

ABSTRACT
Environmental stress contributes to the development of psychiatric disorders, including posttraumatic stress disorder and anxiety. While even acute stress alters gene expression, the molecular mechanisms underlying these changes remain largely unknown. 5-hydroxymethylcytosine (5hmC) is a novel environmentally sensitive DNA modification that is highly enriched in the brain and is associated with active transcription of neuronal genes. Here we examined behavioral and molecular alterations in adult mice that experienced an early-life stress before weaning (postnatal day 12 to 18) and found anxiety-like behaviors in adult female mice that were accompanied by correlated disruptions of hypothalamic 5hmC and gene expression in 118 genes, revealing potentially functional 5hmC (i.e., gene regulation). These genes are known and potentially novel stress-related targets, including Nr3c2, Nrxn1, Nfia, and Clip1, that have a significant enrichment for neuronal ontological functions, such as neuronal development and differentiation. Sequence motif predictions indicated that 5hmC may regulate gene expression by mediating transcription factor binding and alternative splicing of many of these transcripts. Together, these findings represent a critical step toward understanding the effects of early environment on the neuromolecular mechanisms that underlie the risk to develop anxiety disorders.
KEYWORDS: 5-hydroxymethylcytosine, anxiety, DNA methylation, early-life stress, epigenetics, gene expression
Introduction
The development of anxiety and depressive disorders is often exacerbated by environmental stress experienced during critical periods in development.1 Stress activates the hypothalamic-pituitary-adrenal (HPA) axis, which consequently initiates the secretion of corticotropin releasing hormone (CRH), adrenocorticotropic hormone (ACTH), and glucocorticoids. Thus, there is a particular importance for the hypothalamus in response to stress, as it is the brain structure that initiates the response and retains receptors for CRH to provide a method of feedback inhibition to terminate the stress response: efficient binding of glucocorticoids to hypothalamic receptors is crucial for a healthy stress response by the hypothalamus.2 Environmental stress strongly impacts neuronal activity in the hypothalamus and can influence the development of neuropsychiatric disorders.3,4 Environmentally sensitive epigenetic modifications (e.g., DNA methylation) disrupted by stress and traumatic experiences are linked to the origins of psychiatric disorders,5 and an association between altered DNA methylation in the hypothalamus has been shown to modulate stress-related diseases of patients exposed to an early-life environmental stress.6 Thus, further study of environmentally sensitive molecular mechanism(s) is warranted in the hypothalamus during critical developmental periods.
Conventional studies of DNA methylation have focused on the presence or absence of a methyl group on cytosine that is termed 5-methylcytosine (5mC). This DNA modification has a role in experience-dependent plasticity such as an anxious temperament disposition, which is a key risk factor in the development of anxiety and depression.7,8 Moreover, 5mC has been associated with neuropsychiatric disorders, such as schizophrenia, bipolar disorder, and PTSD.9-12 5mC can be oxidized to 5-hydroxymethylcytosine (5hmC) following exposure to environmental stimuli (e.g., oxidative stress).13 5hmC is enriched in the brain (∼10-fold more than in peripheral tissues) and is associated with the regulation of neuronal activity.14-18 Variations in 5hmC have been reported in several neurologic disorders (e.g., Rett syndrome and autism) and neurodegenerative diseases (e.g., Huntington's and Alzheimer).19-24 In addition, we recently reported a sex-specific genome-wide disruption of 5hmC in mice exposed to acute stress followed by a one-hour recovery.25,26 The differential 5hmC significantly resided in known stress-related genes, which served to validate a role for 5hmC in stress response and potentially in the development of neuropsychiatry disorders. Collectively, 5hmC has become a significant focus of neuroscience research due to growing evidence that suggests its critical role in the development of psychiatric disorders, including depression and schizophrenia.27-29
In this current study, we used an established chemical labeling and affinity purification method coupled with high-throughput sequencing technology to generate an unbiased genome-wide profile of hypothalamic 5hmC from adult female mice that were chronically stressed early in life and exhibited anxiety-like behaviors. Integration with transcriptome data generated from the same mice and tissue revealed stress-induced hydroxymethylation that was correlated with differential gene expression and, perhaps more importantly, with isoform production. Together, these findings provide new insight into early-life influences that could epigenetically modulate neuronal circuits that function to shape adult behaviors.
Results
Early-life postnatal stress results in altered adult behaviors in female mice
To test the long-lasting effects of an early-life environmental stress on the development of aberrant adult behaviors, we subjected pregnant females [embryonic day (E) 0.5 to E6.5] and young mice [postnatal day (P) 12 to P18] to early-life stress (Materials and Methods). These developmental time-points were chosen because they have previously been shown to be sensitive to the exposure to environmental stress30, or occur during neuronal migration, following sensory influx and increased behavior interaction. At 3 months of age, prenatal stressed (Pre-S), postnatal stressed (Post-S), and non-stressed (non-S) mice were randomly selected for behavioral testing or sacrificed for molecular analysis. Adult female mice from all behavioral groups (Pre-S, Post-S, and non-S) showed no significant differences in a variety of behavioral tests including open field (time spent in the inner circle: Pre-S, 65 ± 7; Post-S, 60 ± 5; and non-S, 53 ± 4 and number of entries into the inner circle: Pre-S, 24 ± 2; Post-S, 24 ± 2; and non-S, 26 ± 3; P-value > 0.05; Table 1), elevated plus maze (time spent in open arm: Pre-S, 144 ± 7; Post-S, 160 ± 10; and non-S, 129 ± 12; P-value > 0.05 and time spent in closed arm: Pre-S, 217 ± 6; Post-S, 191 ± 11; and non-S, 232 ± 7; P-value > 0.05; Table 1), and forced swim test (time spent floating: Pre-S, 144 ± 7; Post-S, 128 ± 5, and non-S, 133 ± 8, P-value > 0.05; Table 1). However, when the female mice were tested for anxiety in the light and dark box the Post-S mice showed significant alteration in adult behaviors (Fig. 1). The female Post-S mice spent significantly less time in the light compartment of the light and dark box compared with non-S female mice (time spent in the light side: Pre-S, 311 ± 13; Post-S, 298 ± 3; and non-S, 334 ± 17; Kruskal-Wallis One Way ANOVA showed a difference between the groups for time spent in the light compartment, x2 (2) = 6.237, P-value < 0.05; Dunn's multiple comparison indicated a statistical difference between Post-S and non-S, P-value < 0.05). Also, the Post-S group had a significant increase in the number of entries into the dark compartment of the apparatus compared with non-S (number of entries: Pre-S, 34 ± 2; Post-S, 45 ± 2; and non-S, 35 ± 2; Kruskal-Wallis One Way ANOVA showed a difference between the groups for number of entries, x2 (2) = 11.652, P-value < 0.05; Dunn's multiple comparison indicated a statistical difference between Pre-S and non-S, P-value < 0.05). Notably, adult male mice from all behavioral groups (Pre-S, Post-S, and non-PS) showed no significant behavior differences in any of these behavioral tests (Table 1). Together, these data suggest that an early-life stress just before weaning results in stable sex-specific behavioral changes related to anxiety.
Table 1.
Results from the behavioral tests conducted on female and male adult mice.
| Behavioral Test | Female |
Male |
||||
|---|---|---|---|---|---|---|
| Pre-S | Post-S | Non-S | Pre-S | Pre-S | Non-S | |
| Open Field | ||||||
| • Time spent inner circle | 65.7 ± 7.9 | 60.7 ± 5 | 53.9 ± 4 | 66 ± 4 | 61 ± 6 | 69 ± 6 |
| • Number of entries to inner circle | 24.9 ± 2.6 | 24 ± 2 | 26 ± 3 | 25 ± 1 | 23 ± 1 | 26 ± 2 |
| Light/Dark Box | ||||||
| • Time spent in the light compartment | 311.6 ± 13.6 | 298.5 ± 3* | 334.1 ± 17 | 323.6 ± 10.7 | 304 ± 7 | 317.5 ± 12 |
| • Number of entries into the dark compartment | 34 ± 2 | 45.7 ± 2* | 35.7 ± 2 | 34.6 ± 2.8 | 35.5 ± 1.7 | 35.9 ± 0.9 |
| Elevates Plus Maze | ||||||
| • Time spent in open arm | 144 ± 7 | 160 ± 10 | 129 ± 12 | 154 ± 6 | 186 ± 6 | 165 ± 12 |
| • Time spent in closed arm | 217 ± 6 | 191 ± 11 | 232 ± 7 | 204 ± 6 | 172 ± 7 | 186 ± 12 |
| Forced Swim Test | ||||||
| • Time spent floating | 144 ± 7 | 128 ± 5 | 133 ± 8 | 140 ± 10 | 136 ± 9 | 130 ± 6 |
One-way ANOVA, P-value < 0.05
Figure 1.
