

ABSTRACT
Obesity can increase the risk of complex metabolic diseases, including insulin resistance. Moreover, obesity can be caused by environmental and genetic factors. However, the epigenetic mechanisms of obesity are not well defined. Therefore, the identification of novel epigenetic biomarkers of obesity allows for a more complete understanding of the disease and its underlying insulin resistance. The aim of our study was to identify DNA methylation changes in whole-blood that were strongly associated with obesity and insulin resistance. Whole-blood was obtained from lean (n = 10; BMI = 23.6 ± 0.7 kg/m2) and obese (n = 10; BMI = 34.4 ± 1.3 kg/m2) participants in combination with euglycemic hyperinsulinemic clamps to assess insulin sensitivity. We performed reduced representation bisulfite sequencing on genomic DNA isolated from the blood. We identified 49 differentially methylated cytosines (DMCs; q < 0.05) that were altered in obese compared with lean participants. We identified 2 sites (Chr.21:46,957,981 and Chr.21:46,957,915) in the 5’ untranslated region of solute carrier family 19 member 1 (SLC19A1) with decreased methylation in obese participants (lean 0.73 ± 0.11 vs. obese 0.09 ± 0.05; lean 0.68 ± 0.10 vs. obese 0.09 ± 0.05, respectively). These 2 DMCs identified by obesity were also significantly predicted by insulin sensitivity (r = 0.68, P = 0.003; r = 0.66; P = 0.004). In addition, we performed a differentially methylated region (DMR) analysis and demonstrated a decrease in methylation of Chr.21:46,957,915–46,958,001 in SLC19A1 of −34.9% (70.4% lean vs. 35.5% obese). The decrease in whole-blood SLC19A1 methylation in our obese participants was similar to the change observed in skeletal muscle (Chr.21:46,957,981, lean 0.70 ± 0.09 vs. obese 0.31 ± 0.11 and Chr.21:46,957,915, lean 0.72 ± 0.11 vs. obese 0.31 ± 0.13). Pyrosequencing analysis further demonstrated a decrease in methylation at Chr.21:46,957,915 in both whole-blood (lean 0.71 ± 0.10 vs. obese 0.18 ± 0.06) and skeletal muscle (lean 0.71 ± 0.10 vs. obese 0.30 ± 0.11). Our findings demonstrate a new potential epigenetic biomarker, SLC19A1, for obesity and its underlying insulin resistance.
KEYWORDS: Biomarker, DNA methylation, insulin resistance, obesity, whole-blood
Introduction
Obesity is epidemic, and has become the fifth leading risk for global deaths.1 Individuals with obesity show chronic low-grade inflammation.2,3 The expansion of white adipose tissue in obesity has been associated with increased proinflammatory cytokines, such as tumor necrosis factor-α (TNFα) and interleukin 6 (IL-6).2 These inflammatory cytokines circulate in the blood and can have negative effects on peripheral inulin responsive tissues, such as skeletal muscle and liver.4 The activation of Toll-like and interleukin receptors have been proposed to reduce insulin signaling by promoting the signaling cascade of inflammatory kinases.3 The reduction in insulin signaling, in part, is due to phosphorylation of serine residues on the insulin receptor substrate 1 (IRS-1) in skeletal muscle, thus inhibiting its activity.3,5 As such, the majority of individuals with obesity have an underlying insulin resistance.2,6 This state of chronic inflammation associated with obesity can also exacerbate co-morbidities, including type 2 diabetes (T2D), hypertension, dyslipidemia, and cardiovascular disease.1,7
To better understand how to hinder disease progression, it has become important to find reliable biomarkers for early intervention.8 Biomarkers can be any biologic characteristic that can be identified and/or monitored during the progression of a disease.9 This includes non-invasive measurements such as those currently used for identifying risk for the progression to type 2 diabetes and cardiovascular disease, such as high body mass index (BMI) and blood pressure.10 Other traditional biomarkers have included clinical measurements of glucose, hemoglobin A1c (HbA1c), and cholesterol levels from blood.11 However, the progression of obesity and insulin resistance is a consequence of both environmental and genetic factors, and the above mentioned non-invasive traditional measurements do not provide insight into the molecular basis of the disease.12
Our previous work in whole-blood assessed transcriptional changes in a Latino population from the Arizona Insulin Resistance (AIR) Registry.13,14 In one study, we demonstrated transcriptomic changes in genes involved in ribosome, oxidative phosphorylation, and MAPK signaling when analyzing the adults with and without metabolic syndrome.13 In another study, we identified altered expression of genes involved in inflammatory pathways in adolescents with and without obesity.14 Our findings indicate potential biomarkers in whole-blood for inflammation, insulin signaling, and mitochondrial function in obese and metabolic syndrome conditions. We believe that the transcriptomic changes observed in our cohorts are in part due to epigenetic regulation.
Epigenetics is a regulatory process that controls gene expression without altering the nucleotide sequence.15 DNA methylation is the epigenetic process of a methyl addition primarily to a cytosine residue preceding a guanine, termed CpG dinucleotide.15 DNA methylation marks residing in promoter and untranslated regions have been associated with gene silencing.16-18 However, large-scale studies, such as the Human Epigenome Project, have found low correlations between gene expression and differential methylation.19 Specifically, one-third of the differential methylation they identified in 5’ untranslated regions were inversely correlated with transcription.19 Epigenetic mechanisms have become important for determining the molecular basis of diseases, because they are due to both genetic and environmental factors.16 The influence of these factors on DNA methylation has also made it a promising biomarker for disease. The use of DNA methylation as an epigenetic biomarker has become attractive for clinical use due to its covalent bond, making it a robust mark for analysis.9
A number of studies have focused on identifying epigenetic biomarkers in blood that were associated with obesity and insulin resistance.20-23 In our study, we performed reduced representation bisulfite sequencing (RRBS) to assess DNA methylation at the whole-genome level. Here, we set out to identify changes in DNA methylation related to obesity using the most readily available tissue, whole-blood. Based on our previous transcriptomic findings in whole-blood, we hypothesized that there would be alterations in the DNA methylation of genes involved in inflammation, insulin signaling, and mitochondrial function. We could potentially have a low overall correspondence between our DNA methylation data and our previous transcriptomic data, based on the Human Epigenome Project findings.19 Regardless, this study will allow us to identify novel epigenetic biomarkers that are associated with obesity and insulin resistance in blood.
Results
Participants
Table 1 shows the phenotypic characteristics for participants with (n = 10; BMI > 30 kg/m2) and without (n = 10) obesity. By design, lean participants had a significantly lower body mass index (BMI). In addition, lean participants had significantly lower measures of body fat percentage and waist circumference compared with obese participants. As expected, obese participants had higher fasting plasma insulin levels and lower M values (an insulin sensitivity measurement) compared with lean participants.
Table 1.
