

Abstract
The Hippo pathway senses cellular conditions and regulates YAP/TAZ to control cellular and tissue homeostasis, while TBK1 is central for cytosolic nucleic acid sensing and antiviral defense. The correlation between cellular nutrient/physical status and host antiviral defense is interesting but not well understood. Here we find that YAP/TAZ act as natural inhibitors of TBK1 and are vital for antiviral physiology. Independent of transcriptional regulation and through transactivation domain, YAP/TAZ associate directly with TBK1 and abolish virus-induced TBK1 activation, by preventing TBK1 K63-linked ubiquitination and adaptors/substrates binding. Accordingly, YAP/TAZ deletion/depletion or cellular conditions inactivating YAP/TAZ through Lats1/2 kinases relieve TBK1 suppression and boost antiviral responses, whereas expression of the transcriptionally inactive YAP dampens cytosolic RNA/DNA sensing and weakens the antiviral defense in cells and zebrafish. Thus, we describe a function of YAP/TAZ and the Hippo pathway in innate immunity, by linking cellular nutrient/physical status to antiviral host defense.
Keywords: Hippo pathway, YAP/TAZ, TBK1, antiviral response, host defense
INTRODUCTION
Metazoans use innate defense mechanisms to recognize conserved pathogen-associated molecular patterns (PAMPs) and fight against pathogen infections. Cytosolic nucleic acid sensors are crucial components of defense system in vertebrates, particularly for detecting viruses that have breached physical barriers and been replicated within the cell. Viral double-stranded RNA is detected in the cytosol by RIG-I-Like receptors (RLRs)1–2, while viral DNA is recognized by cytosolic sensors including cGAS3–6, IFI16, DDX41, and others7. Facilitated by mitochondrial-associated MAVS (also known as VISA, IPS-1, or Cardif) or endoplasmic reticulum-located STING (also known as ERIS, MITA, MPYS, or TMEM173), viral nucleic acids recognition leads to the activation of TBK1 and/or IKKε kinases that phosphorylate and mobilize IRF3, which then dimerizes and translocates to the nucleus, where it acts as a DNA-binding transcription factor8–9. Assembly of MAVS or STING signaling complex also induces NF-κB activation10–11, which cooperates with IRF3 to drive the expression of type I and III interferons. The antiviral defense of the self and neighboring cells is thus established by coordinating a large number of interferon-stimulated genes (ISGs) through classical JAK-STAT signaling, to clear/prevent viral infection and modulate adaptive immunity12–13.
How cytosolic nucleic acid sensing is affected by cellular conditions, such as nutrient/energy stress or cell-cell contact, is an interesting question that remains to be answered. Self-association of MAVS or STING molecules initiates the recruitment of TRAFs, TBK1/IKKε, and IRF314, where intermolecular trans-phosphorylation, facilitated by K63-linked ubiquitination and adaptor-driven association, leads to TBK1/IKKε activation15. Viral-induced TBK1 activation is a slow process subject to complex regulations involving interacting proteins and posttranslational modifications16–18, including ion metal phosphatase PPM1A19 and kinase Mst120. Conversely, aberrant reactions to own nucleic acids and subsequent IFN production trigger autoimmune and autoinflammatory diseases21–22, thus requires strict regulation. TBK1 and IKKε also serve as key regulators of apoptosis, autophagy, inflammatory responses23–25 and act as important inducers to drive tumorigenesis26–27. Nevertheless, the regulatory mechanism for TBK1/IKKε activation and termination is largely unknown.
The Hippo pathway was originally discovered in Drosophila and is highly conserved28–31. Transcription co-activators YAP and TAZ are the downstream effectors, which are regulated by the Lats1/2 kinases in response to unfavorable growth conditions to retain YAP/TAZ in the cytoplasm for ubiquitination and degradation32–33. Otherwise, YAP/TAZ are localized in the nucleus to bind to and activate the TEAD family transcription factors to transcribe target genes promoting cell proliferation, migration, and survival34. How the Hippo pathway cooperates with other signaling pathways to regulate a variety of physiological processes, such as host defense, is largely unanswered. Regulation of YAP/TAZ is very complex and can be affected via cross-talk with the WNT pathway35, G protein-coupled receptor (GPCR) signaling36–39, and the transforming growth factor-β (TGF-β)40 and Notch pathways41. Both Hippo and cytosolic nucleic acid sensing are ancient and evolutionally highly conserved pathways and are present in all vertebrates. The opposing biological processes, such as growth and survival governed by YAP/TAZ and danger sensing controlled by TBK1, indicate that these factors may influence each other. Pan group recently reported an intriguing crosstalk between the Hippo pathway and Toll-like receptor signaling in Drosophila through Yorkie-mediated induction of the IκB homolog Cactus42. The finding indicates an integral role for YAP/TAZ in anti-bacterial host defense in invertebrate.