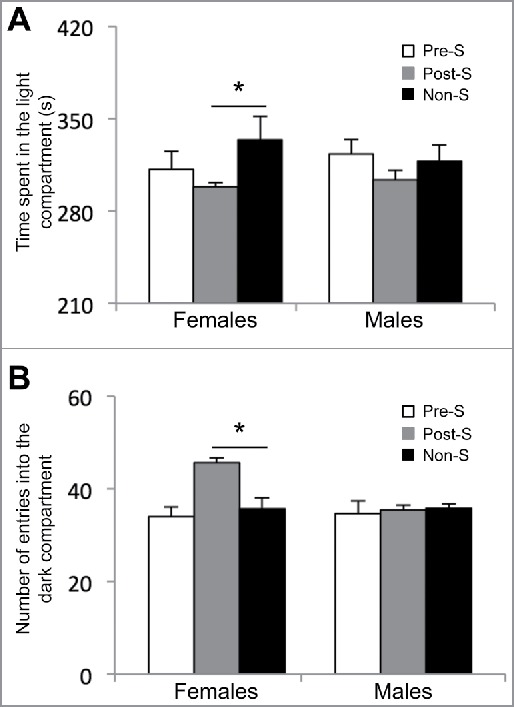
Results from the light and dark box test on female adult mice. (A) The amount of time spent in the light compartment of the apparatus in seconds (s) and (B) the number of entries into the dark compartment of the apparatus for the prenatal stressed (Pre-S), postnatal stressed (Post-S), and non-stressed (Non-S) mice. An asterisk denotes a significant difference (P-value < 0.05).
Stable disruption of 5hmC in female mice following postnatal stress
To determine the effect of postnatal stress on the genome-wide distribution of 5hmC in female mice, we used an established chemical labeling and affinity purification method, coupled with high-throughput sequencing technology.14,25,26,31 Age-matched Post-S and non-S mice were sacrificed at 3 months of age as independent biologic replicates. DNA sequences containing 5hmC were enriched from hypothalamic genomic DNA and high-throughput sequencing technology yielded approximately 17 to 35 million uniquely mapped reads for each genome (Fig. S1a; Materials and Methods). Significant accumulations of uniquely mapped reads represent hydroxymethylated regions throughout the mouse genome. Differentially hydroxymethylated regions (DhMRs) associated with stress were identified in both experimental and control groups (Materials and Methods). DhMRs found in experimental mice (absent in the control mice) were classified as hyper-DhMRs and DhMRs absent in experimental mice (present in control mice) were classified as hypo-DhMRs. A total of 508 hyper- and 531 hypo-DhMRs were identified and these loci were distributed across all chromosomes, with a noticeable depletion on the X chromosome (Fig. 2a; Fig. S1b; data set 1). These data suggest that stress-induced changes in 5hmC are stable throughout life.
Figure 2.
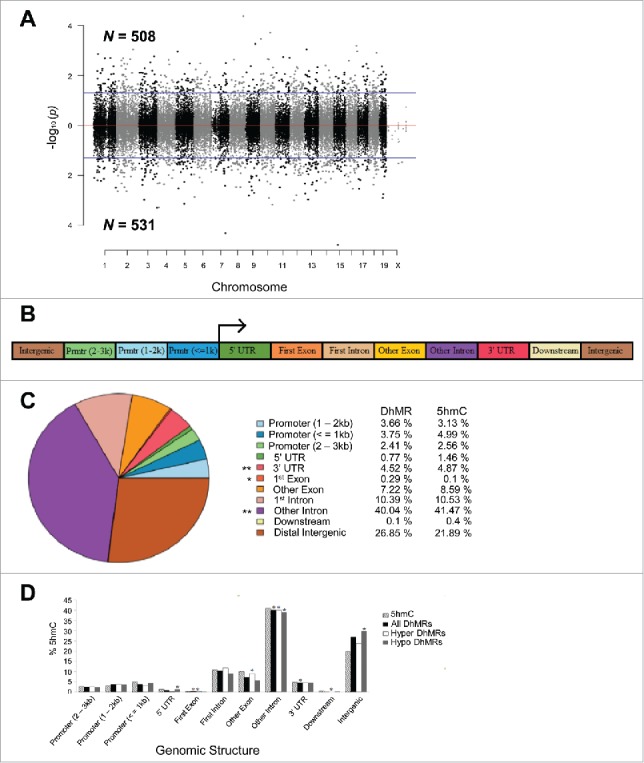
Characterization of DhMRs across standard genomic structures. (A) Modified Manhattan plot of early-life stress-associated DhMRs from the mouse hypothalamus reveals DhMRs to be distributed across the entire genome. Positively and negatively correlated DhMRs are displayed with the -log10 of the P-value. Significant DhMRs are displayed outside the purple lines (P-value < 0.05), while all DhMRs alternate between black and gray to indicate each chromosome. (B) Schematic of the standard genomic structures investigated. DhMR and all 5hmC data were annotated to the following genomic features: 3 promoter (Prmtr) regions located 2–3 kb, 1–2 kb, and up to 1 kb upstream (≤ 1 kb) of the transcription start site; the 5′ untranslated region (UTR); first exon; first intron; other exon; other intron; 3′ UTR; up to 3 kb downstream of transcription end site (≤ 3 kb); and the intergenic regions (> 3 kb upstream or downstream of nearest gene). (C) Pie chart displaying the proportion of all DhMRs falling into each investigated genomic structure. Structures containing a significant over- or under-representation of DhMRs when compared with all 5hmC peaks are indicated (*), which were determined by permutation testing (P-value < 0.05). Columns indicate the percent of all 5hmC peak data (5hmC) and DhMRs in each genomic structure shown. (D) A breakdown of hyper- and hypo-DhMRs in each genomic structure. The percent distribution (y-axis) of all 5hmC peak data (striped), DhMRs (black), hyper-DhMRs (white), and hypo-DhMRs (gray) in each genomic structure is shown. Significant over- and under-representation of DhMRs are indicated (*) for each structure (P-value < 0.05).
Specific regions of the genome are differentially methylated based on the biologic functions of the genes contained within the genomic region; thus, we used permutation testing to determine if the DhMRs are specific to certain regions of the genome by comparing the proportions of DhMRs to the proportions of total detected hydroxymethylated regions in all mice for each chromosome (Materials and Methods). This analysis revealed that chromosomes 9 and 17 have disproportionately less DhMRs than would be expected by chance alone while chromosomes 12 and 18 have disproportionately more DhMRs (Fig. S1b; P-value < 0.05). Together, these data suggest that certain chromosomes may be more susceptible to stable changes in DNA hydroxymethylation following early-life stress.
The DhMRs were next annotated to the nearest genomic structure, which included the following gene-centric regions: distal promoter region (2–3 kb upstream), central promoter region (1–2 kb upstream), proximal promoter region (≤ 1 kb upstream), 5′ UTR, first exon, first intron, other exon, other intron, 3′ UTR, downstream (≤ 3 kb), intergenic (> 3 kb downstream) (Fig. 2b; Materials and Methods). The gross distribution of the data indicates that the largest proportion of DhMRs fall within intronic regions of the genome (∼50%). Permutation testing was conducted to identify over- or under-representation of DhMRs across the genomic structures relative to the total detected hydroxymethylation sites (Materials and Methods). This approach revealed significant over-representations of DhMRs within first exons and under-representations within intronic regions and 3′ UTRs (Fig. 2c; permutated P-value < 0.05). Partitioning the DhMRs into hyper- and hypo-DhMRs found that hyper-DhMRs were over-represented within exonic regions and under-representation within intronic regions and regions downstream of the nearest gene (Fig. 2d). In contrast, the hypo-DhMRs were over-represented within 5′ UTRs and intergenic regions and under-represented within intronic regions. Taken together, these data indicate that alterations to the hypothalamic hydroxymethylome reside within specific genic structures (Fig. 2d), suggesting that such changes brought about by early-life stress are not stochastic throughout the genome.
DhMR-associated genes
Annotation of DhMRs to genes revealed 880 genes with stable alterations in hydroxymethylation following early-life stress, including 124 genes that contain multiple DhMRs and 474 genes that were hyper-hydroxymethylated and 482 genes that were hypo-hydroxymethylated (P-value < 0.05; data set 2). Seventy-six genes were found to have both hyper- and hypo-hydroxymethylated regions, meaning these genes contained regions with both increases (hyper) and decreases (hypo) in 5hmC following early-life stress (data set 2). Notably, the average distance between DhMRs annotated to the same gene was >170 kb, suggesting that when found associated to the same gene DhMRs are not located near each other and may have unique roles in response to early-life stress.
Since these changes in 5hmC were identified following a stress paradigm, we compared DhMR-associated genes with genes known to play a role in stress (Materials and Methods). This comparison found a significant overlap of DhMR-associated genes (n = 338 of 880) with known stress-related genes (n = 3,786; Fig. 3a), indicating that 5hmC marks biologically relevant genes related to stress and supporting a molecular function for 5hmC in stress response. Furthermore, these findings revealed potentially novel stress-related genes in response to early-life stress (n = 542; Fig. 3a). To further characterize the genes and pathways altered by early-life stress, we examined the gene ontological (GO) functions of the 474 and 482 hyper- and hypo-DhMR-associated genes, respectively, and found a significant enrichment of neuronal/immunological-related GO terms for both groups of genes, including nervous system development, generation of neurons, and neurogenesis (Tables 2 and 3; data sets 3–5; Chi-square P-value < 0.01; GO FDR P-value < 0.05; Materials and Methods). Together, these data indicate that the 5hmC changes are stable throughout development and that early-life stress disrupts 5hmC on biologically relevant genes and pathways related to neurodevelopment.
Figure 3.