Characteristics of study participants (n = 20) classified by body mass index.
| Characteristics | Lean | Obese | P value* | P value (age and sex) |
|---|---|---|---|---|
| Sex | 5M/5F | 5M/5F | 1.0 | — |
| Age (y) | 29.9 ± 2.2 | 35.9 ± 3.2 | 0.14 | — |
| Body mass index (kg/m2) | 23.6 ± 0.7 | 34.4 ± 1.3 | <0.001 | <0.001 |
| Body fat (%)╪ | 25.0 ± 1.6 | 36.8 ± 2.2 | <0.001 | <0.001 |
| Waist circumference (cm) | 83.8 ± 2.9 | 103.6 ± 3.4 | <0.001 | 0.0032 |
| Systolic blood pressure (mmHg) | 117.7 ± 2.2 | 119.4 ± 2.5 | 0.62 | 0.57 |
| Diastolic blood pressure (mmHg) | 72.0 ± 1.5 | 75.0 ± 2.0 | 0.24 | 0.52 |
| Triglycerides (mg/dL) | 101.1 ± 15.4 | 105.2 ± 15.0 | 0.85 | 0.77 |
| Cholesterol (mg/dL) | 174.3 ± 10.4 | 182.9 ± 10.7 | 0.57 | 0.61 |
| High density lipoproteins (mg/dL) | 52.5 ± 4.4 | 48.3 ± 3.3 | 0.45 | 0.45 |
| Low density lipoproteins (mg/dL) | 101.7 ± 8.7 | 113.5 ± 9.0 | 0.36 | 0.95 |
| Hemoglobin A1c (%) | 5.2 ± 0.04 | 5.3 ± 0.1 | 0.24 | 0.47 |
| Fasting plasma glucose (mg/dL) | 87.5 ± 2.0 | 90.2 ± 1.7 | 0.31 | 0.34 |
| Fasting plasma insulin (µU/mL) | 6.5 ± 1.3 | 13.4 ± 2.0 | 0.64 | 0.0054 |
| M value (mg/kg.min) | 7.7 ± 0.5 | 3.8 ± 0.5 | <0.001 | <0.001 |
| M value (mg/kg.min·FFM) | 10.2 ± 0.7 | 6.0 ± 0.8 | <0.001 | <0.001 |
Data presented as mean ± SEM, based on independent sample t-tests. Adjusted for age and sex by ANCOVA.
Calculated by Chi-Square Test.
Body fat determined by biometric impedance analysis (BIA). FFM, Fat-free mass.
Genome-wide methylation analysis in human whole-blood
Using next generation RRBS, we identified 5,227,488 methylation sites captured in the blood methylation analysis from lean and obese participants. The methylation sites were categorized by genic regions (Fig. 1a) and CpG island features (Fig. 1b). A large proportion of these sites fell within regulatory regions, with 22% of them in promoter and 18% in 3’ and 5’ untranslated regions (Fig. 1a). When looking at the proportion of each different CpG island feature within each genic region, we found CpG islands to be most concentrated in the promoter and 5’ untranslated region (Fig. 1b).
Figure 1.
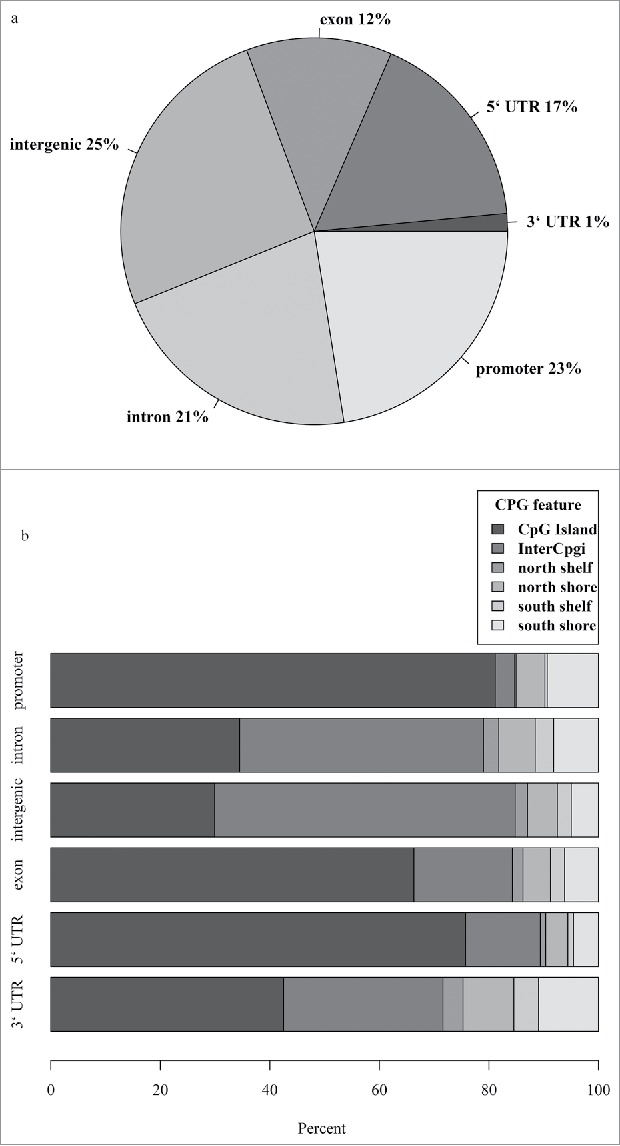
All methylation sites detected in our whole-blood samples using reduced representation bisulfite sequencing technology were mapped (a) in the context of gene regions and (b) CpG island features. Regions were defined using UCSC browser RefGene and CpG island tracks (see methods). The promoter region was defined as 1000 bp upstream of the transcription start site (TSS); CpG island is a 200–3000 bp stretch of DNA with a C+G content of 50% and observed CpG/expected CpG exceeding 0.6; North (N) and South (S) shores flank the CpG island by 0–2000 bp; the North (N) and South (S) shelf flank the shores by 2000 bp (2000–4000 bp from the island). UTR, untranslated region.
Whole-blood differentially methylated cytosines
To identify potential blood biomarkers for obese insulin resistant states, sites within all genomic regions were considered for analysis. Of the 5,227,488 methylation sites captured, 52,995 sites were significantly altered (uncorrected P < 0.05; Table S1) between our groupings. Differentially methylated cytosines (DMC) were corrected by a false discovery rate (FDR; q < 0.05), which identified 49 unique methylation sites (15 decreased and 34 increased; Table 2).
Table 2.
Whole-blood differentially methylated cytosines (DMCs; q < 0.05) between lean and obese groupings.