Here we find that key components of antiviral defense, the TBK1/IKKε kinases, are directly suppressed by YAP/TAZ independent of their transcriptional potential. YAP/TAZ associate with TBK1 and prevent its K63 ubiquitination and adaptor/substrate association. Accordingly, YAP/TAZ knockout or knockdown, or cellular conditions activating Hippo signaling, relieves TBK1 inhibition and boosts the antiviral resistance. Conversely, a transcriptionally-inactive YAP mutant can sensitize cells and zebrafish to virus attack. This work reveals an unexpected function of YAP/TAZ and the Hippo pathway in cytosolic nucleic acid sensing and innate antiviral immunity.
RESULTS
Cellular conditions activating Hippo signaling boost cytosolic RNA/DNA sensing
Understanding the regulation of host antiviral immunity by cellular nutrition/physical status is important but not systemically studied previously. We first evaluated the level of cellular antiviral signaling upon serum starvation by an IRF3-responsive IFNβ reporter. We observed an unanticipated increase in IRF3 transactivation under starvation in response to Sendai virus (SeV) infection (Fig. 1A). Meanwhile we observed an elevated activation of endogenous TBK1 in starved cells upon SeV infection, detected by a phospho-Ser172-specific antibody (Fig. 1B, 1st panel). Likewise, starvation boosted IRF3 transactivation stimulated by ectopic expression of activated RIG-I (caRIG-I) or STING (Fig. 1C and 1D), but did not significantly potentiate signaling such as Wnt, Hedgehog, or TGF-β/Smad (sFig. 1A). Serum starvation is known to activates the Hippo pathway36–37, evidenced by increased YAP Ser127 phosphorylation and TAZ degradation (Fig. 1B). Double deletion of Lats1/2, the upstream kinases of YAP/TAZ, by CRISPR/Cas9 genomic editing27 abolished both effects in response to cellular nutrient/energy stresses (sFig. 1B). Intriguingly, a decrease of IRF3 responsiveness was observed in Lats1/2 dKO HEK293A cells, along with the loss of starvation-induced IRF3 transactivation (Fig. 1C and 1D). This could be partially rescued by re-introduction of Lats1 expression (Fig. 1E). These observations suggest that cellular nutrient status regulate antiviral sensing and it involves the Hippo pathway.
Figure 1. Activation of Hippo signaling enhances cytosolic RNA/DNA sensing.
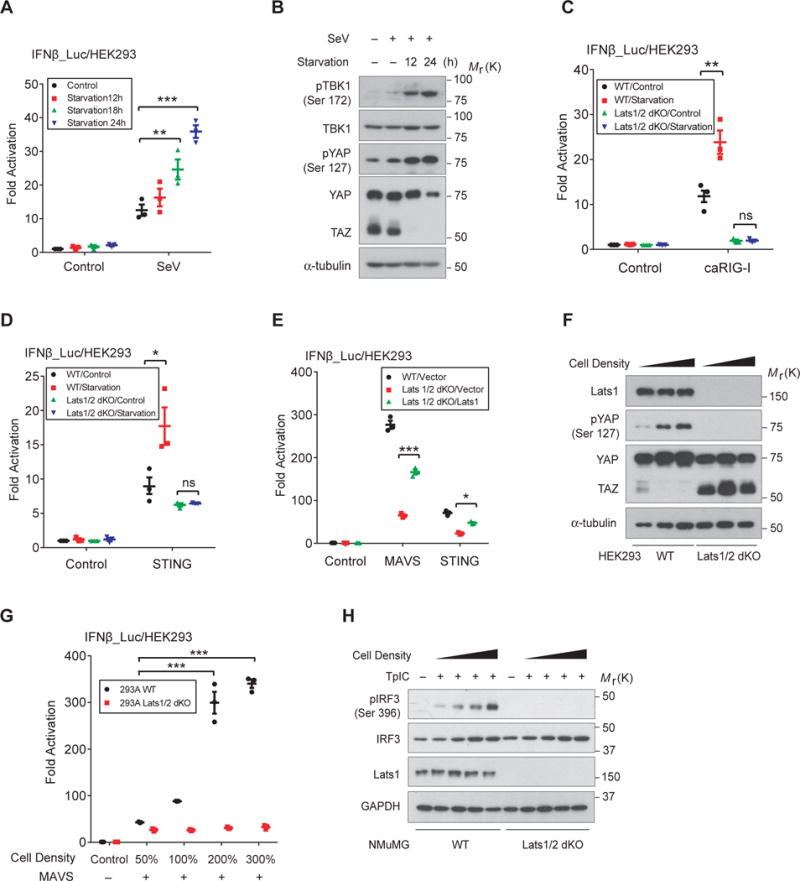
