerol in case glycerol was used as substrate). For glucose measurements following glucagon stimulation cells were incubated for the last 4 h in the same glucose-free medium that also contained glucagon (200nM). The glucose level in the medium was measured using a glucose assay kit from Eton Biosience Inc. by adding 50μl of assay buffer to 50μl of medium followed by 15 min incubation at 37°C. Absorbance was measured at 490nm.
DNA constructs and adenoviruses
Adenoviruses were produced with the pAd-Track/pAd-Easy system. FLAG-HA-PGC-1α and FLAG-GCN5 adenoviruses were made as previously described (Lerin et al., 2006; Rodgers et al., 2005). FLAG-HA-PGC-1α, FLAG-GCN5 pcDNA 3.1 constructs were made as previously described (Lerin et al., 2006; Rodgers et al., 2005). shLacZ was cloned into the BLOCK-iT™ U6 RNAi Entry Vector Kit (Thermo Fisher Scientific) and subsequently cloned into the pAd/BLOCK-iT™ RNAi vector. shPGC-1α was produced using the pAd-Track/pAd-Easy system and was a kind gift from the Montminy lab (Koo et al., 2004).
siRNA mediated knock down of Ppargc1a
siRNA oligos against mouse Ppargc1a were purchased from OriGene (SR420231). Hepatocytes were transfected with a 1:1:1: mixture of 3 unique 27mer siRNA duplexes (20μM each) using Lipofectamin RNAiMax (Invitrogen) immediately upon seeding with plating medium. The following morning cells were washed with PBS and medium was changed to maintenance medium. In the afternoon, cells were transfected again and compound treatment started.
Cell lysis and immunoprecipitation
For PGC-1α acetylation detection in hepatocytes cells were infected with Ad-PGC-1α and Ad-GCN5. Cells were collected in RIPA buffer (containing protease inhibitor cocktail, 5mM NaF, 5mM β-glycerophosphate, 10mM nicotinamide, 1mM DTT and 1 M trichostatin A). FLAG–HA-PGC-1α was immunoprecipitated overnight at 4°C with FLAG beads (Sigma A2220). For GCN5 interaction with PGC-1α, U-2 OS cells were infected with Ad-PGC-1α. Nuclear extracts were obtained by lysing cell in Buffer A (10mM HEPES-KOH (pH 7.9), 10mM KCl, 1.5mM MgCl2, 0.5mM DTT, 0.25% IGEPAL (v/v), 5mM NaF, 5mM β-glycerophosphate, 5mM sodium butyrate and 10mM nicotinamide), supplemented with Protease Inhibitor Cocktail. Once cytoplasmic fractions were separated, nuclear pellets were lysed in Buffer B (20mM HEPES-KOH (pH 7.9), 125mM NaCl, 1mM EDTA, 1mM DTT, 1% IGEPAL (v/v), 10% glycerol (v/v), 5mM NaF, 5mM β-glycerophosphate, 5mM sodium butyrate and 10mM nicotinamide), supplemented with Protease Inhibitor Cocktail. FLAG-HA-PGC-1α was immunoprecipitated with FLAG beads overnight at 4°C. All beads were washed at last 3 times in lysis buffer before eluting in sample loading buffer.
Quantitative real-time PCR analysis
Total RNA was extracted from cells or pulverized liver using TRIzol reagent (Ambion, Life Technologies), followed by cDNA preparation from 2 μg of total RNA with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). cDNA products were quantified by real-time PCR using Power SYBR Green PCR Master Mix (Applied Biosystems) on a CFX384 Real-Time PCR System (Bio-Rad). To calculate IC50 from C-80 dose response, curves were fit to the hill equation in a similar manner used to calculate estimated AC50 from the high-throughput qPCR screen. However, unlike the curves from the qPCR high-throughput screen, values of relative repression were used rather than of delta-Ct.
Transcriptional reporter assays
U-2 OS cells were transfected with a fixed amount of DNA. After 6 h medium was changed and compound treatment started. The following morning medium was changed again and fresh compound was added for additional 3 h after which cells were collected (24 h post transfection) with 1× Passive Lysis Buffer (Promega). Firefly luciferase reporter was determined by addition of Luciferase Assay Substrate (Dual-Luciferase Reporter Assay System, Promega) and quantification of luminescence on a FLUOstar Omega plate reader (BMG Labtech). CMV-driven Renilla luciferase vector was cotransfected as an internal control. Data are presented as firefly luciferase reporter values normalized to Renilla values and are representative of at least two independent experiments.