| DNA Methylation (%) |
|||||||
|---|---|---|---|---|---|---|---|
| Chr. Position | Gene | Lean | Obese | P value | q value | Genic Region | CpG Island Region |
| chr21.46957981 | SLC19A1 | 73.0 ± 11.0 | 9.0 ± 5.0 | <0.001 | 0.01 | 5′UTR | South Shore |
| chr21.46957915 | SLC19A1 | 68.0 ± 10.0 | 9.0 ± 5.0 | <0.001 | 0.01 | 5′UTR | CpG Island |
| chr6.3771940 | — | 76.0 ± 3.0 | 38.0 ± 7.0 | <0.001 | 0.05 | Intergenic | InterCpG |
| chr5.80690459 | ACOT12 | 43.0 ± 4.0 | 15.0 ± 2.0 | <0.001 | <0.001 | Promoter | South Shore |
| chr11.2883716 | — | 52.0 ± 6.0 | 22.0 ± 4.0 | <0.001 | 0.05 | Promoter | CpG Island |
| chr21.46927138 | COL18A1 | 51.0 ± 4.0 | 26.0 ± 3.0 | <0.001 | 0.04 | Intron | North Shelf |
| chr8.145702503 | FOXH1 | 93.0 ± 2.0 | 71.0 ± 2.0 | <0.001 | <0.001 | Promoter | South Shore |
| chr2.121283801 | — | 93.0 ± 2.0 | 79.0 ± 3.0 | <0.001 | 0.05 | Intergenic | South Shelf |
| chr19.46456403 | NOVA2 | 12.0 ± 3.0 | 2.0 ± 1.0 | <0.001 | 0.02 | Intron | CpG Island |
| chr6.146348971 | GRM1 | 16.0 ± 2.0 | 6.0 ± 1.0 | <0.001 | 0.04 | Promoter/Intron | North Shore |
| chr4.2648590 | FAM193A | 99.0 ± 1.0 | 90.0 ± 2.0 | <0.001 | 0.04 | Intron | InterCpG |
| chr1.214504377 | SMYD2 | 92.0 ± 1.0 | 85.0 ± 1.0 | <0.001 | 0.02 | Exon | InterCpG |
| chr20.32856825 | ASIP | 6.0 ± 2.0 | 0.0 ± 0.0 | <0.001 | 0.05 | Exon | CpG Island |
| chr9.139085246 | — | 3.0 ± 1.0 | 0.0 ± 0.0 | <0.001 | <0.001 | Intergenic | CpG Island |
| chr19.55898053 | RPL28 | 5.0 ± 2.0 | 0.0 ± 0.0 | <0.001 | 0.01 | Promoter/Exon | CpG Island |
| chr5.72742630 | FOXD1 | 1.0 ± 0.0 | 5.0 ± 1.0 | <0.001 | 0.05 | 3′UTR | South Shore |
| chr8.132917158 | EFR3A | 0.0 ± 0.0 | 6.0 ± 2.0 | <0.001 | 0.04 | Intron | South Shore |
| chr16.28993311 | LAT/SPNS1 | 94.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.05 | Promoter/Exon/Intron | InterCpG |
| chr17.1184167 | TUSC5 | 92.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.01 | Intron | InterCpG |
| chr7.157655513 | PTPRN2 | 94.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | <0.001 | Exon | North Shelf |
| chr4.843782 | GAK | 92.0 ± 1.0 | 99.0 ± 1.0 | <0.001 | 0.04 | Exon | CpG Island |
| chr11.31827022 | PAX6 | 1.0 ± 0.0 | 7.0 ± 1.0 | <0.001 | <0.001 | Intron | South Shore |
| chr6.158072851 | ZDHHC14 | 95.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.02 | Intron | InterCpG |
| chr12.81444314 | — | 93.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | <0.001 | Intergenic | InterCpG |
| chr7.23646672 | CCDC126 | 92.0 ± 1.0 | 99.0 ± 1.0 | <0.001 | 0.03 | 5′UTR | InterCpG |
| chr6.74371204 | — | 93.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.05 | Intergenic | InterCpG |
| chr2.158272554 | CYTIP | 92.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.01 | Exon | InterCpG |
| chr17.21219144 | — | 87.0 ± 1.0 | 96.0 ± 1.0 | <0.001 | 0.05 | Intergenic | North Shore |
| chr1.87429560 | HS2ST1 | 90.0 ± 2.0 | 99.0 ± 1.0 | <0.001 | 0.03 | Intron | InterCpG |
| chr8.111697045 | — | 91.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | <0.001 | Intergenic | InterCpG |
| chr17.19743530 | ULK2 | 90.0 ± 3.0 | 100.0 ± 0.0 | <0.001 | <0.01 | Intron | InterCpG |
| chr5.61058332 | — | 91.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.04 | Intergenic | InterCpG |
| chr3.50131816 | RBM5 | 90.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.04 | Intron | InterCpG |
| chr9.139093743 | LHX3 | 0.0 ± 0.0 | 9.0 ± 2.0 | <0.001 | <0.01 | Intron | CpG Island |
| chr7.87256217 | ABCB1/RUNDC3B | 88.0 ± 2.0 | 99.0 ± 1.0 | <0.001 | <0.01 | Promoter/Intron | North Shore |
| chr17.3701518 | ITGAE | 92.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.04 | Intron | InterCpG |
| chr9.99791494 | — | 90.0 ± 3.0 | 100.0 ± 0.0 | <0.001 | <0.01 | Intergenic | InterCpG |
| chrX.933751 | — | 86.0 ± 2.0 | 97.0 ± 2.0 | <0.001 | 0.02 | Intergenic | InterCpG |
| chr15.62511245 | — | 89.0 ± 3.0 | 100.0 ± 0.0 | <0.001 | <0.01 | Intergenic | InterCpG |
| chr20.43948457 | — | 2.0 ± 1.0 | 13.0 ± 2.0 | <0.001 | <0.01 | Intergenic | South Shelf |
| chr5.172090073 | NEURL1B | 89.0 ± 2.0 | 100.0 ± 0.0 | <0.001 | 0.04 | Intron | InterCpG |
| chr20.1276940 | SNPH | 86.0 ± 2.0 | 99.0 ± 1.0 | <0.001 | <0.01 | 5′UTR | CpG Island |
| chr12.132871826 | GALNT9 | 86.0 ± 2.0 | 98.0 ± 1.0 | <0.001 | <0.001 | Intron | North Shore |
| chr7.140180051 | MKRN1 | 81.0 ± 3.0 | 98.0 ± 1.0 | <0.001 | 0.05 | Promoter | South Shore |
| chr19.1289934 | EFNA2 | 73.0 ± 3.0 | 90.0 ± 2.0 | <0.001 | 0.03 | Intron | North Shore |
| chr9.129088683 | FAM125B | 19.0 ± 2.0 | 36.0 ± 3.0 | <0.001 | <0.01 | Promoter | CpG Island |
| chr1.103319604 | — | 62.0 ± 7.0 | 86.0 ± 3.0 | <0.001 | 0.04 | Intergenic | InterCpG |
| chr8.22560981 | — | 18.0 ± 2.0 | 39.0 ± 5.0 | <0.001 | 0.02 | Intergenic | CpG Island |
| chr7.1659260 | — | 74.0 ± 6.0 | 98.0 ± 1.0 | <0.001 | 0.02 | Intergenic | CpG Island |
Methylation data presented as mean ± SEM q value generated by Benjamini-Hochberg multiple testing correction. CpG island is a 200–3000 bp stretch of DNA with a C+G content of 50% and observed CpG/expected CpG exceeding 0.6; North (N) and South (S) shores flank the CpG island by 0–2000 bp; the North (N) and South (S) shelf flank the shores by 2000 bp (2000–4000 bp from the island). InterCpG are locations between CpG islands.
Insulin sensitivity regression analysis of DMCs
We identified 49 DMCs that were altered with obesity. There is a strong association between obesity and insulin resistance.7 In this study, we observed that the M value, as measured by the euglycemic hyperinsulinemic clamp, and BMI measurements were significantly correlated (r = −0.778; P = 0.00004). Therefore, we further aimed to identify which DMCs were significantly associated with insulin sensitivity (i.e., M value). In multiple regression analyses with age, sex, and M value as the independent variables and methylation ratio of DMCs as the dependent variables, we found that M value independently explained a range of 25–54% of variance in 36 of the 49 DMCs (all P = 0.05; Table 3).
Table 3.