(A), Cellular nutrient stress by serum starvation potentiated the IRF3 responsiveness in HEK293T cells stimulated by the infection of Sendai virus (SeV). n=3 independent experiments. Mean ± SEM. *P=0.029, and ***P<0.001, by ANOVA test and Bonferroni correction. (B), Endogenous TBK1 activation, revealed by immunoblotting TBK1 Ser172 phosphorylation, but not its expression, was profoundly increased upon nutrient stress in response to the infection of SeV. As expected, serum starvation activated Hippo signaling, evidenced by enhanced YAP Ser127 phosphorylation and TAZ degradation. (C and D), Serum starvation boosted IRF3 transactivation in wild-type HEK293A cells, but not in cells with Lats1/2 deletion (dKO), stimulated by the expression of either activated RIG-I (caRIG-I) (C) or STING (D). IRF3 transactivation was also lower in Lats1/2 dKO cells. n=3 independent experiments. Mean ± SEM. **P=0.0038, and * P=0.022, by ANOVA test and Bonferroni correction. (E), Re-introduction of Lats1 expression in Lats1/2 dKO cells partially rescued the defect of cytosolic RNA/DNA sensing signaling stimulated by coexpression of MAVS or STING. n=3 independent experiments. Mean ± SEM. ***P<0.001, and *P=0.016, by ANOVA test and Bonferroni correction. (F), Strong TAZ accumulation and YAP Ser127 phosphorylation was detected in Lats1/2 dKO HEK293A cells, which failed to respond to cell density for YAP phosphorylation and TAZ degradation. (G and H), Wild-type or Lats1/2 dKO HEK293A or NMuMG cells were transfected with MAVS (G) or poly(I:C) (H) to activate signaling of cytosolic RNA sensing, then seeded into different confluence to activate the Hippo pathway. Markedly enhanced IRF3 activation was detected by IFNβ reporter in wild-type cells with high cell density (G), or by immunoblotting of endogenous IRF3 Ser396 phosphorylation after poly(I:C) stimulation (H). In contrast, Lats1/2 dKO cells failed largely to boost IRF3 activation in response to increased cell density (G and H). n=3 independent experiments. Mean ± SEM. ***P<0.001, byANOVA test and Bonferroni correction. Unprocessed images of blots are shown in Supplementary Figure 6. Statistics source data are provided in Supplementary Table 1.
High cell confluence is known to activate Hippo signaling and lead to YAP/TAZ inactivation and degradation (33,43 and Fig. 1F). We thus examined the effect of high cell confluence on IRF3 activation. When stimulated by MAVS, we detected a robust enhancement of IRF3 transactivation in cells at high confluency, which was absent in Lats1/2 dKO cells (Fig. 1G). Likewise, poly(I:C) transfection (TpIC)-induced endogenous IRF3 activation, which simulates cytosolic RNA sensing, was also diminished in Lats1/2 dKO cells (Fig. 1H). Together, these observations verify that the Hippo pathway is a potent regulator of cellular antiviral response.