Chromatin immunoprecipitation
Primary hepatocytes were fixed in 1% formaldehyde for 10 minutes at room temperature. Crosslinking was quenched by adding glycine to a final concentration of 125mM and rinsing twice with PBS. Cells were collected in PBS containing protease inhibitors followed by lysis in chromatin immunoprecipitation (ChIP) buffer (50mM HEPES pH, 7.9; 140mM NaCl; 1mM EDTA; 1% Triton X-100; 0.1% NaDOC, 0.1% sodium dodecyl sulfate (SDS) and protease inhibitors). Chromatin was sheared by sonication with a Diagenode Biorupter for three cycles of 5 minutes (30 seconds on, 30 seconds off). Samples were clarified and chromatin immunoprecipitated overnight at 4 °C with the indicated antibody. Immunecomplexes were recovered with Protein A magnetic beads (Dynabeads; Novex, Life Technologies) preblocked with salmon sperm DNA (Invitrogen, Life Technologies). Following extensive washes, immunoprecipitated DNA was then isolated with a Chelex. Input DNA was prepared from 10% of respective chromatin prior to precipitation. Immunoprecipitated DNA and input DNA were analyzed by quantitative real-time PCR with primers specific for the indicated region.
Adenovirus Injection to Mice
Virus was introduced to mice through the tail vein injection. Virus was thawed before injection and the desired amount of virus was diluted with saline to a final volume of 100 L per mouse. Each mouse was injected with 6*107 pfu/g. SR-18292 treatment started on day 5 post infection.
Hyperinsulinemic-Euglycemic Clamp Studies
Hyperinsulinemic-euglycemic clamps were performed on mice fed HFD for 16 weeks as previously described with minor modifications (Jurczak et al., 2012). Briefly, an indwelling catheter was surgically implanted in the right jugular vein 7 days prior to study. Three days before study, mice were injected with SR-18292 and vehicle control following the same protocol described above. Mice were fasted overnight prior to clamps and then infused with a fixed amount of insulin [4 mU/(kg-min)] and a variable amount of 20% dextrose to maintain euglycemia. [3-H]glucose was included in the infusate to allow for the calculation of whole-body rates of glucose metabolism.
GCN5 activity assay
For determination of GCN5 HAT activity U-2 OS cells overexpressing Ad-GCN5 were treated with SR-18292 (10μM) for 18 h. Cells were lysed with buffer B (20mM HEPES-KOH (pH 7.9), 125mM NaCl, 1mM EDTA, 1mM DTT, 1% IGEPAL (v/v), 10% glycerol (v/v), 5mM NaF, 5mM β-glycerophosphate, 5mM sodium butyrate and 10mM nicotinamide), supplemented with Protease Inhibitor Cocktail. FLAG-GCN5 was immunoprecipitated with FLAG beads overnight at 4°C following multiple wash es with lysis buffer. GCN5 was then eluted using 3× FLAG peptide and the purified protein was used to determine HAT activity using the HAT Inhibitor Screening Assay Kit (Cayman Chemicals) per manufacturer instructions. To determine the potential effect of SR-18292 on GCN5 activity in vitro, GCN5 was purified from U-2 OS cells in a similar manner but without treatment with SR-18292.