Regression analysis of the differentially methylated cytosines (DMCs; q < 0.05) predicted by M value after adjusting for age and sex.
| Chromosome Position | Gene | Genic Region | CpG Island Region | partial r | P value |
|---|---|---|---|---|---|
| chr21.46957981 | SLC19A1 | 5′UTR | South Shore | 0.68 | 0.003 |
| chr21.46957915 | SLC19A1 | 5′UTR | CpG Island | 0.66 | 0.004 |
| chr6.3771940 | — | Intergenic | InterCpG | 0.615 | 0.009 |
| chr5.80690459 | ACOT12 | Promoter | South Shore | 0.563 | 0.019 |
| chr11.2883716 | — | Promoter | CpG Island | 0.657 | 0.004 |
| chr21.46927138 | COL18A1 | Intron | North Shelf | 0.705 | 0.002 |
| chr8.145702503 | FOXH1 | Promoter | South Shore | 0.683 | 0.002 |
| chr2.121283801 | — | Intergenic | South Shelf | 0.711 | 0.001 |
| chr19.46456403 | NOVA2 | Intron | CpG Island | 0.51 | 0.037 |
| chr6.146348971 | GRM1 | Promoter | North Shore | 0.616 | 0.009 |
| chr4.2648590 | FAM193A | Intron | InterCpG | 0.59 | 0.013 |
| chr1.214504377 | SMYD2 | Exon | InterCpG | 0.558 | 0.02 |
| chr20.32856825 | ASIP | Exon | CpG Island | 0.569 | 0.017 |
| chr5.72742630 | FOXD1 | 3′UTR | South Shore | −0.565 | 0.018 |
| chr16.28993311 | LAT | Exon | InterCpG | −0.497 | 0.043 |
| chr17.1184167 | TUSC5 | Intron | InterCpG | −0.577 | 0.015 |
| chr4.843782 | GAK | Exon | CpG Island | −0.556 | 0.021 |
| chr11.31827022 | PAX6 | Intron | South Shore | −0.581 | 0.014 |
| chr12.81444314 | — | Intergenic | InterCpG | −0.498 | 0.042 |
| chr7.23646672 | CCDC126 | 5′UTR | InterCpG | −0.546 | 0.023 |
| chr17.21219144 | — | Intergenic | North Shore | −0.589 | 0.013 |
| chr1.87429560 | HS2ST1 | Intron | InterCpG | −0.674 | 0.003 |
| chr5.61058332 | — | Intergenic | InterCpG | −0.644 | 0.005 |
| chr3.50131816 | RBM5 | Intron | InterCpG | −0.689 | 0.002 |
| chr9.139093743 | LHX3 | Intron | CpG Island | −0.589 | 0.013 |
| chr7.87256217 | ABCB1 | Promoter | North Shore | −0.67 | 0.003 |
| chr9.99791494 | — | Intergenic | InterCpG | −0.506 | 0.038 |
| chrX.933751 | — | Intergenic | InterCpG | −0.692 | 0.002 |
| chr15.62511245 | — | Intergenic | InterCpG | −0.568 | 0.017 |
| chr5.172090073 | NEURL1B | Intron | InterCpG | −0.637 | 0.006 |
| chr7.140180051 | MKRN1 | Promoter | South Shore | −0.498 | 0.042 |
| chr19.1289934 | EFNA2 | Intron | North Shore | −0.737 | 0.001 |
| chr9.129088683 | FAM125B | Promoter | CpG Island | −0.721 | 0.001 |
| chr1.103319604 | — | Intergenic | InterCpG | −0.568 | 0.017 |
| chr8.22560981 | — | Intergenic | CpG Island | −0.58 | 0.015 |
| chr7.1659260 | — | Intergenic | CpG Island | −0.637 | 0.006 |
Whole-blood differentially methylated regions
DNA methylation regulation can be mediated by a single CpG or by a group of CpGs in close proximity to each other. Therefore, a regional analysis was performed on the 52,995 blood DMCs that were significantly altered (uncorrected P < 0.05). This analysis identified 74 differentially methylated regions (DMRs; Table S2). When the 74 blood DMRs were compared with the 49 blood DMCs (q < 0.05), 2 genes [solute carrier family 19 member 1 (SLC19A1) and ephrin-A2 (EFNA2)] were in common between the 2 analyses. The DMR (Chr.21:46,957,915–46,958,001) in SLC19A1 was decreased in methylation by −34.9% (70.4% methylation in lean vs. 35.5% methylation in obese) and the DMR for EFNA2 (Chr.19:1287,750–1287,781) was increased by +14.3% (28.4% methylation in lean vs. 42.7% methylation in obese) with obesity.
Potential blood-based biomarkers of skeletal muscle
Skeletal muscle is the major site for insulin-stimulated glucose disposal, making it an important target tissue for understanding insulin resistance.24 However, accessibility to this tissue is more difficult compared with blood. Therefore, we set out to identify blood-based biomarkers of methylation sites in genes that were also identified in skeletal muscle using our previously published data.25 The skeletal muscle results are based on 11 lean and 9 obese (BMI > 30 kg/m2; Table S3) patients. Of these individuals, 9 lean and 6 obese were in common with the individuals included in the whole-blood analyses. When we compared the significantly changing whole-blood DMCs (FDR; q < 0.05) with our previously published skeletal muscle data,25 we identified 3 sites that were in common. One site (Chr.21:46,927,138) was in collagen, type XVIII, α 1 (COL18A1) and the other 2 sites (Chr.21:46,957,915 and Chr.21:46,957,981) were upstream of SLC19A1 (Fig. 2).
Figure 2.
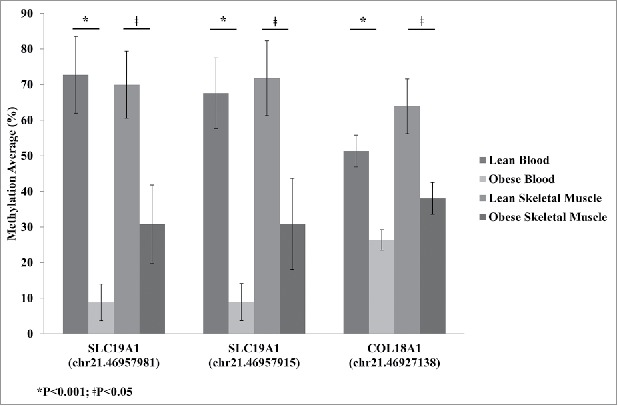
Average methylation detected by reduced representation bisulfite sequencing (RRBS) for SLC19A1 sites Chr.21:46,957,981 and Chr.21:46,957,915 and COL18A1 site Chr.21:46,927,138 for lean and obese in both blood and skeletal muscle. Significance based on independent sample t-tests.
SLC19A1 correlation analysis
The 2 significant (q < 0.05) SLC19A1 methylation sites, Chr.21:46,957,981 and Chr.21:46,957,915, are located downstream of 2 transcription start sites (TSSs) Chr.21:46,964,325 and Chr.21:46,962,385, based on the UCSC genome browser. The distances from the TSS are 6,410 bp and 4,470 bp for Chr.21:46,957,915, and 6,344 bp and 4,404 bp for Chr.21:46,957,981, respectively. These methylation sites were found in both whole-blood and skeletal muscle methylation. To determine the relationship of methylation at those sites between tissues, Pearson correlation analysis was performed. We found that methylation levels between blood and skeletal muscle were significantly and positively correlated at both SLC19A1 sites (Fig. 3).
Figure 3.
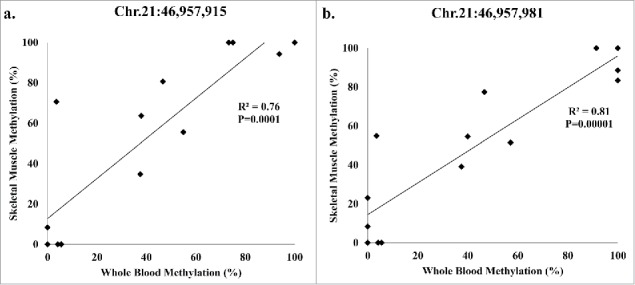
Pearson correlation analysis of the participants (8 lean; 6 obese) present in both whole-blood and skeletal muscle methylation for SLC19A1 (a) Chr.21:46,957,915 and (b) Chr.21:46,957,981.