YAP/TAZ attenuate cytosolic nucleic acid sensing and the antiviral response
YAP/TAZ are Lats1/2 substrates and are key effectors of the Hippo pathway. The level of TAZ protein and/or YAP phosphorylation are related with antiviral signaling (Fig. 1B–D, and 1F–G). We thus examined the potential effect of YAP/TAZ. Reporter assays with IRF3-responsive IFNβ or ISRE promoter revealed that antiviral responses stimulated by activated RIG-I (caRIG-I) (Fig. 2A and 2B), STING (Fig. 2C), or TBK1 and IKKε (sFig. 2A and 2B), were all strongly inhibited by ectopic expression of YAP or TAZ in a dose-dependent manner. Similarly, RIG-I-induced IRF3 Ser396 phosphorylation was abolished by YAP cotransfection in a dose-dependent manner (Fig. 2D). Conversely, RIG-I or STING stimulated IFNβ reporter was significantly higher, when either YAP or TAZ or both was knocked down by siRNA (Fig. 2E and 2F), similar to MAVS-induced IRF3 transactivation (sFig. 2C). We also detected an enhanced TBK1 auto-activation in YAP/TAZ knockdown cells (sFig. 2D). All these observations suggest a negative regulation of YAP/TAZ in antiviral signaling. We observed a similar suppression of YAP/TAZ in TRIF-stimulated IRF3 transactivation or MyD88-mediated pathway (sFig. 2E and 2F). However, since YAP/TAZ proteins were often at a very low level in variety of immune cells (44 and sFig. 2G), their regulation on TRIF/MyD88 pathways requires further validation.
Figure 2. YAP/TAZ attenuate cytosolic RNA/DNA sensing and antiviral responses.
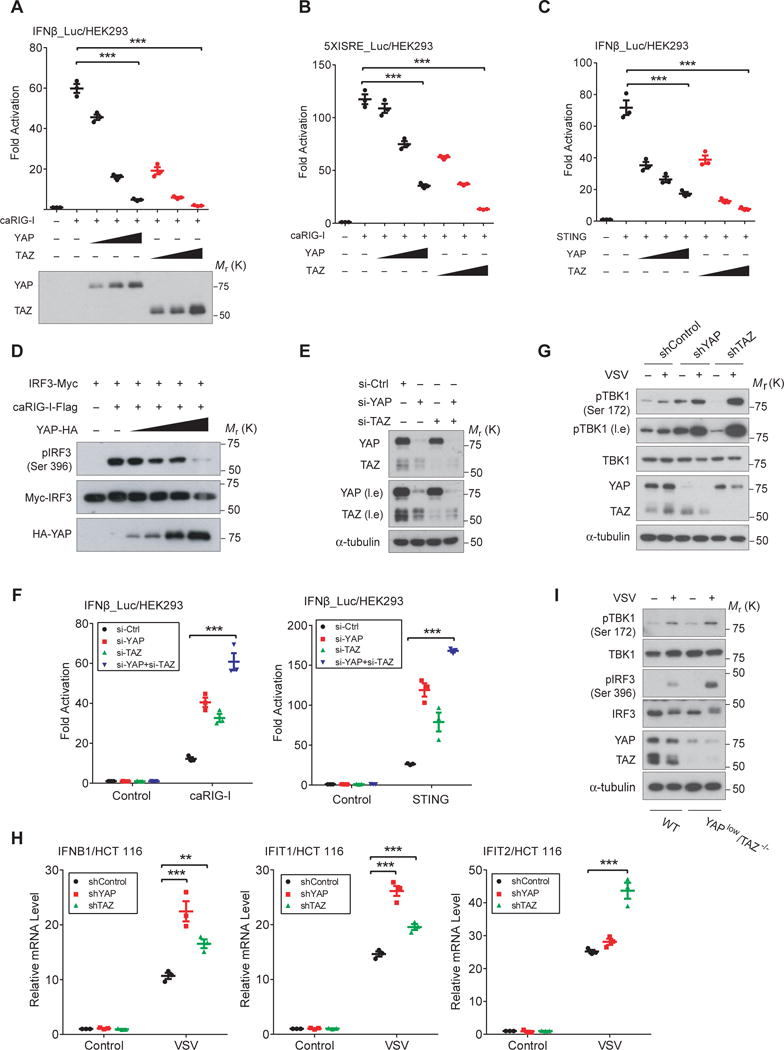
(A and B), Ectopic expression of YAP or TAZ inhibited IRF3 transactivation, which was stimulated by caRIG-I and examined by the IFNβ reporter (A) or 5xISRE reporter (B), in a dose-dependent manner. n=3 independent experiments. Mean ± SEM. ***P<0.001, by ANOVA test and Bonferroni correction. (C), STING-induced IRF3 activation, which stimulates cytosolic DNA sensing, was suppressed by the cotransfection of YAP or TAZ in a dose-dependent manner. n=3 independent experiments. Mean ± SEM. ***P<0.001, by ANOVA test and Bonferroni correction. (D), IRF3 activation, stimulated by caRIG-I and detected by immunoblotting of IRF3 phospho-Ser396, was abolished by the coexpression of YAP in a dose-dependent manner. (E and F), siRNA-mediated knockdown of YAP and/or TAZ in HEK293A cells potentiated the caRIG-I or STING-stimulated IRF3 responsiveness. Higher IRF3 transactivation was detected when both YAP and TAZ were depleted. The efficiency of YAP/TAZ depletion was verified by immunoblotting (E). n=3 independent experiments. Mean ± SEM. ***P <0.001, by ANOVA test and Bonferroni correction. (G), YAP or TAZ in HCT 116 cells was knocked down by shRNA and verified by immunoblotting (4th panel). VSV infection-induced activation of endogenous TBK1 was more robust after YAP or TAZ depletion. (H), VSV infection-induced mRNA expression of IFNβ and ISGs were boosted in HCT 116 cells with YAP or TAZ depletion, as evaluated by qRT-PCR assays at 12 hpi. n=3 independent experiments. Mean ± SEM. *** P<0.001, and **P=0.0059 by ANOVA test and Bonferroni correction. (I), YAPlow/TAZ−/− NMuMG cells was generated by CRISPR/Cas9-mediated genomic editing and verified by immunoblotting (5th panel), which exhibited an enhanced level of activation for endogenous TBK1 and IRF3 upon VSV infection. Unprocessed images of blots are shown in Supplementary Figure 6. Statistics source data are provided in Supplementary Table 1.
We subsequently examined endogenous TBK1 activation upon VSV infection in HCT 116 colon carcinoma cells, which had shRNA-mediated knockdown of YAP or TAZ. The shRNA-mediated knockdown was efficient (Fig. 2G), and a marked enhancement of VSV-induced activation of endogenous TBK1 was detected (Fig. 2G), as well as an enhanced expression of IFNβ and ISGs (Fig. 2H). Since YAP/TAZ dKO cells grow extremely slowly and were not practical for use in experiments, we generated YAPlow/TAZ−/− NMuMG cells by CRISPR/Cas9 genomic editing and verified the expression of YAP/TAZ (Fig. 2I). YAPlow/TAZ−/− NMuMG cells exhibited a significant enhancement of endogenous TBK1 and IRF3 activation upon VSV infection (Fig. 2I). These consistent observations suggest that YAP/TAZ negatively regulate cytosolic antiviral sensing and antiviral response.
YAP/TAZ inhibit TBK1 activation independent of transcriptional regulation
To dissect the molecular basis for YAP/TAZ-mediated TBK1 inhibition, we first examined effects of the transcriptionally active (5SA) and inactive (6SA) form of YAP45–47. The S94A mutation in the YAP 6SA mutant abolishes its interaction with TEADs and thus is transcriptionally inactive (48–49 and Fig. 3A). Measured by the IFNβ reporter, we unexpectedly observed a profound inhibition of IRF3 transactivation by YAP 6SA, similar to or even stronger than wild-type or active YAP (Fig. 3B). This observation suggests that YAP-mediated suppression might be a direct effect rather than through its transcriptional target(s).
Figure 3. YAP and TAZ abrogate TBK1 activation independent of their transcriptional potential.