Mass spectrometry
Gel bands were dehydrated with acetonitrile, dried and resuspended in 50 mM ammonium bicarbonate (pH 8.0) containing 500 ng of sequencing-grade trypsin (Promega) and incubated at 37°C for 8 hours. Digested samples were then loaded onto StageTips and desalted. Peptides were eluted with 50% acetonitrile, 5% formic acid, dried using a Speed-Vac apparatus, and resuspended in 25 l of HEPES, pH 8.5. Peptides were labeled with 6-plex tandem mass tag (TMT) reagents (Thermo Scientific, Rockford, IL). After 1 hour of incubation at room temperature, the reaction was quenched with hydroxylamine and acidification with formic acid to pH ~2. TMT labeled peptides were combined, desalted by StageTips, dried and resuspended in 10 l of 1% formic acid. All spectra were acquired using an Oribtrap Fusion mass spectrometer (Thermo Scientific) in line with an Easy-nLC 1200 (Thermo Fisher Scientific) ultra-high pressure liquid chromatography (UHPLC) pump. TMT labeled peptides (4 l) were separated onto a 75 μM inner diameter column containing 1 cm of Magic C4 resin (5 m, 100 Å, Michrom Bioresources) followed by 35 cm of Sepax Technologies GP-C18 resin (1.8 m, 120 Å) with a gradient consisting of 4–30% (ACN, 0.125% FA) over 90 min at ~250 nl/min. For all LC-MS/MS experiments, the mass spectrometer was operated in the data-dependent mode where the MS1 spectra was set at a resolution of 120,000, with an AGC target of 150,000 and a max injection time of 100 ms. The ten most intense ions were selected for MS2. MS1 precursor ions were excluded using a dynamic window (60 seconds +/− 10 ppm) and the MS2 precursors were isolated with a quadrupole mass filter set to a width of 0.5 DA. For MS3 based TMT quantitation, MS2 spectra were collected at an AGC of 4000, max injection time of 150 ms, and CID collision energy set at 35%. MS3 spectra were acquired in the Orbitrap parameters where the HCD collision energy was increased to 55%. Synchronous-precursor-selection (SPS) was enabled to include up to six MS2 fragment ions for the MS3 spectrum. A compendium of in-house software was used to convert raw files to mzXML format that adjusted monoisotopic m/z measurements and corrected erroneous peptide charge state assignments (Huttlin et al., 2010). Assignment of MS2 spectra was performed using the SEQUEST algorithm. All searches utilized the human UniProt database (downloaded 6/10/2016) where reversed protein sequences and known contaminants (keratins, etc.) were included. SEQUEST searches were performed using a 10 ppm precursor ion tolerance and requiring each peptide’s N/C terminus to have trypic specificity and allowing up to three missed cleavages. TMT tags on lysine residues and the N-terminus (+229.162932 Da) was set as static modifications as well, while methionine oxidation (+15.99492 Da) and acetylation on lysine (−187.1523673 DA) was set as variable modifications. An MS spectra assignment false discovery rate (FDR) of less than 1% was achieved by applying the target-decoy database search strategy3 and acetylated peptides were also manually validated. For quantification, a 0.03 m/z window centered on the theoretical DA value of each reporter ion was utilized for the nearest maximal signal intensity. Reporter ion intensities were adjusted to correct for the isotopic impurities from the different TMT reagents as per manufacturer’s specifications. The signal to noise values for all peptides for PGC-1α were summed within each TMT channel and all acetylated peptides were normalized to that PGC-1α summed signal. For each peptide, a total minimum sum signal to noise value of 200 and an isolation purity greater than 70% was required.
Serum and liver biochemistry measurements
Serum samples were gathered from blood collected by cardiac puncture. Insulin was measured with Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, 90080). Kits used to measure alanine transaminase (EALT-100), aspartate transaminase (EASTR-100), lactate dehydrogenase (DLDH-100) were purchased from Bioassay Systems. Kits used to measure lactate (MAK 064), β- hydroxybutyrate (MAK 041), Triglycerides (TR0100) were purchased from Sigma. Kit used to measure cholesterol (C7510) was purchased from Pointe Scientific. Kit used to measure non-esterified fatty acids was purchased from Wako.
Cytotoxicity determination in primary hepatocytes
For cell viability determination using MTT, primary hepatocytes were seeded on a 96-well plate at 20,000 cells/well. The following day cells were treated at different doses, as indicated, for 18 h following similar protocol described above for drug treatment of primary hepatocytes. 5μl of MTT reagent (5mg/ml) was then added to each well (n=4/dose) and cells were incubated for 1h at 37°C. Medium was discarded and dye was extracted by adding 100μl DMSO to each well. For cytotoxicity determination using ToxiLight Non-destructive Cytotoxicity Bioassay, hepatocytes were seeded on a 6-well plate and treated with either SR-18292 (20μM) or cisplatin (50μM) for 18 h. 50 μl of medium was collected and used to measure cellular toxicity by adding 100 of adenylate kinase detection reagent (AKDR) and incubating 5 min at RT before measuring luminescence.
Quantification and statistical analysis
Data were analyzed using Prism software (GraphPad Software, Inc.) and are expressed as mean ± SEM. Two-tailed Student t tests, one-way ANOVA with either Tuckey or Dunnet or Two-way ANOVA with Sidak comparison test were used to compare means between groups as indicated; P < 0.05 was considered significant.