SLC19A1 predicted transcription factor binding
We analyzed the sequences containing DMCs and the DMR associated with SLC19A1 using the program PROMO.26 Transcription factor binding motifs did not overlap with our most significant DMCs, Chr.21:46,957,981 and Chr.21:46,957,915. However, using the DMR sequence (containing 4 CpGs: Chr.21:46,957,915, Chr.21:46,957,981, Chr.21:46,957,988, and Chr.21:46,958,001), we found 2 predicted transcription factor genes, forkhead box P3 (FOXP3) and glucocorticoid receptor (GR), to overlap a CpG site at position Chr.21:46,957,988.
SLC19A1 validation
The SLC19A1 DMCs were significantly altered in both whole-blood and skeletal muscle. This may indicate that SLC19A1 is tightly associated with obesity. We confirmed the methylation changes between the lean and obese groups at Chr.21:46,957,915 using pyrosequencing. DNA methylation was significantly decreased in obese participants compared with lean in both whole-blood and skeletal muscle (Fig. 4). The changes observed in both tissues were comparable to the decreased methylation detected using RRBS.
Figure 4.
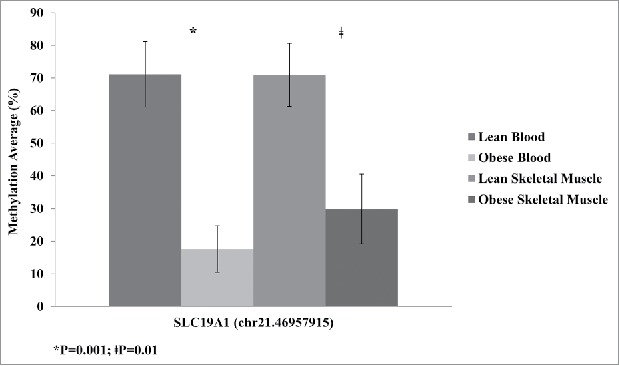
Average methylation of SLC19A1 site Chr.21: 46,957,915 detected by pyrosequencing validation for lean and obese samples in both blood and skeletal muscle. Significance based on independent sample t-tests.
Discussion
The present study was undertaken with the purpose of identifying whole-blood biomarkers of DNA methylation that were altered in obesity and insulin resistance. Our genome-wide RRBS analysis demonstrated that the promoter region was most concentrated with CpG islands, which is well established in the field.27 Interestingly, the distribution pattern of all detected CpG sites were similar to the patterns observed in our previous study in skeletal muscle.25 The consistent coverage of the genome regardless of tissue type presents RRBS as a viable technique for cross-tissue analysis.
Other recent studies have provided useful findings of DNA methylation differences in blood from obese compared with lean individuals.28,29 Wang et al. identified methylation changes in obesity associated with UBASH3A and TRIM3 in blood leukocytes from participants 14–30 years old.28 A study by Ronn et al. identified changes in methylation impacted by age, BMI, and HbA1c levels in blood and adipose tissue using multiple cohorts whose ages collectively spanned ages 23–83 years.29 We did not observe an overlap between our 49 corrected DMCs and the most significant sites identified from the aforementioned studies. The lack of overlap may be attributed to blood tissue type, age of cohorts, and methylation detection technology used.
We determined that the most reliable biomarker of obesity and its underlying insulin resistance would be identified by using several analyses. First, SLC19A1 was identified as significantly decreased in obesity in both whole-blood DMC and DMR analyses. Furthermore, we set out to identify similarities between whole-blood and the insulin responsive tissue, skeletal muscle. By using skeletal muscle methylation changes assessed in our previous study,25 we found SLC19A1 to be a blood-based DNA methylation biomarker for that tissue. Not only were these DMCs decreased in methylation in both blood and skeletal muscle, but also the level of change was to a similar extent. The confirmation of this finding through pyrosequencing leads us to believe that the altered SLC19A1 methylation is strongly associated with obesity and its underlying insulin resistance, regardless of tissue. Others have also identified methylation marks that are similar across different tissue types in association with a trait.9,30 Moreover, regression analyses of the SLC19A1 sites demonstrated a significant relationship with insulin sensitivity. The utility of the euglycemic hyperinsulinemic clamp is considered a gold standard for measuring insulin sensitivity.31 This was a key measurement to correlate with DNA methylation to identify epigenetic biomarkers of obese insulin resistance in whole-blood.
SLC19A1 codes for the protein reduced folate carrier (RFC), which contributes to methionine and de novo purine synthesis.32 Folate is a methyl donor, and is suggested to have an important role in fetal programming by providing a substrate for DNA methylation.33 Imbalances in folate levels, specifically high levels, have been predictive of adiposity and insulin resistance.33 This observation has provided evidence for potential epigenetic influences on the risk of disease development. Rupasree et al.34 conducted a study in systemic lupus erythematosus (SLE) cases and identified differential methylation in blood lymphocytes of genes involved in one-carbon metabolism. They found a decrease in SLC19A1 promoter methylation in SLE cases that were positive for anti-ribonucleoprotein (RNP) antibodies. The detection of anti-RNP in SLE patients is used for further classification of connective tissue diseases, such as Raynaud's phenomenon.35 Chronic inflammation in SLE may contribute to the similarities in decreased SLC19A1 methylation found in both obese, insulin resistant states from our study and the anti-RNP positive SLE cases.3,36 Increased levels of TNFα and IL-6 have been associated with both obesity and SLE.2,37 However, these measurements were not taken in this study, so inflammation cannot be confirmed in our participants. He et al.38 found a significant decrease in methylation in the promoter of SLC19A1 in the placenta of intrauterine growth restricted (IUGR) samples. This study speculated that the change in methylation may play a role in in utero development. IUGR has been associated with increased risk for type 2 diabetes, metabolic syndrome, and cardiovascular and heart disease.39 It is interesting to speculate that the decreased SLC19A1 methylation associated with obesity identified in our study may stem from a developmental origin. Although our study identified different chromosomal positions from the above mentioned studies, we believe the methylation status of both Chr.21:46,957,915 and Chr.21:46,957,981 provide new potential epigenetic biomarkers for better understanding obesity-related insulin resistance.
Our study focused on the identification of novel epigenetic biomarkers. Based on previous transcriptomic findings from our laboratory, we hypothesized that we would identify altered methylation of genes involved in inflammation, insulin signaling, and mitochondrial function.13,14 We identified 3 genes, integrin α E (ITGAE), RNA binding motif protein 5 (RBM5), and SLC19A1, in common with our previous findings.13 We expected to find more genes in common between the transcriptomic and epigenomic data sets. The lack of concordance across the data sets could be explained by differences in ethnicity and lower number of subjects study.40 However, the occurrence of SLC19A1 in the transcriptomic data set13 with the present study solidifies its connection to obesity.
In this study, we have focused primarily on SLC19A1; however, there are other genes from the list of 49 DMCs (q < 0.05) that could potentially be relevant to obesity. One such gene is EFNA2, which codes for the glycosylphosphatidylinositol (GPI)-linked ephrin-A ligand. EFNA2 interacts with Eph receptor tyrosine kinases thereby affecting the activities of actin cytoskeleton, cell motility, proliferation, and secretion.41 We have previously found a reduction in actin cytoskeleton proteins with insulin resistance.3,42 Another gene was COL18A1, which showed similar methylation changes in both whole-blood and skeletal muscle. COL18A1 codes for a multiplexin localized at the basal lamina.43 We3,44,45 and others46 have shown increased collagen content in insulin resistant skeletal muscle. Taken together, the changes in methylation for EFNA2 and COL18A1 suggest that these genes may be relevant epigenetic biomarkers of blood in obesity.