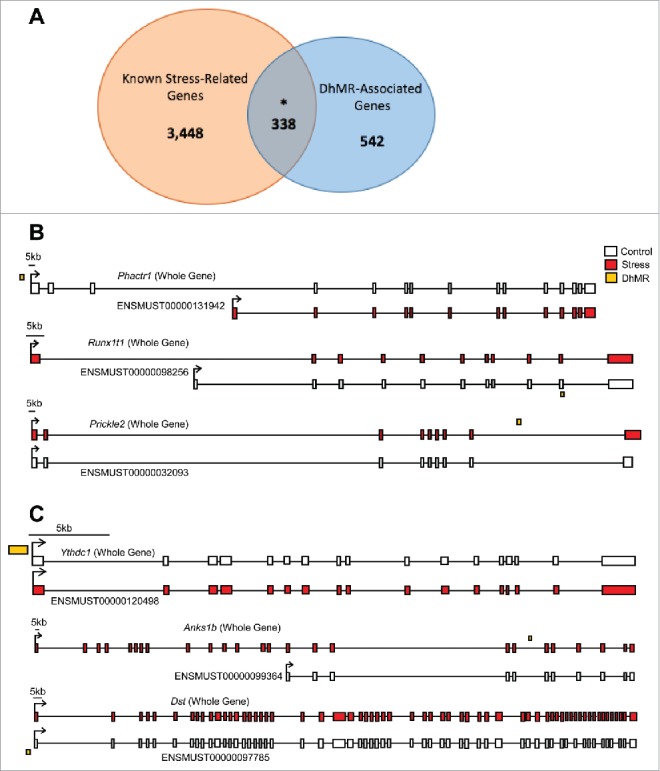
Annotation of DhMRs to genes. (A) Venn diagram displaying the proportion of DhMR-associated genes (blue; n = 880) overlapping with genes that have a known role in stress-response (yellow; n = 4,348). The overlap (n = 338) is significant, as indicated by and asterisk (Chi-square P-value < 0.001). (B) Gene schematics of the top 3 statistically significant differentially expressed isoforms from known stress-related genes containing a DhMR. Transcripts are depicted as either the whole-gene transcripts or differentially expressed isoforms from control (white) and stressed (red) mice. The relative genomic location of the DhMR (gold box) is shown, hyper-DhMRs are near stressed (red) mice transcripts and hypo-DhMRs are near control (white) mice transcripts. (C) Gene schematics of the top 3 statistically significant differentially expressed isoforms from genes containing a DhMR and that are known to play a role in psychiatric-disorders. Transcripts are depicted as either the whole-gene transcripts or differentially expressed isoforms from control (white) and stressed (red) mice. The relative genomic location of the DhMR (gold box) is shown, hyper-DhMRs are near stressed (red) mice transcripts and hypo-DhMRs are near control (white) mice transcripts. An asterisk indicates which exons have been either partially or completely excluded from the isoform shown.
Table 2.
Top 5 Gene Ontology Terms for Hyper-DhMRs.
| Gene Ontology Term | FDR P-value | **Gene Symbols |
|---|---|---|
| *Generation of neurons | 0.00000000509 | Mycbp2, Foxp1, Braf, Arhgef28, Ank3, Rnd2, Arsb, Atoh1, Prdm1, Bmp2, Bmpr1a, Runx2, Cdh11, Epha3, Epha4, Etv1, Gdnf, Gfra1, Lpar1, Hgf, Hhip, Ifrd1, Il2, Itgb1, Kif5b, Lama2, Man2a1, Mllt4, Trpm1, Map2, Ncam1, Ctnnd2, Ntrk3, Nr4a2, Enpp2, Prkg1, Prox1, Ptprd, Ptprm, Rheb, Robo1, Sema6a, Slit2, Sox14, Sox5, Cpeb3, Stxbp1, Arfgef1, Tcf4, Zeb1, Zfp521, Psd3, Tenm2, Tenm3, Kif20b, Prickle2, Rgma, Lsamp, Negr1, Slitrk3, Rgs6, Magi2, Park2, Corin, Golga4, Dfna5, Rapgef4, Gsk3b, Cdon, Sall1, Dleu2, Hmg20a, Zswim6, Agtpbp1, Rere, Tmem106b, Prmt3, Fryl, Ispd, Sdccag8, Fgf20, Sirt1 |
| *Nervous system development | 0.0000000186 | Mycbp2, Adgrb1, Foxp1, Braf, Arhgef28, Ank3, Rnd2, Arsb, Atoh1, Prdm1, Bmp2, Bmpr1a, Runx2, Cdh11, Epha3, Epha4, Etv1, Gdnf, Gfra1, Lpar1, Grik1, Hgf, Hhip, Hspg2, Ifrd1, Il2, Itgb1, Kif5b, Lama2, Man2a1, Mllt4, Trpm1, Map2, Ncam1, Nfia, Ctnnd2, Nrxn1, Nrxn3, Musk, Ntrk3, Nr4a2, Otx1, P2ry1, Enpp2, Prkg1, Prox1, Ptprd, Ptprm, Rheb, Robo1, Nptn, Sema6a, Slit2, Sox14, Sox5, Cpeb3, Stxbp1, Arfgef1, Tcf4, Zeb1, Alx1, Ttn, Zfp521, Psd3, Tenm2, Tenm3, Kif20b, Prickle2, Rgma, Mcph1, Rpgrip1l, Rbfox1, Lsamp, Negr1, Slitrk3, Rgs6, Magi2, Park2, Corin, Golga4, Dfna5, Cadm1, Rapgef4, Gsk3b, Cdon, Sall1, Dleu2, Hmg20a, Zswim6, Agtpbp1, Rere, Dpf3, Tmem106b, Prmt3, Fryl, Kif1bp, Ispd, Sdccag8, Hdac9, Fgf20, Sirt1 |
| *Neurogenesis | 0.0000000186 | Braf, Agtpbp1, Ank3, Arfgef1, Arhgef28, Arsb, Atoh1, Bmp2, Bmpr1a, Cdh11, Cdon, Corin, Cpeb3, Ctnnd2, Dfna5, Dleu2, Enpp2, Epha3, Epha4, Etv1, Fgf20, Foxp1, Fryl, Gdnf, Gfra1, Golga4, Gsk3b, Hgf, Hhip, Hmg20a, Ifrd1, Il2, Ispd, Itgb1, Kif20b, Kif5b, Lama2, Lpar1, Lsamp, Magi2, Man2a1, Map2, Mllt4, Mycbp2, Ncam1, Negr1, Nr4a2, Ntrk3, P2ry1, Park2, Prdm1, Prickle2, Prkg1, Prmt3, Prox1, Psd3, Ptprd, Ptprm, Rapgef4, Rere, Rgma, Rgs6, Rheb, Rnd2, Robo1, Runx2, Sall1, Sdccag8, Sema6a, Sirt1, Slit2, Slitrk3, Sox14, Sox5, Stxbp1, Tcf4, Tenm2, Tenm3, Tmem106b, Trpm1, Zeb1, Zfp521 |
| Cell Development | 0.0000000366 | Camk2d, Abi1, Agfg1, Akap6, Alx1, Ank3, Arfgef1, Arhgef28, Arid4a, Arsb, Atoh1, Bin3, Bmp2, Bmpr1a, Braf, Cdh11, Cdon, Cnot2, Col11a1, Cpeb3, Ctnnd2, Dleu2, Enpp2, Epha3, Epha4, Etv1, Fermt2, Foxp1, Fryl, Gdnf, Gfra1, Golga4, Gsk3b, Hdac9, Hgf, Hmg20a, Hspa2, Ifrd1, Il2, Impad1, Ispd, Itga4, Itgb1, Kif20b, Kif5b, Klf5, Lama2, Lims1, Lpar1, Lsamp, Magi2, Maml1, Man2a1, Map2, Megf10, Mllt4, Mycbp2, Ncam1, Negr1, Nr4a2, Ntrk3, Park2, Prdm1, Prickle2, Prkg1, Prmt3, Prox1, Ptprd, Ptprm, Rap1b, Rapgef4, Rdx, Rere, Rgma, Rgs6, Rheb, Rnd2, Robo1, Runx2, S1pr3, Sall1, Sema6a, Sirt1, Slc8a1, Slit2, Slitrk3, Smyd3, Sox14, Sox5, Stxbp1, Tcf4, Tenm2, Tenm3, Tmem106b, Trpm1, Trps1, Ttn, Zeb1, Zfp42, Zswim6 |
| *Neuron differentiation | 0.0000000366 | Mycbp2, Foxp1, Braf, Arhgef28, Ank3, Rnd2, Arsb, Atoh1, Prdm1, Bmp2, Runx2, Cdh11, Epha3, Epha4, Etv1, Gdnf, Gfra1, Lpar1, Hgf, Ifrd1, Il2, Itgb1, Kif5b, Lama2, Trpm1, Map2, Ncam1, Ctnnd2, Ntrk3, Nr4a2, Prkg1, Prox1, Ptprd, Ptprm, Robo1, Sema6a, Slit2, Sox5, Cpeb3, Stxbp1, Arfgef1, Tcf4, Zeb1, Zfp521, Psd3, Tenm2, Tenm3, Kif20b, Prickle2, Rgma, Negr1, Slitrk3, Rgs6, Magi2, Park2, Corin, Golga4, Dfna5, Rapgef4, Gsk3b, Cdon, Sall1, Dleu2, Hmg20a, Zswim6, Agtpbp1, Rere, Tmem106b, Prmt3, Fryl, Ispd, Fgf20, Sirt1 |
Indicates neuronal-related pathway as previously determined by Geigman et al., 2010
Bolded gene symbols indicate known stress-related genes from the GeneCards database.
Table 3.