Data and software availability
All data related to highthroughput studies (AlphaLisa assay and qPCR) are available through pubchem (https://pubchem.ncbi.nlm.nih.gov/bioassay/651721, https://pubchem.ncbi.nlm.nih.gov/bioassay/651723, https://pubchem.ncbi.nlm.nih.gov/bioassay/720513, https://pubchem.ncbi.nlm.nih.gov/bioassay/743465 and https://pubchem.ncbi.nlm.nih.gov/bioassay/743463).
Supplementary Material
Figure S1. High throughput screens to identify small molecules that can potentially inhibit gluconeogenesis. Related to Figure 1.
(A) Work flow of screen starting with a high-throughput AlphaLisa screen through secondary screens.
(B) Dose response acetylation curves of final hits presented in Figure 1E.
(C) Dose response curves showing estimated inhibitory activity toward Pck1 and G6pc gene expression following forskolin treatment. To calculate estimated inhibitory AC50 values (eAC50) that were comparable between compounds, curves were forced through 100% inhibitory activity (which was determined to be the DMSO control). AC50 were calculated as described in STAR methods.
(D) Chemical structures of screen hits that are able to reduce glucose production in hepatocytes.
Figure S2. Characterization of SR-18292 effects in hepatocytes. Related to Figure 2.
(A) Western blot analysis of CREB phosphorylation (S133) and total TORC2 upon glucagon stimulation (200nM) and SR-18292 treatment (20μM). Glucagon-stimulated dephosphorylation of TORC2 results in a shift in gel band.
(B) Western blot analysis of Akt phosphorylation (S473) upon insulin stimulation (100nM) and SR-18292 treatment (20μM).
(C) Culture medium was collected from hepatocytes treated with either SR-18292 or cisplatin for 18 hrs. Cellular viability was assessed by using Toxilight bioassay kit (Lonza) (n=3) and by MTT assay (n=4) (D). ***P<0.001
(E) Total protein levels are not affected by SR-18292 treatment. n=3.
(F) PEPCK protein levels are reduced in hepatocytes treated with SR-18292 and infected with Ad-PGC-1α.
(G) mRNA expression level of Ppargc1a, Pck1 and G6pc following 18 h treatment with the SR-18292 (20μM). Hepatocytes were infected with Ad-PGC-1α to induce expression of Pck1 and G6pc genes and to induce glucose release from either Pyruvate/lactate (2mM/20mM) or glycerol (10mM) (H).
Figure S3. Mechanistic studies with SR-18292. Related to Figure 3.
(A) HAT activity assay showing that the acetyl transferase activity of GCN5 toward H3 peptide is not increased in cells treated with SR-18292. n=3.
(B) HAT activity assay showing that the acetyl transferase activity of GCN5 toward H3 peptide is not affected by in-vitro treatment with SR-18292. n=3.
(C) Interaction of PGC-1α with ectopically expressed GCN5 is increased after treatment with SR-18292. IP, immunoprecipitation.
(D) U-2 OS cells were infected with Ad-PGC-1α and Ad-GCN5 and treated with SR-18292 (10μM) for 18 h. Mass spectrometry analysis was performed to identify lysine residues whose acetylation is being increased following treatment.
(E) Promoter occupancy of FoxO1 over Pck1 and G6pc promoter regions is not affected by drug treatment. n=3. **, P<0.01. Related to Figure 3.
Figure S4. Structure-Activity Relationship studies with SR-18292 analogs. Related to Figures 2 and 3.
(A) Chemical structure of several analogs of SR-18292.
(B) mRNA expression level of Ppargc1a, Pck1 and G6pc following 18 h treatment with the indicated compounds (20μM). Glucagon stimulation (200nM) was used to induce the expression of Pck1 and G6pc genes.
(C) mRNA expression level of Ppargc1a, Pck1 and G6pc following 18 h treatment with the indicated compounds (20μM). Hepatocytes were infected with Ad-PGC-1α to induce expression of Pck1 and G6pc genes.
Data presented as mean +/− S.E.M. n=3, one-way ANOVA with Dunnet posttest.
**P<0.01, ***P<0.001.
(D) Western blot analysis to detect acetylation status of PGC-1α following treatment with SR-18292 (10μM) and SR-19138 (10μM). U-2 OS cells were infected with Ad-PGC-1α and Ad-GCN5 and treated with the indicated compound for 18 h. PGC-1α was immunoprecipitated using FLAG beads. IP, immunoprecipitation.