Although we have described novel epigenetic biomarkers of blood associated with obesity and its underlying insulin resistance, we acknowledge the shortcomings of our study. Whole blood has a heterogeneous cell composition, and potential differences in inflammation between our groups could confound the DNA methylation results.47 Furthermore, we identified new potential biomarkers for obesity-related insulin resistance within a limited sample size. Future studies could fractionate blood cell types to avoid confounding composition effects, and will need to replicate our findings in larger cohorts to be considered candidate biomarkers. Our study was novel in that we identified epigenetic changes in whole-blood using RRBS. Specifically, we identified SLC19A1 from obese participants as a potential epigenetic biomarker that is significantly predicted by insulin sensitivity (i.e., M value). Moreover, blood SLC19A1 methylation was positively correlated with skeletal muscle methylation. Our transcription factor binding analysis found potential binding within the SLC19A1 DMR, but not at the most significant DMC sites. However, we speculate that methylation at those sites may have a regulatory effect through the recruitment of methylcytosine-binding proteins.48 These proteins can associate with protein complexes that contain co-repressors and histone deacetylases, and could influence chromatin structure.47 Our findings demonstrate that the DNA methylation status associated with SLC19A1 is a promising biomarker for obesity and its underlying insulin resistance, as it is present in both skeletal muscle and blood.
Material and methods
Participants
Ten participants with obesity (BMI ≥ 30 kg/m2; 5 male/5 female; ages: 23–52 years) and 10 participants without obesity (BMI < 25 kg/m2; 5 male/5 female; ages: 21–43 years) took part in this study. Metabolic data for some of these participants were included in a previous publication.25 Demographics, anthropometric measurements, and screening blood tests were obtained on all participants. Body impedance analysis (BIA) was used to assess percent body fat. A 75 g oral glucose tolerance test following a 10–12 h overnight fast was used to assess normal glucose tolerance. No subject was taking any medication known to affect glucose metabolism. Written consent was obtained from all study participants. The study was approved by the Institutional Review Boards at Mayo Clinic in Arizona and Arizona State University.
Study design
Fasted participants reported to the Clinical Studies Infusion Unit at the Mayo Clinic in Arizona. Blood was collected into PAXgene Blood DNA and RNA tubes (BD Diagnostics, Franklin Lakes, NJ) and stored at −80°C until processed. Following blood collection, a 2-hour euglycemic hyperinsulinemic clamp (80 mU.m−2.min−1) to measure insulin sensitivity was performed as described previously.25
Substrate and hormone determinations
Fasted blood samples for comprehensive metabolic, lipid, and hemogram panels were performed by the Biospecimens Accessioning and Processing (BAP) Core at Mayo Clinic in Scottsdale. Plasma glucose concentration was determined by the glucose oxidase method on an YSI 2,300 STAT plus (YSI INC., Yellow Springs, OH, USA). Plasma insulin was measured by a 2-site immunoenzymatic assay performed on the DxI 800 automated immunoassay system (Beckman Instruments, Chaska, MN, USA).
Whole-blood processing for DNA isolation
Genomic DNA was isolated using the PAXgene Blood DNA Kit, as per the manufacturer's instructions (Qiagen, Valencia, CA). DNA quantity and quality was assessed using agarose gel electrophoresis and spectrophotometer A260/A280 values were determined using the NanoVue (GE Healthcare, United Kingdom).
Reduced Representation Bisulfite Sequencing
RRBS was performed on whole-blood genomic DNA at the Mayo Clinic Genotyping Shared Resource facility, and library preparation was performed as described previously.25 Sequencing data was analyzed using a streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing, SAAP-RRBS.25,49 The methylation data set supporting the conclusions of this article are available in the Gene Expression Omnibus repository, GSE85928 (http://www.ncbi.nlm.nih.gov/geo/). Furthermore, bigwig files were used to create a custom track on the UCSC genome browser (https://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=rlcolett&hgS_otherUserSessionName=blood%20Methylatio).
Whole-blood differentially methylated cytosines analysis
To determine differences in methylation sites between groups, the aligned (Hg19) data was imported into the free open source R package, MethylSig.50 A minimum of 5 reads and the recovery of the site in all 10 participants from each group were required for the inclusion of a cytosine in subsequent analyses. The mean methylation values were adjusted by a β binomial approach to account for biologic variation among the groups being compared.50 A comparison of the DNA methylation between groups with and without obesity at each site was based on a likelihood ratio test (nominal P value) and a Benjamini-Hochberg multiple testing correction was applied. Regional annotations for each DMC were imported from the University of California, Santa Cruz (UCSC) Genome Browser's RefSeq Genes and CpG Island tracks. Priority was given to annotating the site as a promoter or untranslated region if available in another transcript of the gene or in a different gene.
Whole-blood differentially methylated region analysis
Differences in methylated regions between groups were identified using the open source R package, dispersion shrinkage for sequencing data (DSS).51 The analysis included the BSmooth algorithm, which determined the level of methylation in a region for each sample and accounted for biologic variation. The criteria for inclusion was: 1) each region contained 3 CpGs supported with a read coverage of 5X; 2) recovery of the site in all 10 participants from each group; and 3) significance of P<0.05 (uncorrected) from the DMC analysis. DMRs were created by a sliding-window of 500 bp and a t-statistic cutoff of 2.5. The significance of a DMR was determined by the weight of the Area Stat, which is the sum of t-statistic values in each DMR. Regional annotations for the DMRs were imported from the UCSC Genome Browser's RefSeq Genes and CpG Island tracks. Priority was given to annotating the region as a promoter or untranslated region if available in another transcript of the gene or in a different gene.
Blood-based biomarkers of skeletal muscle DMC analysis
Skeletal muscle RRBS data from 11 lean (7 females/4 males; ages: 21–43 years) and 9 obese (BMI > 30 kg/m2; 4 females/5 males; ages: 32–52 years) in our previous study25 was used for comparative analysis. Both whole-blood and skeletal muscle were analyzed using the program MethylSig.50 There were 9 lean and 6 obese that were the same as the individuals included in the whole-blood analyses. Promoter and untranslated region DMCs from both whole-blood and skeletal muscle were merged based on matching chromosomal positions. The merged DMCs were then filtered for analogous direction and level of methylation in each tissue by grouping.
SLC19A1 predictive transcription factor binding analysis
Prediction of transcription factor binding was performed using the program PROMO version 3.0.2.26 Analyses were performed with a 5% maximum matrix dissimilarity rate using TRANSFAC version 8.3 database. The sequence from Chr.21:46,957,905,–46,957,991 was used to assess binding at the 2 SLC19A1 DMCs. Furthermore, transcription factor binding was assessed for the SLC19A1 DMR using the sequence from Chr.21:46,957,905,–46,958,011.
Pyrosequencing
Confirmation of DNA methylation detected in both whole-blood and skeletal muscle was performed using pyrosequencing, as described previously.25 To assess the SLC19A1 DMC (Chr.21:46,957,915), bisulfite-converted DNA was amplified by PCR using the following primers: forward 5’-GTTGGGTTGGAGGGTATTAT-3’ and biotinylated reverse 5’-CCATCTTCCAAAATACCCTAACT-3’. Pyrosequencing was performed using the PyroMark Q96 MD system and the Gold Q96 kit with sequencing primers 5’-GGTTGGAGGGTATTATT-3’ according to the manufacturer's instructions (Qiagen, Valencia, CA). Sequence analysis was performed using the PyroMark CpG SW 1.0 software (Qiagen, Valencia, CA).