Top 5 Gene Ontology Terms for Hypo-DhMRs.
| Gene Ontology Term | FDR P-value | **Gene Symbols |
|---|---|---|
| *Nervous system development | 0.0000141 | Bcr, Abl1, Rims2, Apc, Nr2f2, App, Atoh1, Bmp5, Runx2, Runx1, Cdh11, Chl1, Cntn1, Hapln1, Vcan, Dab1, Dclk1, Dct, Dst, Epha4, Fzd1, Gap43, Htr7, Kcnma1, Kdr, Lama2, Lrp6, Crb1, Nme7, Map2, Ncoa1, Nfia, Ctnnd2, Nrxn3, Otx1, Otx2, Pax3, Prkd1, Prkg1, Nlgn1, Ptprd, Ptprm, Ptpro, Pura, Robo1, Sema3c, Sema6a, Slit2, Sox11, Sox6, Stxbp3, Tcf4, Nav1, Lrrtm3, Tiam1, Twist1, Dpysl3, Unc5c, Ush2a, Zfp521, Btbd3, Picalm, Helt, Psd3, Dlg2, Tenm3, Lrrc7, Grin3a, Ppp1r9a, Grhl2, Map3k7, Sema4g, Pclo, Lsamp, Robo2, Luzp1, Fat4, Flrt2, Grxcr1, Ccdc141, Clstn2, Bzw2, Zswim6, Rere, Trim32, Ttll7, Nptxr, Lrrtm1, Kidins220, Stxbp5, Hdac9, Nedd4l, Rbfox2, Klf7, Adgrl2 |
| *Neuron Projection Morphogenesis | 0.0000466 | Abl1, Apc, App, Atoh1, Btbd3, Cdh11, Chl1, Ctnnd2, Dab1, Dclk1, Dst, Epha4, Flrt2, Gap43, Kdr, Kidins220, Klf7, Lama2, Map2, Nedd4l, Nlgn1, Otx2, Picalm, Ppp1r9a, Ptprd, Ptprm, Ptpro, Rbfox2, Rere, Rims2, Robo1, Robo2, Sema3c, Sema6a, Slit2, Stxbp5, Tiam1, Unc5c, Zswim6 |
| *Neuron differentiation | 0.0000546 | Abl1, Rims2, Apc, App, Atoh1, Bmp5, Runx2, Runx1, Cdh11, Chl1, Cntn1, Dab1, Dclk1, Dst, Epha4, Gap43, Htr7, Kcnma1, Kdr, Lama2, Lrp6, Crb1, Map2, Ncoa1, Ctnnd2, Otx2, Pax3, Prkd1, Prkg1, Nlgn1, Ptprd, Ptprm, Ptpro, Robo1, Sema3c, Sema6a, Slit2, Sox11, Tcf4, Tiam1, Dpysl3, Unc5c, Ush2a, Zfp521, Btbd3, Picalm, Helt, Psd3, Dlg2, Tenm3, Lrrc7, Grin3a, Ppp1r9a, Robo2, Fat4, Flrt2, Grxcr1, Zswim6, Rere, Trim32, Nptxr, Kidins220, Stxbp5, Nedd4l, Rbfox2, Klf7 |
| *Generation of neurons | 0.0000546 | Abl1, Rims2, Apc, Nr2f2, App, Atoh1, Bmp5, Runx2, Runx1, Cdh11, Chl1, Cntn1, Dab1, Dclk1, Dct, Dst, Epha4, Gap43, Htr7, Kcnma1, Kdr, Lama2, Lrp6, Crb1, Map2, Ncoa1, Ctnnd2, Otx2, Pax3, Prkd1, Prkg1, Nlgn1, Ptprd, Ptprm, Ptpro, Robo1, Sema3c, Sema6a, Slit2, Sox11, Tcf4, Nav1, Tiam1, Twist1, Dpysl3, Unc5c, Ush2a, Zfp521, Btbd3, Picalm, Helt, Psd3, Dlg2, Tenm3, Lrrc7, Grin3a, Ppp1r9a, Lsamp, Robo2, Fat4, Flrt2, Grxcr1, Zswim6, Rere, Trim32, Nptxr, Kidins220, Stxbp5, Nedd4l, Rbfox2, Klf7 |
| *Cell morphogenesis involved in neuron differentiation | 0.0000642 | Abl1, Apc, App, Atoh1, Cdh11, Chl1, Dab1, Dclk1, Dst, Epha4, Gap43, Lama2, Map2, Ctnnd2, Otx2, Nlgn1, Ptprd, Ptprm, Ptpro, Robo1, Sema3c, Sema6a, Slit2, Tiam1, Unc5c, Btbd3, Picalm, Ppp1r9a, Robo2, Flrt2, Rere, Kidins220, Stxbp5, Nedd4l, Rbfox2, Klf7 |
Bolded gene symbols indicate known stress-related genes from the GeneCards database
Discovery of potentially functional DhMRs
To understand the potential mechanism(s) for DhMRs resulting from early-life stress, we used RNA sequencing (RNAseq) to profile gene expression in the hypothalamus of the same mice and tissue surveyed for 5hmC (Materials and Methods). Comparison of transcript levels in experimental and control mice revealed 1,716 unique gene that are differentially expressed at the whole-gene and/or isoform level (P-value < 0.05; data set 6; Materials and Methods). Several of the differentially expressed genes previously were shown to be highly dynamic in response to stress, as we found upregulation of Gria1 and Bdnf, and downregulation of Nf3c1. Notably, significant changes in the expression of genes in the DNA methylation pathway were not found, such as Tet1–3 or Dnmt1–3a/b. Examination of the gene ontologies of these 1,716 differentially expressed genes found a significant enrichment of neuronal ontological terms, including regulation of neurogenesis, nervous system development, and neuron projection morphogenesis (Chi-squared P-value < 0.01; GO FDR P-value < 0.05; data sets 7–9; Materials and Methods). The identification of DhMRs that are associated with differentially expressed genes functions as an independent validation of stress-induced molecular changes of both 5hmC and gene expression; moreover, these associations reveal candidate functional DhMRs that may have a direct role in gene regulation. An overlay of the DhMR data with the differentially expressed (DE) genes and isoforms revealed 151 potentially functional DhMRs corresponding to 118 unique genes. While many of the genes associated to potentially functional DhMRs have documented roles in stress and/or stress-induced psychiatric related disorders, including Nr3c2, Nrxn1, Nfia, and Clip1, it is perhaps most interesting that several of the potentially functional DhMRs were associated with genes that express stress-induced isoforms (Fig. 3b and 3c; data set 6). In fact, DE isoforms were nearly 5-times more prevalent than DE whole-genes, further supporting a role for 5hmC in RNA processing such as alternative splicing, which also is consistent with previous findings.26,32,33 This finding prompted us to investigate whether there was an enrichment of potentially functional DhMRs at genomic structures that may facilitate alternative splicing (e.g., intron/exon boundaries). While a significant enrichment was not found at intron/exon boundaries, these DhMRs did contain branch point sequences, polyadenylation signals, and microRNA seed sequences (data not shown). Clearly further work is needed to understand the role of 5hmC in RNA processing. These data also revealed potentially functional DhMRs in genes that have an uncharacterized role in stress but have been implicated in mental illness, such as Anks1b, Kynu, Ythdc1, Foxp1, and Ptprd (DhMR, P-value < 0.05; DE, P-value < 0.05; Fig. 3; data set 6).34-36 Examination of the gene ontologies of these potentially functional DhMR-associated genes found a significant enrichment of neuronal ontological terms related to neuronal development and differentiation, as well as neuron apoptotic process and death (Chi-squared P-value < 0.01; GO FDR P-value < 0.05; data sets 10–12). Together, these data demonstrate that 5hmC is correlated with altered gene expression in neurogenesis and neuronal apoptotic pathways in response to early-life stress and suggest that differential 5hmC may play a role in stress-induced isoform production.
To gain a general understanding of 5hmC function, we next investigated if there was a consistent correlation between 5hmC abundance and stress-induced changes in gene expression and found that both hyper- and hypo-DhMRs were associated with up- and downregulation of gene expression. When we subsequently examined whether the location of the potentially functional DhMR was specifically correlated with up- or downregulation of gene expression, in regards to gene structure (e.g., promoter or gene body), we did not find any consistent correlations. Thus, there is not a clear relationship between stress-induced changes in hydroxymethylation level and the direction of altered gene expression.
Since we previously showed that 5hmC may regulate the expression of neuronal genes following exposure to stress by modulating the binding and/or function of transcription factors,26 we examined whether early-life stress-related DhMRs were enriched with transcription factor (TF) binding sites using the DREME (Discriminative Regular Expression Motif Elicitation) software.37 Forty-six TF sequence motifs (23 for hyper-DhMRs, 23 for hypo-DhMRs) significantly associated with the DhMRs (Figs. 4a and 4b). Many of the transcription factors predicted to bind to these motifs have links to stress-related behaviors and disorders, such as SP1 and Tfap2c, which are associated with schizophrenia, bipolar disorder, and major depressive disorder (see discussion and ref. 38, 39). Subsequent examination of each DhMR residing in a stress-induced differentially expressed gene revealed that nearly 90% of the potentially functional DhMR-associated genes contained at least one of the enriched transcription factor binding sequence motifs (data set 6). Moreover, when comparing the putative TF sequence motifs from the full set of potentially functional DhMRs, the majority of motifs overlapped with those found from either the hyper- or hypo-DhMRs (Fig. 4c). Together, these data support a role for 5hmC in the modulation of TF binding or function and the direct regulation of gene expression following an early-life stress.
Figure 4.