Figure S5. Characterization of in-vivo effects of SR-18292. Related to Figures 4 and 5.
(A) Plasma insulin levels after GTT and PTT (B) presented in Fig. 4H and 5A, respectively.
*, P<0.05, **, P<0.01.
(C) Serum lactate and β-hydroxybutyrate levels are not altered with SR-18292 treatment. Mice were fed HFD for 4 weeks. n=7.
(D) mRNA expression level of lipid metabolic genes is not changed with SR-18292 treatment. Mice were fed HFD for 13 weeks. n=10.
(E) Serum TG, free fatty acids and cholesterol levels are not altered with SR-18292 treatment. Mice were fed HFD for 13 weeks. NEFA, non-esterified fatty acids. n=10.
(F) mRNA expression level of mitochondrial genes in liver is not changed with SR-18292 treatment. Mice were fed HFD for 13 weeks. n=10.
(G) Mice fed HFD for 6 weeks were injected daily with SR-18292 for a total of 14 days and body weight was monitored during the treatment. Mice were fasted at day 3 and 13 and fasting blood glucose was measured at day 4 and 14 (H). Fed blood glucose was also measured at day 10. n=10, *P<0.05.
(I) Serum ALT and AST levels were not altered after 14 days of SR-18292 treatment. n=10
(J) H&E staining of liver slices obtained from livers harvested after 14 days of SR-18292 treatment. 3 representative images are presented for each group. All samples (n=10/vehicle, n=10/SR-18292) were assessed by an expert mouse pathologist and no sign of toxicity was observed. scale bar, 50μm.
(K) Expression level of inflammatory genes from livers harvested after 14 days of SR-18292 treatment. n=10
(L) Liver triglycerides and cholesterol levels are not altered after 14 days of SR-18292 treatment. n=10
(M) Serum lactate, β-hydroxybutyrate and cholesterol levels are not altered after 14 days of SR-18292 treatment. n=10
Table S1. Serum biochemistry parameters following treatment with SR-18292. Related to Figures 4 and 5.
Table S2. A list of primers used for ChIP assays. Related to Figures 3 and S3.
Table S3. A list of primers used for qPCR analysis. Related to Figures 2, 4, S2, S4 and S5.
Data S1. HRMS of SR-18292 and its intermediates. Related to Figures 2, 3, 4, 5, S2, S3, S4 and S5.
Highlights.
High-throughput screen identified small molecules that increase PGC-1α acetylation.
Small molecules inhibit PGC-1α-dependent gluconeogenic activity.
SR-18292 suppresses HNF4α/PGC-1α gluconeogenic transcriptional function.
SR-18292 improves insulin sensitivity and reduces blood glucose in T2D mice.
Acknowledgments
K.S. was partially funded by a post-doctoral fellowship from the AHA (15POST22880002). C.D.J.T. was partially funded by a post-doctoral fellowship from the ADA (1-16-PDF-111). A.K.R. received a post-doctoral fellowship from NIH/NIDDK (F32 DK102293-01). The HTS was funded by a grant from NIH/NIDDK to the Broad Institute as a MLPCN Center (S.L.S) (U54HG005032). P.P. and P.R.G. were funded by the NIH/NIDDK (R24DK080261). G.I.S was funded by (NIH/NIDDK R01DK-40936 9) and 2U2CDK059635. P.P. was funded by R03DA032468, R01 DK069966 and the ADA (7-12-MN-68).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors contributions
The overall project was originally conceived by P.P. with initial technical discussions with J.E.D. and R.S. The experiments, except for the AlphaLisa high-throughput (HTS) experiments, were designed, performed and analyzed by K.S. and C.D.J.T. and aided with discussions and advise by P.P. HTS studies were designed, performed and analyzed by R.S., M.H., P.P.N., M.P., J.A.B. and J.P., aided with discussions and advise by S.L.S., J.E.D. and P.P. Small molecules synthesis and the chemistry was designed and performed by H.L., T.M.K. and P.R.G. Hyperinsulinemic-euglycemic clamp studies were designed and performed by G.I.S, J.P.C and R.P. Tail-vein injections of adenoviruses were designed and performed by U.O and J.L. Mass spectrometry experiments were designed and performed by M.P.J. and S.P.G. Manuscript was written by K.S and P.P with input from J.E.D, T.M.K, C.D.J.T, G.I.S. and A.K.R.