Statistical analysis
Independent sample t-tests and chi-square were used to compare physical and metabolic characteristics between lean and obese groups. Non-normally distributed data were log10 or square root transformed. However, untransformed data are presented as a mean ± standard error of the mean (SEM) for ease of interpretation. Multiple regression analyses were performed with the purpose of adjustments for age, sex, and/or BMI to estimate bivariate relationships between insulin sensitivity (i.e., M value) and significantly altered methylation. Pearson correlation analysis was performed to determine the relationship between whole-blood and skeletal muscle methylation data. The SPSS 23.0 statistical software package was used. See above for the statistical analysis of the methylation data.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the volunteers of the study and are grateful for their participation and cooperation. We thank the Clinical Studies Infusion Unit nurses and research staff for their excellent care of the participants. We thank the Mayo Clinic Genotyping Shared Resource facility for the RRBS next generation methylation analysis. We thank Kara Peterson and Dr. Melanie Carless for their assistance with the pyrosequencing experiments.
Funding
This study was supported by the National Institutes of Health Grants R01DK094013 (DKC).
Author contributions
DKC conceived the experiments. SED, LAG, LEC, RLC and DKC performed the experiments. TRB, LRR and EADF performed the euglycemic hyperinsulinemic clamps. SED, RLC, JYK and DKC performed the analysis of the data with assistance from LJM. SED and DKC wrote the article. JYK, LAG, LEC, RLC, LRR, TRB, EADF, and LJM read the manuscript and provided comments. DKC is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Declarations
Ethical approval statements are listed in the methods. All authors read and approved the manuscript.
References
- 1.Kyrou I, Randeva HS, Weickert MO. Clinical Problems Caused by Obesity. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, et al., eds. Endotext, South Dartmouth (MA), 2000; PMID:2590520723964268 [Google Scholar]
- 2.Tateya S, Kim F, Tamori Y. Recent advances in obesity-induced inflammation and insulin resistance. Front Endocrinol 2013; 4:93; PMID:23964268; http://dx.doi.org/ 10.3389/fendo.2013.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coletta DK, Mandarino LJ. Mitochondrial dysfunction and insulin resistance from the outside in: extracellular matrix, the cytoskeleton, and mitochondria. Am J Physiol Endocrinol Metab 2011; 301:E749–55; PMID:21862724; http://dx.doi.org/ 10.1152/ajpendo.00363.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verdile G, Keane KN, Cruzat VF, Medic S, Sabale M, Rowles J, Wijesekara N, Martins RN, Fraser PE, Newsholme P. Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer's disease. Mediators Inflamm 2015; 2015:105828; PMID:26693205; http://dx.doi.org/ 10.1155/2015/105828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010; 72:219–46; PMID:20148674; http://dx.doi.org/ 10.1146/annurev-physiol-021909-135846 [DOI] [PubMed] [Google Scholar]
- 6.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 2006; 6:772–83; PMID:16998510; http://dx.doi.org/ 10.1038/nri1937 [DOI] [PubMed] [Google Scholar]
- 7.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007; 132:2169–80; PMID:17498510; http://dx.doi.org/ 10.1053/j.gastro.2007.03.059 [DOI] [PubMed] [Google Scholar]
- 8.Dayeh T, Tuomi T, Almgren P, Perfilyev A, Jansson PA, de Mello VD, Pihlajamaki J, Vaag A, Groop L, Nilsson E, et al.. DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics 2016; 11:482–8; PMID:27148772; http://dx.doi.org/ 10.1080/15592294.2016.1178418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mikeska T, Craig JM. DNA methylation biomarkers: cancer and beyond. Genes 2014; 5:821–64; PMID:25229548; http://dx.doi.org/ 10.3390/genes5030821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh S, Dhingra S, Ramdath DD, Vasdev S, Gill V, Singal PK. Risk factors preceding type 2 diabetes and cardiomyopathy. J Cardiovasc Transl Res 2010; 3:580–96; PMID:20593256; http://dx.doi.org/ 10.1007/s12265-010-9197-3 [DOI] [PubMed] [Google Scholar]
- 11.Niswender K. Diabetes and obesity: therapeutic targeting and risk reduction - a complex interplay. Diabetes Obes Metab 2010; 12:267–87; http://dx.doi.org/ 10.1111/j.1463-1326.2009.01175.x [DOI] [PubMed] [Google Scholar]
- 12.O'Connell TM, Markunas CA. DNA methylation and MicroRNA-based biomarkers for risk of type 2 diabetes. Curr Diabetes Revi 2016; 12:20–9; PMID:25981498; http://dx.doi.org/ 10.2174/1573399811666150515125557 [DOI] [PubMed] [Google Scholar]
- 13.Tangen SE, Tsinajinnie D, Nunez M, Shaibi GQ, Mandarino LJ, Coletta DK. Whole blood gene expression profiles in insulin resistant Latinos with the metabolic syndrome. PloS One 2013; 8:e84002; PMID:24358323; http://dx.doi.org/ 10.1371/journal.pone.0084002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JY, Campbell LE, Shaibi GQ, Coletta DK. Gene expression profiling and association of circulating lactoferrin level with obesity-related phenotypes in Latino youth. Pediatr Obes 2015; 10:338–44; PMID:25394788; http://dx.doi.org/ 10.1111/ijpo.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429:457–63; PMID:15164071; http://dx.doi.org/ 10.1038/nature02625 [DOI] [PubMed] [Google Scholar]
- 16.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 2009; 58:2718–25; PMID:19940235; http://dx.doi.org/ 10.2337/db09-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu B, Russanova VR, Gravina S, Hartley S, Mullikin JC, Ignezweski A, Graham J, Segars JH, DeCherney AH, Howard BH. DNA methylome and transcriptome sequencing in human ovarian granulosa cells links age-related changes in gene expression to gene body methylation and 3′-end GC density. Oncotarget 2015; 6:3627–43; PMID:25682867; http://dx.doi.org/ 10.18632/oncotarget.2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maussion G, Yang J, Suderman M, Diallo A, Nagy C, Arnovitz M, Mechawar N, Turecki G. Functional DNA methylation in a transcript specific 3′UTR region of TrkB associates with suicide. Epigenetics 2014; 9:1061–70; PMID:24802768; http://dx.doi.org/ 10.4161/epi.29068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al.. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 2006; 38:1378–85; PMID:17072317; http://dx.doi.org/ 10.1038/ng1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aslibekyan S, Demerath EW, Mendelson M, Zhi D, Guan W, Liang L, Sha J, Pankow JS, Liu C, Irvin MR, et al.. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity 2015; 23:1493–501; PMID:26110892; http://dx.doi.org/ 10.1002/oby.21111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toperoff G, Aran D, Kark JD, Rosenberg M, Dubnikov T, Nissan B, Wainstein J, Friedlander Y, Levy-Lahad E, Glaser B, et al.. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum Mol Genet 2012; 21:371–83; PMID:21994764; http://dx.doi.org/ 10.1093/hmg/ddr472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S, Meduri E, Morange PE, Gagnon F, Grallert H, et al.. DNA methylation and body-mass index: a genome-wide analysis. Lancet 2014; 383:1990–8; PMID:24630777; http://dx.doi.org/ 10.1016/S0140-6736(13)62674-4 [DOI] [PubMed] [Google Scholar]