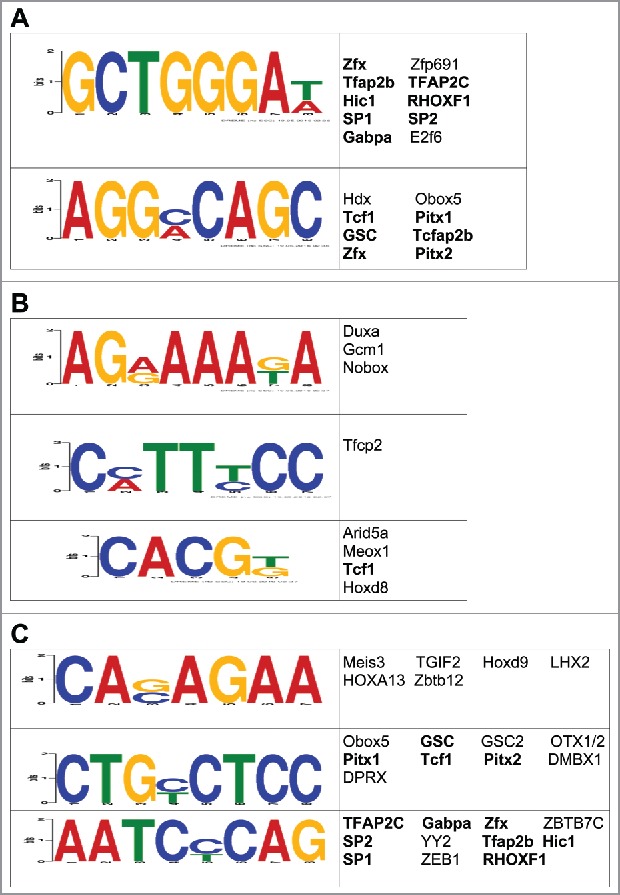
Characterization of the potential role(s) of DhMRs in gene expression. (A-C) Identification of DhMR-associated transcription factor sequence motifs that were predicted by the DREME suite (E-value < 10e-3) in hyper- (a), hypo- (b), and potentially functional (c) DhMRs. The putative transcription binding factors were predicted using SpaMo directly from the DREME suite and are shown next to each sequence motifs. Transcription factors associated with hyper- and/or hypo-DhMRs (a-b) that are shared between those found in the potentially functional DhMR sequences (c) are indicated by a bold font.
Discussion
The rediscovery of 5-hydroxymethylcytosine (5hmC) has raised new questions regarding the role of DNA methylation in response to stress. Here, we present evidence that an early-life stress results in stable changes in 5hmC that are correlated with gene expression. The majority of stress-induced changes in 5hmC were located within genic structures, predominantly within intronic regions (∼50%) that undergo a significant decrease in 5hmC in response to stress. The annotation of stress-induced changes in 5hmC to genes identified known and potentially novel stress-related genes, further validating a role for 5hmC in the response to stress. Together, these data provide insights into novel treatment targets for individuals that have already developed clinically significant anxiety disorders.
Furthermore, the presence of stable changes in 5hmC following an early-life stress that are linked to the development of anxiety-like behavior reveals a role for 5hmC in stress-induced behavioral outcomes. This association is further corroborated with the significant overlap of DhMR-associated genes and known stress-related genes (n = 338/880). Thus, the DhMRs reveal genes and pathways contributing to the stress-induced behavior changes, including genes involved in neurotransmission (e.g., glutamate receptors: Gria2, Grik1, Grik2, and Grin3a, and a serotonin receptor, Htr7) and ion channels, which are known to undergo distinct changes during development and may regulate the onset of anxiety-like behaviors (e.g., Kcnc2, Kcnj2, Kcnj3, and Cacnb4). Indeed, inhibitory interneurons undergo major transcriptional and electrophysiological changes between P7 and P40 in mice40 including Kcnc2 between P10 and P25. Finally, several members of the Kruppel-like factor (KLF) family of transcription factors, known to regulate gene expression, were found to contain DhMRs (e.g., Klf5, Klf6, Klf7, and Klf12). These genes encode proteins that are required for different stages of neuronal maturation in the developing brain.41 Together, these data suggest that the long-lasting disruption of 5hmC following early-life stress is targeting pathways known to function in the highly dynamic development of the nervous system. Furthermore, it is noteworthy that these findings implicate several potentially novel genes that may be associated with stress-response (at least a subset of n = 542). The stress-related potential of these genes is supported by the biologically relevant GO terms (e.g., neurogenesis and neuron projection morphogenesis). Further study of these novel stress-related genes may improve our understanding of stress response and recovery.
The generation of transcriptome data from the same mice and tissue reveals 678 differentially expressed whole-genes and 1,242 differentially expressed isoforms. This is consistent with a previous study that reported a significant number of differentially expressed isoforms associated with cocaine action.32 Together, these studies suggest a role for 5hmC in alternative splicing and isoform production. The overlay of hydroxymethylome and transcriptome data found genes with potentially functional DhMRs, which provides evidence for stable changes in 5hmC regulating stress-induced gene expression that is related to the observed behavioral outcomes. Here we identified 151 potentially functional DhMRs that are associated with 118 differentially expressed genes. Interestingly, the potentially functional DhMRs preferentially associated with differentially expressed isoforms (5-times over whole-genes), suggesting that 5hmC may have a more dynamic role in stress-induced expression of alternatively spliced isoforms rather than whole-genes. More than half of these potentially functional DhMR-associated genes (n = 61/118) are linked to stress, including Gdnf, Clip1, Dgkh, Nrxn1, Nfia, and Nr3c2, and several others have been implicated in developmental brain disorders such as autism, schizophrenia, and bipolar disorder (e.g., Nrxn1, Dgkh, and Gdnf).36,42,43 Since the genetic etiology of developmental brain disorders remain largely unknown, it remains possible that alterations in 5hmC following early-life stress may contribute to some forms of these disorders. Together, these data support a functional role for 5hmC in response to early-life stress and that stress-induced changes in 5hmC target genes in the brain that are influenced by the environment and are involved in the etiology of developmental brain disorders, including anxiety.
Since nearly 90% of the potentially functional DhMRs contained an enrichment of a transcription factor (TF) sequence motif(s), the functional role for stress-induced changes in 5hmC likely involves the modulation of TF binding/function. These enrichments located within DhMRs revealed the motifs of several TFs that are implicated in stress-related behaviors and disorders. Specificity Protein 1 (Sp1) is involved in many cellular processes, including cell differentiation and apoptosis. Recently it was shown that Sp1 is disrupted in schizophrenia and depression.44,45 The transcription factor AP-2 Gamma (Tfap2c) activates several developmental genes during retinoic acid-mediated differentiation in the development of the neural tube. Tfab2b has been implicated in the development of depression and bipolar disorders.46,47 Together, these data suggest that 5hmC may modulate transcription factor binding on developmentally important genes in the brain. It will be important that future studies verify this link and determine the nucleotide-specific 5hmC modifications in DhMRs that are necessary and sufficient for TF function.
The stress paradigm implemented in this study likely affects the development of emotion, cognition, and social behaviors.48 While all of these domains were not tested in this study, we did find disruptions in anxiety-related behaviors in the postnatal stressed mice. It is notable that we did not find altered adult behaviors in the prenatal stressed mice because it was previously reported that prenatal exposure to this early-life stress paradigm resulted in aberrant adult behaviors.49 However, it should be noted that the previously published study examined different behaviors using different behavioral tests, suggesting the prenatally stressed mice examined here may have cognitive and other stress coping deficits. Thus, it is warranted to examine the 5hmC levels in the prenatally stressed mice generated here, despite not finding anxiety-related deficits.
These studies parallel those of previous work that showed that acute stress disrupted 5hmC at known stress-related genes.26 It is notable that we find an overlap of 10 potentially functional genes between both studies and that the majority of these genes are known stress-related genes [8/10 (bold): Nfia, Map3k7, Ank2, Anks1b, Dock4, Erc2, Lpin1, Ndfip2, Nav1, and Abca8a]. While these data suggest that potentially functional stress-induced disruptions in 5hmC can be independent of stress paradigm and the timing of postnatal development exposure, paradigm and timing specific effects clearly exist. Nonetheless, the molecular factors influencing vulnerability and resilience to environmental stress clearly involve environmentally sensitive epigenetic mechanisms such as DNA methylation. The stable potentially functional stress-related variations in 5hmC identified here are associated with significant changes in gene expression, providing an independent validation of these data and a foundation that will aid in the future study of environmental factors affecting stress response and/or recovery. It will be important for future studies to characterize the developmental timing of behavior-related effects on the epigenome and transcriptome in a biologically relevant cell-type (e.g., GABAergic interneurons) to improve early therapeutic interventions. It is of great interest to harness the diagnostic and therapeutic power of these substrates toward healthy outcomes.
Materials and methods
Mice and stress paradigm
Mice were purchased from the Jackson laboratories (Bar Harbor, ME) and maintained on C57BL/6J background. The mice were housed under uniform conditions in a pathogen-free mouse facility with a 12- hour light/dark cycle. Food and water were available ad libitum. All experiments were approved by the University of Wisconsin — Madison Institutional Animal Care and Use Committee (M02529). To minimize for the stress of animal handling, all of the following were conducted by a single researcher: animal colony maintenance; administration of the stress paradigm; and behavioral tests.