References
- Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nature reviews Molecular cell biology. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. Options for combination therapy in type 2 diabetes: comparison of the ADA/EASD position statement and AACE/ACE algorithm. The American journal of medicine. 2013;126:S10–20. doi: 10.1016/j.amjmed.2013.06.009. [DOI] [PubMed] [Google Scholar]
- Bodo M, Dobrossy L, Rahoty P, Daubner K. Diagnosis of carcinoma of the breast by aspiration biopsy cytology. Archiv fur Geschwulstforschung. 1977;47:624–626. [PubMed] [Google Scholar]
- Burgess SC, He T, Yan Z, Lindner J, Sherry AD, Malloy CR, Browning JD, Magnuson MA. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab. 2007;5:313–320. doi: 10.1016/j.cmet.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SC, Leone TC, Wende AR, Croce MA, Chen Z, Sherry AD, Malloy CR, Finck BN. Diminished hepatic gluconeogenesis via defects in tricarboxylic acid cycle flux in peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha)-deficient mice. The Journal of biological chemistry. 2006;281:19000–19008. doi: 10.1074/jbc.M600050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrington AD. Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48:1198–1214. doi: 10.2337/diabetes.48.5.1198. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. The Medical clinics of North America. 2004;88:787–835, ix. doi: 10.1016/j.mcna.2004.04.013. [DOI] [PubMed] [Google Scholar]
- DiTullio NW, Berkoff CE, Blank B, Kostos V, Stack EJ, Saunders HL. 3-mercaptopicolinic acid, an inhibitor of gluconeogenesis. The Biochemical journal. 1974;138:387–394. doi: 10.1042/bj1380387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy JE, Gerhart-Hines Z, Puigserver P. Nutrient-dependent acetylation controls basic regulatory metabolic switches and cellular reprogramming. Cold Spring Harbor symposia on quantitative biology. 2011;76:203–209. doi: 10.1101/sqb.2012.76.010843. [DOI] [PubMed] [Google Scholar]
- Dominy JE, Jr, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan HB, Feldman J, Pierce K, Mostoslavsky R, et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Molecular cell. 2012;48:900–913. doi: 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estall JL, Kahn M, Cooper MP, Fisher FM, Wu MK, Laznik D, Qu L, Cohen DE, Shulman GI, Spiegelman BM. Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression. Diabetes. 2009;58:1499–1508. doi: 10.2337/db08-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. The American journal of clinical nutrition. 2011;93:884S–890. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- He L, Wondisford FE. Metformin action: concentrations matter. Cell Metab. 2015;21:159–162. doi: 10.1016/j.cmet.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. The New England journal of medicine. 2008;359:1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF, Landau BR, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. The Lancet. 2014;383:1068–1083. doi: 10.1016/S0140-6736(13)62154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Saraf A, Florens L, Washburn M, Workman JL. Gcn5 regulates the dissociation of SWI/SNF from chromatin by acetylation of Swi2/Snf2. Genes & development. 2010;24:2766–2771. doi: 10.1101/gad.1979710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510:547–551. doi: 10.1038/nature13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lustig Y, Ruas JL, Estall JL, Lo JC, Devarakonda S, Laznik D, Choi JH, Ono H, Olsen JV, Spiegelman BM. Separation of the gluconeogenic and mitochondrial functions of PGC-1{alpha} through S6 kinase. Genes & development. 2011;25:1232–1244. doi: 10.1101/gad.2054711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–1327. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr. 2012;3:286–294. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Zhang XM, Zhang D, Kumashiro N, Camporez JP, Cline GW, Rothman DL, Shulman GI. Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Med. 2014;20:759–763. doi: 10.1038/nm.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Ramnanan CJ, Edgerton DS, Rivera N, Irimia-Dominguez J, Farmer B, Neal DW, Lautz M, Donahue EP, Meyer CM, Roach PJ, et al. Molecular characterization of insulin-mediated suppression of hepatic glucose production in vivo. Diabetes. 