- 23.Nilsson EK, Ernst B, Voisin S, Almen MS, Benedict C, Mwinyi J, Fredriksson R, Schultes B, Schioth HB. Roux-en Y gastric bypass surgery induces genome-wide promoter-specific changes in DNA methylation in whole blood of obese patients. PloS One 2015; 10:e0115186; PMID:25710379; http://dx.doi.org/ 10.1371/journal.pone.0115186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdul-Ghani MA, DeFronzo RA. Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol 2010; 2010:476279; PMID:20445742; http://dx.doi.org/27437034 10.1155/2010/476279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Day SE, Coletta RL, Kim JY, Campbell LE, Benjamin TR, Roust LR, De Filippis EA, Dinu V, Shaibi GQ, Mandarino LJ, et al.. Next-generation sequencing methylation profiling of subjects with obesity identifies novel gene changes. Clin Epigenet 2016; 8:77; PMID:27437034; http://dx.doi.org/ 10.1186/s13148-016-0246-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, Alba MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002; 18:333–4; PMID:11847087; http://dx.doi.org/ 10.1093/bioinformatics/18.2.333 [DOI] [PubMed] [Google Scholar]
- 27.Deaton AM, Bird A. CpG islands and the regulation of transcription. Gen Dev 2011; 25:1010–22; PMID:21576262; http://dx.doi.org/21176133 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Zhu H, Snieder H, Su S, Munn D, Harshfield G, Maria BL, Dong Y, Treiber F, Gutin B, et al.. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med 2010; 8:87; PMID:21176133; http://dx.doi.org/ 10.1186/1741-7015-8-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ronn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, Jorgensen SW, Brons C, Jansson PA, Eriksson KF, et al.. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet 2015; 24:3792–813; PMID:25861810; http://dx.doi.org/ 10.1093/hmg/ddv124 [DOI] [PubMed] [Google Scholar]
- 30.Gillberg L, Ling C. The potential use of DNA methylation biomarkers to identify risk and progression of type 2 diabetes. Front Endocrinol 2015; 6:43; PMID:25870586; http://dx.doi.org/ 10.3389/fendo.2015.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979; 237:E214–23; PMID:382871 [DOI] [PubMed] [Google Scholar]
- 32.Desmoulin SK, Hou Z, Gangjee A, Matherly LH. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol Ther 2012; 13:1355–73; PMID:22954694; http://dx.doi.org/24333266 10.4161/cbt.22020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yajnik CS, Deshmukh US. Maternal nutrition, intrauterine programming and consequential risks in the offspring. Rev Endocr Metab Dis 2008; 9:203–11; PMID:18661241; http://dx.doi.org/24333266 10.1007/s11154-008-9087-z [DOI] [PubMed] [Google Scholar]
- 34.Rupasree Y, Naushad SM, Rajasekhar L, Kutala VK. Epigenetic modulation of RFC1, MHC2TA and HLA-DR in systemic lupus erythematosus: association with serological markers and six functional polymorphisms of one-carbon metabolic pathway. Gene 2014; 536:45–52; PMID:24333266; http://dx.doi.org/ 10.1016/j.gene.2013.11.094 [DOI] [PubMed] [Google Scholar]
- 35.Migliorini P, Baldini C, Rocchi V, Bombardieri S. Anti-Sm and anti-RNP antibodies. Autoimmunity 2005; 38:47–54; PMID:15804705; http://dx.doi.org/ 10.1080/08916930400022715 [DOI] [PubMed] [Google Scholar]
- 36.Podolska MJ, Biermann MH, Maueroder C, Hahn J, Herrmann M. Inflammatory etiopathogenesis of systemic lupus erythematosus: an update. J Inflammat Res 2015; 8:161–71; PMID:26316795; http://dx.doi.org/ 10.2147/JIR.S70325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agha-Hosseini F, Moosavi MS, Hajifaraj Tabrizi M. Comparison of oral lichen planus and systemic lupus erythematosus in interleukins level. Arch Iranian Med 2015; 18:703–12; PMID:26443253; http://dx.doi.org/0151810/AIM.0011 [PubMed] [Google Scholar]
- 38.He Z, Lu H, Luo H, Gao F, Wang T, Gao Y, Fang Q, Wang J. The promoter methylomes of monochorionic twin placentas reveal intrauterine growth restriction-specific variations in the methylation patterns. Sci Rep 2016; 6:20181; PMID:26830322; http://dx.doi.org/ 10.1038/srep20181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chernausek SD. Update: consequences of abnormal fetal growth. J Clin Endocrinol Metab 2012; 97:689–95; PMID:22238390; http://dx.doi.org/ 10.1210/jc.2011-2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cossrow N, Falkner B. Race/ethnic issues in obesity and obesity-related comorbidities. J Clin Endocrinol Metab 2004; 89:2590–4; PMID:15181028; http://dx.doi.org/ 10.1210/jc.2004-0339 [DOI] [PubMed] [Google Scholar]
- 41.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008; 133:38–52; PMID:18394988; http://dx.doi.org/ 10.1016/j.cell.2008.03.011 [DOI] [PubMed] [Google Scholar]
- 42.Hwang H, Bowen BP, Lefort N, Flynn CR, De Filippis EA, Roberts C, Smoke CC, Meyer C, Hojlund K, Yi Z, et al.. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes 2010; 59:33–42; PMID:19833877; http://dx.doi.org/ 10.2337/db09-0214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halfter W, Dong S, Schurer B, Cole GJ. Collagen XVIII is a basement membrane heparan sulfate proteoglycan. J Biol Chem 1998; 273:25404–12; PMID:9738008; http://dx.doi.org/ 10.1074/jbc.273.39.25404 [DOI] [PubMed] [Google Scholar]
- 44.Richardson DK, Kashyap S, Bajaj M, Cusi K, Mandarino SJ, Finlayson J, DeFronzo RA, Jenkinson CP, Mandarino LJ. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem 2005; 280:10290–7; PMID:15598661; http://dx.doi.org/ 10.1074/jbc.M408985200 [DOI] [PubMed] [Google Scholar]
- 45.Berria R, Wang L, Richardson DK, Finlayson J, Belfort R, Pratipanawatr T, De Filippis EA, Kashyap S, Mandarino LJ. Increased collagen content in insulin-resistant skeletal muscle. Am J Physiol Endocrinol Metab 2006; 290:E560–5; PMID:16249255; http://dx.doi.org/ 10.1152/ajpendo.00202.2005 [DOI] [PubMed] [Google Scholar]
- 46.Kang L, Ayala JE, Lee-Young RS, Zhang Z, James FD, Neufer PD, Pozzi A, Zutter MM, Wasserman DH. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes 2011; 60:416–26; PMID:21270253; http://dx.doi.org/ 10.2337/db10-1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Houseman EA, Kim S, Kelsey KT, Wiencke JK. DNA methylation in whole blood: uses and challenges. Curr Environ Health Rep 2015; 2:145–54; PMID:26231364; http://dx.doi.org/ 10.1007/s40572-015-0050-3 [DOI] [PubMed] [Google Scholar]
- 48.Attwood JT, Yung RL, Richardson BC. DNA methylation and the regulation of gene transcription. Cell Mol Life Sci 2002; 59:241–57; PMID:11915942; http://dx.doi.org/ 10.1007/s00018-002-8420-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Z, Baheti S, Middha S, Kanwar R, Zhang Y, Li X, Beutler AS, Klee E, Asmann YW, Thompson EA, et al.. SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics 2012; 28:2180–1; PMID:22689387; http://dx.doi.org/ 10.1093/bioinformatics/bts337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park Y, Figueroa ME, Rozek LS, Sartor MA. MethylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics 2014; 30:2414–22; PMID:24836530; http://dx.doi.org/ 10.1093/bioinformatics/btu339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu H, Xu T, Feng H, Chen L, Li B, Yao B, Qin Z, Jin P, Conneely KN. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res 2015; 43:e141; PMID:26184873; http://dx.doi.org/ 10.1093/nar/gkv715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.