For the prenatal stress cohort, a 3-month-old C57BL/6J male mouse was placed together with a virgin 3-month-old C57BL/6J female mouse at 6 p.m. (one hour before lights off); every morning before 9 a.m. (2 h after lights on) female mice were checked for vaginal copulation plug and separated from the male. Presence of a copulation plug denoted day 0.5 of gestation and the pregnant female was individually housed and given a cotton nestlet. At embryonic day (E) 0.5, pregnant females were randomly assigned to either variable stress (Pre-S) or to a non-stressed control (non-S) group. Pregnant mice assigned to the Pre-S group experienced a different daily stressor on each of the 7 d during early pregnancy (E0.5 to E6.5). These variable stressors included: 36 h of constant light, 15 min of fox odor (Cat# W332518) beginning 2 h after lights on, novel objects (8 marbles) exposure overnight, 5 min of restraint stress (beginning 2 h after lights on), novel noise (white noise, nature sound-sleep machine®, Brookstone) overnight, 12 cage changes during light period, and saturated bedding (700 mL, 23°C water) overnight.30,49 These mild stressors were previously published and were selected because they do not include pain or directly influence maternal food intake or weight gain. Litter sizes of less than 5 and more than 8 pups were removed from the experiment. Offspring were left undisturbed until weaning day (P18), at which time the mice were group housed with same sex mice. Females were left intact, and were not cycled.49 Finally, to minimize the effect of parent-to-offspring interaction per litter, 2 pups/litter were randomly selected for behavioral experiments and the other 2 littermates were selected for molecular experiments.
For the postnatal stress (Post-S) group, each litter was randomly assigned to either variable stress or a non-S group at P12. Mice assigned to the Post-S group experienced a different daily stressor on each of 7 d (P12 to P18). These were the same mild stressors that were received by the prenatal stress cohort and are described above. Litter sizes of less than 5 and more than 8 pups were removed from the experiment. At weaning day (P18) mice were group housed with same sex littermates and 2 pups/litter were randomly selected for behavioral experiments and the other 2 littermates were selected for molecular experiments.
Behavioral tests
Adult stressed and non-stressed mice (n = 10–12 for each sex and group, 12–17 weeks of age) were submitted to tests of anxiety and depression. All testing was performed during a period of 4 h in dark period (beginning 2 h after lights off). With one-week between tests, the same group of mice was subjected to the following sequence of behavioral tests: open field; light/dark box; elevated plus maze; and forced swim test. An experimenter who was blind to the animal's group scored video recordings of each test.
Open Field Test - The open field apparatus consisted of a circular arena (104 cm diameter). A marker was used to inscribe a smaller circle 25 cm from the walls. Each mouse was placed in the inner circle of the apparatus and allowed to explore for 10 min. The time spent in the center and numbers of entries to inner circle were scored for each mouse.
Light/Dark Box Test - The light/dark box test was performed in a rectangular box divided in 2 compartments (light and dark). The walls of the light and dark compartments were constructed of Plexiglas (same areas 319.3 cm2). A removable dark Plexiglas partition was used to divide the box into light and dark sides. Each animal was placed into the light side of the box, facing away from the dark side and allowed to explore both chambers of the apparatus for 10 min. The time spent in the light side and the number of entries into the dark compartment was scored for each mouse.
Elevated Plus Maze - The elevated plus maze consisted of 2 open and 2 closed arms, elevated 52 cm above the floor, with each arm projecting 50 cm from the center (a 10 × 10 cm area). Each mouse was individually placed in the center area, facing the open arm, and allowed to explore for 7 min. The measurements scored for each mouse were as follows: time spent in closed arms, time spent in open arms.
Forced Swim Test -The mice were individually placed into a glass cylinder (14-cm internal diameter, 38 cm high) filled with water (28-cm deep, 25–26 °C) for 6 min. During the last 4 min of the test, the time spent floating was scored for each mouse. Floating was defined as immobility or minimal movements necessary to maintain the head above the water.
Statistical analysis for behavioral tests
Data is reported as mean ± the standard error of the mean (SEM) Homogeneity of variance was assessed using the Levene test and the normal distribution of the data was tested by the Shapiro-Wilk test. One-way ANOVA was used to detect differences within treatment (Pre-S, Post-S, and non-S) for time spent in the inner circle and number of entries into that circle (Open field test); time spent in closed arm and time spent in open arm (Elevated plus maze); time spent in light compartment and number of entries into the dark compartment (Light/Dark box test) and time spent floating (Forced swim test). Post hoc analysis was performed using a Bonferroni correction. Kruskal-Wallis One Way ANOVA followed by Dunn's post-hoc test was performed when the data did not meet the criteria above.
Tissue acquisition and DNA/RNA extractions
The long-lasting effects of early-life stress on DNA methylation and gene expression was obtained from postnatally stressed and non-stressed 3-month old female mice (n = 4 per group). Mice were killed (2 h after lights on) and whole brains were extracted and immediately flash frozen in 2-methylbutane and dry ice. Hypothalamus tissue was excised by micropunch (0.37 to −2.53 mm posterior to bregma) and approximately 30 mg of tissue was homogenized with glass beads (Sigma) and DNA/RNA were extracted using AllPrep DNA/RNA mini kit (Qiagen).
5hmC enrichment of genomic DNA
Chemical labeling-based 5hmC enrichment was described previously.17,25,31 Briefly, a total of 10 μg of hypothalamus DNA was sonicated to 300 bp and incubated for 1 h at 37°C in the following labeling reaction: 1.5 μl of N3-Uridine diphosphate Glucose (2 mM); 1.5 μl β-Glucosyltransferase (β-GT; 60 μM); and 3 μl of 10× β-GT buffer, in a total of 30 μl. Biotin was added and the reaction was incubated at 37°C for 2 h before capture on streptavidin-coupled Dynabeads (Invitrogen, 65001). Enriched DNA was released from the beads during a 2-hour incubation at room temperature with 75 mM Dithiothreitol (DTT; Invitrogen, 15508013), which was removed using a Bio-Rad column (Bio-Rad, 732–6227). Capture efficiency was approximately 5–7% for each sample.
Library preparation and high-throughput sequencing of genomic DNA
5hmC-enriched libraries were generated using the NEBNext ChIP-Seq Library Prep Reagent Set for Illumina sequencing, according to the manufacturer's protocol. Briefly, the 5hmC-enriched DNA fragments were purified after the adaptor ligation step using AMPure XP beads (Agencourt A63880). An Agilent 2100 BioAnalyzer was used to quantify the amplified library DNA and 20-pM of diluted libraries were used for sequencing. A 50-cycle single-end sequencing reaction, which was performed by Beckman Coulter Genomics, generated 50 nucleotides from each read. Image processing and sequence extraction were done using the standard Illumina Pipeline.
Sequence alignment of 5hmC data and peak calling
Alignment of ChiP-seq data was performed using Bowtie v0.12.750. Reads were aligned to the NCBI37v1/mm9 genome by using the following parameters in Bowtie: only uniquely mapping reads with no more than 2 mismatches throughout the entire read that were within the ‘best stratum’ were kept for further analysis for each sample. Fragment lengths were estimated using R package SPP for each separate sample.51 To identify peaks for control and experimental samples, R package MOSAiCS was used.52 Using MOSAiCS, each sample was separately analyzed using default parameters with the exception of the following: bin size was increased to 300 bp, fragment length as estimated by SPP, capping at most 5 reads starting at the same nucleotide, FDR cutoff of 0.05, ‘matchLow’ background estimation, a maximum gap of 300 bp between peaks, threshold of ChiP-seq reads at the 95th quantile, and the addition of mappability, GC content and ambiguity scores of mm9 preprocessed data. MOSAiCS was also used to find the summit of peaks. Peaks for each sample were redefined by extending +/− 500 bp of the summit.
DhMR identification and genomic annotations
Peaks from control and stressed samples were converted to Granges objects and peaks for each group were defined as the following from the 2 grouped samples: peaks from all samples were pooled together, overlapping peaks were merged to form one region, and such overlapping peaks were used for further analysis if at least 2 samples had such overlap. To form the candidate regions for DhMR identification, peaks from the control and stressed groups were similarly pooled and merged together. This produced 48,452 candidate region peaks. To identify DhMRs, R package edgeR was used.53 Reads for each sample were extended to their estimated fragment length as determined by SPP. Normalization factors, common dispersion and tagwise dispersion were calculated for the data, aiding to alleviate differences in library size per sample. DhMRs were found using an exact test in edgeR using a P-value cutoff of 0.05, which produced 1,039 DhMRs. To annotate DhMRs to genomic structures and gene symbols, R package ChIPseeker was used using Bioconductor packages TxDb.Mmusculus.UCSC.mm9.knownGene and org.Mm.eg.db for the annotation of genomic feature and gene symbol, respectively.54
RNA library preparation and sequencing
Approximately 100 ng of total RNA was used for sequence library construction following instructions of NuGen mRNA sample prep kit (cat# 0348). In brief, total RNA was copied into first strand cDNA using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA Polymerase I and RNaseH. These cDNA fragments went through an end repair process, the addition of a single ‘A’ base, and then ligation of the adapters. These products were gel purified and enriched with PCR to create the final cDNA libraries. The library constructs were run on the bioanalyzer to verify the size and concentration before sequencing on the Illumina HiSeq2500 machine where 100-cycle single-end sequencing was performed by the University of Wisconsin Biotechnology Center. In short, libraries from the same mice and combinations that were used to generate the 5hmC data were sequenced for each experimental condition.
RNA-sequencing analysis
Each sample was separately run using RSEM which used Bowtie v0.12.7 in its analysis to calculate expression.55 All parameters of RSEM were set to default with the exception of the addition of the Gencode M1 Release reference genome annotation (http://www.gencodegenes.org/mouse_releases/). This produced read counts at both the whole-gene level and at the isoform level. Two matrices of read counts (i.e., at whole-gene and isoform level) for all samples were created using RSEM and read counts were rounded for analysis of differential expression using edgeR. Similar to methods used to identify DhMRs, normalization factors, common and tagwise dispersions of the data were calculated in edgeR and an exact test was used to identify cases of differential expression between control and stressed groups using a P-value cutoff of 0.05 and found 678 cases of differentially expressed whole-genes and 1,242 differentially expressed isoforms originating from 1,115 unique genes. By overlapping these 2 lists found 77 instances of differential whole-gene expression that coincided with differential isoform expression.