2010;59:1302–1311. doi: 10.2337/db09-1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravnskjaer K, Hogan MF, Lackey D, Tora L, Dent SY, Olefsky J, Montminy M. Glucagon regulates gluconeogenesis through KAT2B- and WDR5-mediated epigenetic effects. J Clin Invest. 2013;123:4318–4328. doi: 10.1172/JCI69035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, Spiegelman BM. 2003 doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 100:4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson LC, Pollack LA. Therapy Insight: influence of type 2 diabetes on the development, treatment and outcomes of cancer. Nat Clin Prac Oncol. 2005;2:48–53. doi: 10.1038/ncponc0062. [DOI] [PubMed] [Google Scholar]
- Rizza RA. Pathogenesis of Fasting and Postprandial Hyperglycemia in Type 2 Diabetes: Implications for Therapy. Diabetes. 2010;59:2697–2707. doi: 10.2337/db10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Haas W, Gygi SP, Puigserver P. Cdc2-like kinase 2 is an insulin-regulated suppressor of hepatic gluconeogenesis. Cell Metab. 2010;11:23–34. doi: 10.1016/j.cmet.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Letters. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Beddow SA, Iwasaki T, Zhang XM, Chu X, Still CD, Gerhard GS, Shulman GI. Fasting hyperglycemia is not associated with increased expression of PEPCK or G6Pc in patients with Type 2 Diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12121–12126. doi: 10.1073/pnas.0812547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Wang JC, Scott DK, Granner DK. Transcription activation by the orphan nuclear receptor, chicken ovalbumin upstream promoter-transcription factor I (COUP-TFI). Definition of the domain involved in the glucocorticoid response of the phosphoenolpyruvate carboxykinase gene. The Journal of biological chemistry. 2000;275:3446–3454. doi: 10.1074/jbc.275.5.3446. [DOI] [PubMed] [Google Scholar]
- Sun C, Wang M, Liu X, Luo L, Li K, Zhang S, Wang Y, Yang Y, Ding F, Gu X. PCAF Improves Glucose Homeostasis by Suppressing the Gluconeogenic Activity of PGC-1α. Cell Reports. 2014;9:2250–2262. doi: 10.1016/j.celrep.2014.11.029. [DOI] [PubMed] [Google Scholar]
- Yasgar A, Jadhav A, Simeonov A, Coussens NP. AlphaScreen-Based Assays: Ultra-High-Throughput Screening for Small-Molecule Inhibitors of Challenging Enzymes and Protein-Protein Interactions. Methods Mol Biol. 2016;1439:77–98. doi: 10.1007/978-1-4939-3673-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. High throughput screens to identify small molecules that can potentially inhibit gluconeogenesis. Related to Figure 1.
(A) Work flow of screen starting with a high-throughput AlphaLisa screen through secondary screens.
(B) Dose response acetylation curves of final hits presented in Figure 1E.
(C) Dose response curves showing estimated inhibitory activity toward Pck1 and G6pc gene expression following forskolin treatment. To calculate estimated inhibitory AC50 values (eAC50) that were comparable between compounds, curves were forced through 100% inhibitory activity (which was determined to be the DMSO control). AC50 were calculated as described in STAR methods.
(D) Chemical structures of screen hits that are able to reduce glucose production in hepatocytes.
Figure S2. Characterization of SR-18292 effects in hepatocytes. Related to Figure 2.
(A) Western blot analysis of CREB phosphorylation (S133) and total TORC2 upon glucagon stimulation (200nM) and SR-18292 treatment (20μM). Glucagon-stimulated dephosphorylation of TORC2 results in a shift in gel band.
(B) Western blot analysis of Akt phosphorylation (S473) upon insulin stimulation (100nM) and SR-18292 treatment (20μM).
(C) Culture medium was collected from hepatocytes treated with either SR-18292 or cisplatin for 18 hrs. Cellular viability was assessed by using Toxilight bioassay kit (Lonza) (n=3) and by MTT assay (n=4) (D). ***P<0.001
(E) Total protein levels are not affected by SR-18292 treatment. n=3.
(F) PEPCK protein levels are reduced in hepatocytes treated with SR-18292 and infected with Ad-PGC-1α.
(G) mRNA expression level of Ppargc1a, Pck1 and G6pc following 18 h treatment with the SR-18292 (20μM). Hepatocytes were infected with Ad-PGC-1α to induce expression of Pck1 and G6pc genes and to induce glucose release from either Pyruvate/lactate (2mM/20mM) or glycerol (10mM) (H).
Figure S3. Mechanistic studies with SR-18292. Related to Figure 3.
(A) HAT activity assay showing that the acetyl transferase activity of GCN5 toward H3 peptide is not increased in cells treated with SR-18292. n=3.
(B) HAT activity assay showing that the acetyl transferase activity of GCN5 toward H3 peptide is not affected by in-vitro treatment with SR-18292. n=3.
(C) Interaction of PGC-1α with ectopically expressed GCN5 is increased after treatment with SR-18292. IP, immunoprecipitation.
(D) U-2 OS cells were infected with Ad-PGC-1α and Ad-GCN5 and treated with SR-18292 (10μM) for 18 h. Mass spectrometry analysis was performed to identify lysine residues whose acetylation is being increased following treatment.
(E) Promoter occupancy of FoxO1 over Pck1 and G6pc promoter regions is not affected by drug treatment. n=3. **, P<0.01. Related to Figure 3.
Figure S4. Structure-Activity Relationship studies with SR-18292 analogs. Related to Figures 2 and 3.
(A) Chemical structure of several analogs of SR-18292.
(B) mRNA expression level of Ppargc1a, Pck1 and G6pc following 18 h treatment with the indicated compounds (20μM). Glucagon stimulation (200nM) was used to induce the expression of Pck1 and G6pc genes.
(C) mRNA expression level of Ppargc1a, Pck1 and G6pc following 18 h treatment with the indicated compounds (20μM). Hepatocytes were infected with Ad-PGC-1α to induce expression of Pck1 and G6pc genes.
Data presented as mean +/− S.E.M. n=3, one-way ANOVA with Dunnet posttest.
**P<0.01, ***P<0.001.
(D) Western blot analysis to detect acetylation status of PGC-1α following treatment with SR-18292 (10μM) and SR-19138 (10μM). U-2 OS cells were infected with Ad-PGC-1α and Ad-GCN5 and treated with the indicated compound for 18 h. PGC-1α was immunoprecipitated using FLAG beads. IP, immunoprecipitation.
Figure S5. Characterization of in-vivo effects of SR-18292. Related to Figures 4 and 5.
(A) Plasma insulin levels after GTT and PTT (B) presented in Fig. 4H and 5A, respectively.
*, P<0.05, **, P<0.01.
(C) Serum lactate and β-hydroxybutyrate levels are not altered with SR-18292 treatment. Mice were fed HFD for 4 weeks. n=7.
(D) mRNA expression level of lipid metabolic genes is not changed with SR-18292 treatment. Mice were fed HFD for 13 weeks. n=10.
(E) Serum TG, free fatty acids and cholesterol levels are not altered with SR-18292 treatment. Mice were fed HFD for 13 weeks. NEFA, non-esterified fatty acids. n=10.
(F) mRNA expression level of mitochondrial genes in liver is not changed with SR-18292 treatment. Mice were fed HFD for 13 weeks. n=10.
(G) Mice fed HFD for 6 weeks were injected daily with SR-18292 for a total of 14 days and body weight was monitored during the treatment. Mice were fasted at day 3 and 13 and fasting blood glucose was measured at day 4 and 14 (H). Fed blood glucose was also measured at day 10. n=10, *P<0.05.
(I) Serum ALT and AST levels were not altered after 14 days of SR-18292 treatment. n=10
(J) H&E staining of liver slices obtained from livers harvested after 14 days of SR-18292 treatment. 3 representative images are presented for each group. All samples (n=10/vehicle, n=10/SR-18292) were assessed by an expert mouse pathologist and no sign of toxicity was observed. scale bar, 50μm.
(K) Expression level of inflammatory genes from livers harvested after 14 days of SR-18292 treatment. n=10
(L) Liver triglycerides and cholesterol levels are not altered after 14 days of SR-18292 treatment. n=10
(M) Serum lactate, β-hydroxybutyrate and cholesterol levels are not altered after 14 days of SR-18292 treatment. n=10
Table S1. Serum biochemistry parameters following treatment with SR-18292. Related to Figures 4 and 5.
Table S2. A list of primers used for ChIP assays. Related to Figures 3 and S3.
Table S3. A list of primers used for qPCR analysis. Related to Figures 2, 4, S2, S4 and S5.
Data S1. HRMS of SR-18292 and its intermediates. Related to Figures 2, 3, 4, 5, S2, S3, S4 and S5.