Identification of gene ontological (GO) enrichments
Genes associated with DhMRs or found to be differentially expressed at either the whole-gene or isoform level were separately studied for ontological enrichment using R package clusterProfiler.56 Gene symbols were converted to their Entrez IDs using clusterProfiler and were then used to find significant enrichment of ‘biologic processes’ (BP). Notably, as nearly a third (N = 209) of differentially expressed whole-genes were predicted/pseudo/Riken genes, these were excluded from ontological analysis, as they provide little to no relevant information for such ontological analyses. A P-value cutoff of 0.05 was initially used during the clusterProfiler analysis, then a FDR cutoff of 0.05 for the produced GO terms was used to filter out GO terms for all analyses except for whole-gene analyses which used a P-value cutoff of 0.05. To further filter GOs, terms were kept for further analysis if the ratio of ‘GeneRatio’ over ‘BgRatio’ (as determined by clusterProfiler) either exceeded a fold-change threshold of 1.5 or was smaller than a fold-change threshold of 1/1.5. The gene universe for DhMRs consisted of all genes associated with 5hmC peaks (n = 14,320) while the gene universe for differential gene/isoform expression and potentially functional DhMRs consisted of all genes tested for expression by RSEM (n = 16,518).
Transcription factor binding site motifs
The DREME suite37 was used to identify enrichments of transcription factor sequence motifs in the hyper- and hypo-DhMRs. Motifs that were not found in control-only 5hmC regions, experimental-only 5hmC regions, and all 5hmC regions and that had an E-value < 10e−3 were considered to be significant. Putative binding factors were predicted using SpaMo directly from the DREME suite software package and are listed in the tables shown with their putative transcription factors.
Statistical analysis for molecular testing
To study over- and under-representation of DhMRs (n = 1,039) across genomic structures, permutation testing was used using a perl script. The number of DhMRs falling into each genomic structure was tallied and this number was termed the “actual number” for each genomic structure. Notably, DhMRs that fell across multiple genomic structures were counted twice. All 5hmC peaks from all samples were pooled and annotated using R package ChIPseeker, giving a full set of peaks (n = 289,098). The same number of DhMRs (n = 1,039) were randomly selected from this full set and the number of times each peak fell into each genomic structure was tallied, termed the “permutated number” for each genomic structure. This “permutated number” was generated 10e5 times. To achieve the permutated P-value, the number of times the “permutated number” of each genomic structure surpassed the “actual number” for the same structure was divided by 10e5.
Similar to permutation procedures used for genomic structure mentioned above, over- and under-representation of DhMRs across chromosomes was also studied using permutation methods using a perl script. Unlike methods used for genomic structures, to correct for multiple hypotheses (i.e., 19 autosomes and the X chromosome), proportions of DhMRs falling on each chromosome were calculated and the “actual proportion” of each chromosome was compared with the “permutated proportions” of any chromosome for each permutation. Again, 10e5 permutations were performed and the number of times the “actual proportion” of a chromosome was surpassed by the “permutated proportion” of any other chromosome was divided by 10e5 to achieve the permutated P-value.
To identify if ontological findings of BP of DhMRs showed a significant enrichment for neuronal-related terms, a Pearson's chi-square test with Yates' continuity correction was conducted in R using a published list of neuronal/immunological-related GO BP terms (n = 3,071).57 Similarly, a chi-square test was used to compare DhMR-associated genes (n = 880) and known stress-related genes (n = 3,786) extracted from the GeneCards database using the following terms: anxiety; bipolar; fear; depression; major depressive disorder; posttraumatic stress disorder; psychosis; schizophrenia; stress; and trauma. Notably, the gene universe used for the chi-square test consisted of all the genes associated with peaks from sequenced data of all animals (n = 14,320).
Supplementary Material
Disclosure of potential conflicts of interest
The coauthors of this manuscript do not have any conflicts of interest regarding the contents of this manuscript.
Acknowledgments
The authors would like to thank the WISPIC animal facility, the University of Wisconsin Biotechnology Center, and Dr. Rupa Sridharan and her group for RNAseq assistance.
Funding
This work was supported in part by the University of Wisconsin-Madison department of Psychiatry, University of Wisconsin Vilas Cycle Professorships #133AAA2989 and University of Wisconsin Graduate School #MSN184352 (all to RSA), NARSAD Young Investigator Grant from the Brain & Behavioral Research Foundation #22669 (LP), National Science Foundation under Grant No. 1400815 (AM), and the University of Wisconsin Neuroscience training grant T32-GM007507 (SL).
References
- 1.Eiland L, Romeo RD. Stress and the developing adolescent brain. Neuroscience 2013; 249:162-71; PMID:23123920; http://dx.doi.org/ 10.1016/j.neuroscience.2012.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev 2007; 87:873-904; PMID:17615391; http://dx.doi.org/ 10.1152/physrev.00041.2006 [DOI] [PubMed] [Google Scholar]
- 3.Vreeburg SA, Hoogendijk WJ, van Pelt J, Derijk RH, Verhagen JC, van Dyck R, Smit JH, Zitman FG, Penninx BW. Major depressive disorder and hypothalamic-pituitary-adrenal axis activity: results from a large cohort study. Arch Gen Psychiatry 2009; 66:617-26; PMID:19487626; http://dx.doi.org/ 10.1001/archgenpsychiatry.2009.50 [DOI] [PubMed] [Google Scholar]
- 4.Stokes PE, Sikes CR. Hypothalamic-pituitary-adrenal axis in psychiatric disorders. Annu Rev Med 1991; 42:519-31; PMID:2035992; http://dx.doi.org/ 10.1146/annurev.me.42.020191.002511 [DOI] [PubMed] [Google Scholar]
- 5.Klengel T, Pape J, Binder EB, Mehta D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacol 2014; 80:115-32; PMID:24452011; http://dx.doi.org/25069392 10.1016/j.neuropharm.2014.01.013 [DOI] [PubMed] [Google Scholar]
- 6.Bekdash R, Zhang C and Sarkar D. Fetal alcohol programming of hypothalamic proopiomelanocortin system by epigenetic mechanisms and later life vulnerability to stress. Alcohol Clin Exp Res 2014; 38:2323-30; PMID:25069392; http://dx.doi.org/ 10.1111/acer.12497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alisch RS, Chopra P, Fox AS, Chen K, White AT, Roseboom PH, Keles S, Kalin NH. Differentially methylated plasticity genes in the amygdala of young primates are linked to anxious temperament, an at risk phenotype for anxiety and depressive disorders. J Neurosci 2014; 34:15548-56; PMID:25411484; http://dx.doi.org/ 10.1523/JNEUROSCI.3338-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pidsley R, Viana J, Hannon E, Spiers H, Troakes C, Al-Saraj S, Mechawar N, Turecki G, Schalkwyk LC, Bray NJ, et al.. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol 2014; 15:483; PMID:25347937; http://dx.doi.org/ 10.1186/s13059-014-0483-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A 2005; 102:9341-6; PMID:15961543; http://dx.doi.org/ 10.1073/pnas.0503736102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdolmaleky HM, Cheng KH, Faraone SV, Wilcox M, Glatt SJ, Gao F, Smith CL, Shafa R, Aeali B, Carnevale J, et al.. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet 2006; 15:3132-45; PMID:16984965; http://dx.doi.org/ 10.1093/hmg/ddl253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuratomi G, Iwamoto K, Bundo M, Kusumi I, Kato N, Iwata N, Ozaki N, Kato T. Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol Psychiatry 2008; 13:429-41; PMID:17471289; http://dx.doi.org/ 10.1038/sj.mp.4002001 [DOI] [PubMed] [Google Scholar]
- 12.Poulter MO, Du L, Weaver IC, Palkovits M, Faludi G, Merali Z, Szyf M, Anisman H. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol Psychiatry 2008; 64:645-52; PMID:18639864; http://dx.doi.org/ 10.1016/j.biopsych.2008.05.028 [DOI] [PubMed] [Google Scholar]
- 13.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013; 502:472-9; PMID:24153300; http://dx.doi.org/ 10.1038/nature12750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, et al.. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 2011; 14:1607-16; PMID:22037496; http://dx.doi.org/ 10.1038/nn.2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szulwach KE, Li X, Li Y, Song CX, Han JW, Kim S, Namburi S, Hermetz K, Kim JJ, Rudd MK, et al.. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet 2011; 7:e1002154; PMID:21731508; http://dx.doi.org/ 10.1371/journal.pgen.1002154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009; 324:929-30; PMID:19372393; http://dx.doi.org/ 10.1126/science.1169786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, et al.. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol 2011; 29:68-72; PMID:21151123; http://dx.doi.org/ 10.1038/nbt.1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao B, Jin P. Cytosine modifications in neurodevelopment and diseases. Cell Mol Life Sci 2014; 71:405-18; PMID:23912899; http://dx.doi.org/ 10.1007/s00018-013-1433-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Mahdawi S, Virmouni SA, Pook MA. The emerging role of 5-hydroxymethylcytosine in neurodegenerative diseases. Front Neurosci 2014; 8:397; PMID:25538551; http://dx.doi.org/ 10.3389/fnins.2014.00397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012; 151:1417-30; PMID:23260135; http://dx.doi.org/ 10.1016/j.cell.2012.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhubi A, Chen Y, Dong E, Cook EH, Guidotti A, Grayson DR. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl Psychiatry 2014; 4:e349; PMID:24448211; http://dx.doi.org/ 10.1038/tp.2013.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villar-Menendez I, Blanch M, Tyebji S, Pereira-Veiga T, Albasanz JL, Martin M, Ferrer I, Perez-Navarro E, Barrachina M. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington's disease. Neuromol Med 2013; 15:295-309; PMID:23385980; http://dx.doi.org/23669348 10.1007/s12017-013-8219-0 [DOI] [PubMed] [Google Scholar]
- 23.Wang F, Yang Y, Lin X, Wang JQ, Wu YS, Xie W, Wang D, Zhu S, Liao YQ, Sun Q, et al.. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington's disease. Hum Mol Genet 2013; 22:3641-53; PMID:23669348; http://dx.doi.org/ 10.1093/hmg/ddt214 [DOI] [PubMed] [Google Scholar]
- 24.Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, Steinbusch HW, Coleman PD, Rutten BP, van den Hove DL. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol Aging 2013; 34:2091-9; PMID:23582657; http://dx.doi.org/ 10.1016/j.neurobiolaging.2013.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, Papale LA, Zhang Q, Madrid A, Chen L, Chopra P, Keles S, Jin P, Alisch RS. Genome-wide alterations in hippocampal 5-hydroxymethylcytosine links plasticity genes to acute stress. Neurobiol Dis 2016; 86:99-108; PMID:26598390; http://dx.doi.org/ 10.1016/j.nbd.2015.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papale LA, Li S, Madrid A, Zhang Q, Chen L, Chopra P, Jin P, Keles S, Alisch RS. Sex-specific hippocampal 5-hydroxymethylcytosine is disrupted in response to acute stress. Neurobiol Dis 2016; 96:54-66; PMID:27576189; http://dx.doi.org/ 10.1016/j.nbd.2016.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao B, Lin L, Street RC, Zalewski ZA, Galloway JN, Wu H, Nelson DL, Jin P. Genome-wide alteration of 5-hydroxymethylcytosine in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum Mol Genet 2014; 23:1095-107; PMID:24108107; http://dx.doi.org/ 10.1093/hmg/ddt504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong E, Dzitoyeva SG, Matrisciano F, Tueting P, Grayson DR, Guidotti A. Brain-derived neurotrophic factor epigenetic modifications associated with schizophrenia-like phenotype induced by prenatal stress in mice. Biol Psychiatry 2015; 77:589-96; PMID:25444166; http://dx.doi.org/ 10.1016/j.biopsych.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishioka M, Bundo M, Kasai K, Iwamoto K. DNA methylation in schizophrenia: progress and challenges of epigenetic studies. Genome Med 2012; 4:96; PMID:23234572; http://dx.doi.org/ 10.1186/gm397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller BR, Bale TL. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol Behav 2006; 88:605-14; PMID:16828814; http://dx.doi.org/ 10.1016/j.physbeh.2006.05.019 [DOI] [PubMed] [Google Scholar]
- 31.Papale LA, Zhang Q, Li S, Chen K, Keles S, Alisch RS. Genome-wide disruption of 5-hydroxymethylcytosine in a mouse model of autism. Hum Mol Genet 2015; 24:7121-31; PMID:26423458; http://dx.doi.org/ 10.1093/hmg/ddv411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng J, Shao N, Szulwach KE, Vialou V, Huynh J, Zhong C, Le T, Ferguson D, Cahill ME, Li Y, et al.. Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nat Neurosci 2015; 18:536-44; PMID:25774451; http://dx.doi.org/ 10.1038/nn.3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khare T, Pai S, Koncevicius K, Pal M, Kriukiene E, Liutkeviciute Z, Irimia M, Jia P, Ptak C, Xia M, et al.. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol 2012; 19:1037-43; PMID:22961382; http://dx.doi.org/ 10.1038/nsmb.2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kahler A, et al.. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014; 506:185-90; PMID:24463508; http://dx.doi.org/ 10.1038/nature12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al.. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012; 338:1619-22; PMID:23160955; http://dx.doi.org/ 10.1126/science.1227764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez-Mantilla AJ, Moreno-De-Luca A, Ledbetter DH, Martin CL. A Cross-Disorder Method to Identify Novel Candidate Genes for Developmental Brain Disorders. JAMA Psychiatry 2016; 73:275-83; PMID:26817790; http://dx.doi.org/ 10.1001/jamapsychiatry.2015.2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011; 27:1653-9; PMID:21543442; http://dx.doi.org/ 10.1093/bioinformatics/btr261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet 2006; 7:415-26; PMID:16708070; http://dx.doi.org/ 10.1038/nrg1878 [DOI] [PubMed] [Google Scholar]
- 39.Gharani N, Benayed R, Mancuso V, Brzustowicz LM, Millonig JH. Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol Psychiatry 2004; 9:474-84; PMID:15024396; http://dx.doi.org/ 10.1038/sj.mp.4001498 [DOI] [PubMed] [Google Scholar]
- 40.Okaty BW, Miller MN, Sugino K, Hempel CM, Nelson SB. Transcriptional, electrophysiological maturation of neocortical fast-spiking GABAergic interneurons. J Neurosci 2009; 29:7040-52; PMID:19474331; http://dx.doi.org/ 10.1523/JNEUROSCI.0105-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scobie KN, Hall BJ, Wilke SA, Klemenhagen KC, Fujii-Kuriyama Y, Ghosh A, Hen R, Sahay A. Kruppel-like factor 9 is necessary for late-phase neuronal maturation in the developing dentate gyrus and during adult hippocampal neurogenesis. J Neurosci 2009; 29:9875-87; PMID:19657039; http://dx.doi.org/ 10.1523/JNEUROSCI.2260-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kittel-Schneider S, Wobrock T, Scherk H, Schneider-Axmann T, Trost S, Zilles D, Wolf C, Schmitt A, Malchow B, Hasan A, et al.. Influence of DGKH variants on amygdala volume in patients with bipolar affective disorder and schizophrenia. Eur Arch Psychiatry Clin Neurosci 2015; 265:127-36; PMID:24958494; http://dx.doi.org/ 10.1007/s00406-014-0513-9 [DOI] [PubMed] [Google Scholar]
- 43.Tunca Z, Kivircik Akdede B, Ozerdem A, Alkin T, Polat S, Ceylan D, Bayin M, Cengizcetin Kocuk N, Simsek S, Resmi H, et al.. Diverse glial cell line-derived neurotrophic factor (GDNF support between mania and schizophrenia: a comparative study in four major psychiatric disorders. Eur Psychiatry 2015; 30:198-204; PMID:25543333; http://dx.doi.org/ 10.1016/j.eurpsy.2014.11.003 [DOI] [PubMed] [Google Scholar]
- 44.Ben-Shachar D, Karry R. Sp1 expression is disrupted in schizophrenia; a possible mechanism for the abnormal expression of mitochondrial complex I genes, NDUFV1 and NDUFV2. PLoS One 2007; 2:e817; PMID:17786189; http://dx.doi.org/ 10.1371/journal.pone.0000817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bayles R, Baker EK, Jowett JB, Barton D, Esler M, El-Osta A, Lambert G. Methylation of the SLC6a2 gene promoter in major depression and panic disorder. PLoS One 2013; 8:e83223; PMID:24312678; http://dx.doi.org/ 10.1371/journal.pone.0083223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, Steel G, Nestadt G, Liang KY, Huganir RL, et al.. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet 2005; 77:918-36; PMID:16380905; http://dx.doi.org/ 10.1086/497703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nilsson KW, Sjoberg RL, Leppert J, Oreland L, Damberg M. Transcription factor AP-2 beta genotype and psychosocial adversity in relation to adolescent depressive symptomatology. J Neural Transm (Vienna) 2009; 116:363-70; PMID:19184334; http://dx.doi.org/ 10.1007/s00702-009-0183-3 [DOI] [PubMed] [Google Scholar]
- 48.Li M, Xue X, Shao S, Shao F, Wang W. Cognitive, emotional and neurochemical effects of repeated maternal separation in adolescent rats. Brain Res 2013; 1518:82-90; PMID:23623774; http://dx.doi.org/ 10.1016/j.brainres.2013.04.026 [DOI] [PubMed] [Google Scholar]
- 49.Mueller BR, Bale TL. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol Behav 2007; 91:55-65; PMID:17367828; http://dx.doi.org/ 10.1016/j.physbeh.2007.01.017 [DOI] [PubMed] [Google Scholar]
- 50.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/ 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 2008; 26:1351-9; PMID:19029915; http://dx.doi.org/ 10.1038/nbt.1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuan PF, Chung D, Pan G, Thomson JA, Stewart R, Keles S. A Statistical Framework for the Analysis of ChIP-Seq Data. J Am Stat Assoc 2011; 106:891-903; PMID:26478641; http://dx.doi.org/ 10.1198/jasa.2011.ap09706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139-40; PMID:19910308; http://dx.doi.org/ 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015; 31:2382-3; PMID:25765347; http://dx.doi.org/ 10.1093/bioinformatics/btv145 [DOI] [PubMed] [Google Scholar]
- 55.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011; 12:323; PMID:21816040; http://dx.doi.org/ 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012; 16:284-7; PMID:22455463; http://dx.doi.org/ 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geifman N, Monsonego A, Rubin E. The Neural/Immune Gene Ontology: clipping the Gene Ontology for neurological and immunological systems. BMC Bioinformatics 2010; 11:458; PMID:20831831; http://dx.doi.org/ 10.1186/1471-2105-11-458 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.