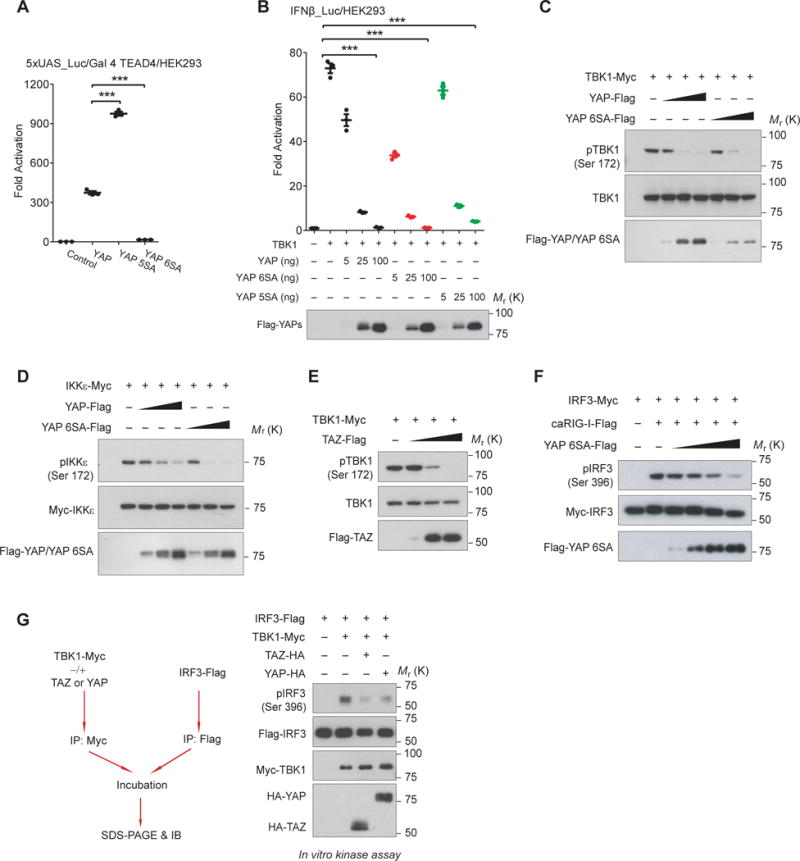
(A and B), Ectopic expression of YAP wild-type, the transcriptionally active (5SA), or the transcriptionally inactive (6SA) mutant elicited similar inhibition on TBK1-induced IRF3 transactivation (B, upper panel). Transcriptional potential and expression of YAPs were revealed by TEAD4-responsive reporter (A) and by immunoblotting (B, lower panel), respectively. n=3 independent experiments. Mean ± SEM. ***P<0.001, by ANOVA test and Bonferroni correction. (C and D), Cotransfection of wild-type or the transcriptionally inactive (6SA) YAP with TBK1/IKKε resulted in the failure of auto-phosphorylation and activation of TBK1 (C) or IKKε (D), in a dose-dependent manner. (E), TAZ expression led to the inactivation of TBK1 in a dose-dependent manner. (F), YAP 6SA prevented RIG-I-stimulated activation of IRF3 in a dose-dependent pattern. (G), TBK1 isolated from HEK293T cells with YAP or TAZ cotransfection failed to phosphorylate IRF3 in vitro, suggesting the loss of TBK1 kinase activity in the presence of YAP or TAZ. Unprocessed images of blots are shown in Supplementary Figure 6. Statistics source data are provided in Supplementary Table 1.
We detected a marked decrease of TBK1 and IKKε activation by cotransfecting of wild-type YAP or YAP 6SA (Fig. 3C and 3D), or TAZ (Fig. 3E), in a dose-dependent manner. Likewise, YAP 6SA abolished caRIG-I-stimulated IRF3 phosphorylation (Fig. 3F). In an in vitro kinase assay with purified TBK1 and using IRF3 as the substrate, we detected a significantly lower catalytic activity of TBK1 when TBK1 was coexpressed with either YAP or TAZ (Fig. 3G). All these data suggest that YAP/TAZ inhibit the activation and/or activity of TBK1 in cells.
YAP/TAZ associate with and prevent TBK1 K63 ubiquitination and adaptor/substrate interaction
In elucidating YAP/TAZ-mediated TBK1 inhibition, we detected an endogenous complex of YAP/TAZ and TBK1 in NMuMG cells by co-immunoprecipitation (Fig. 4A), and verified this interaction with transfected proteins (Fig. 4B). We also noticed an obvious mobility shift of YAP in the presence of wild-type TBK1/IKKε (Fig. 4B), suggesting a potential modification of YAP by TBK1/IKKε kinases, although the significance of this regulation was not investigated in this study.
Figure 4. YAP and TAZ associate with and disrupt TBK1 signaling complex and K63 ubiquitination.
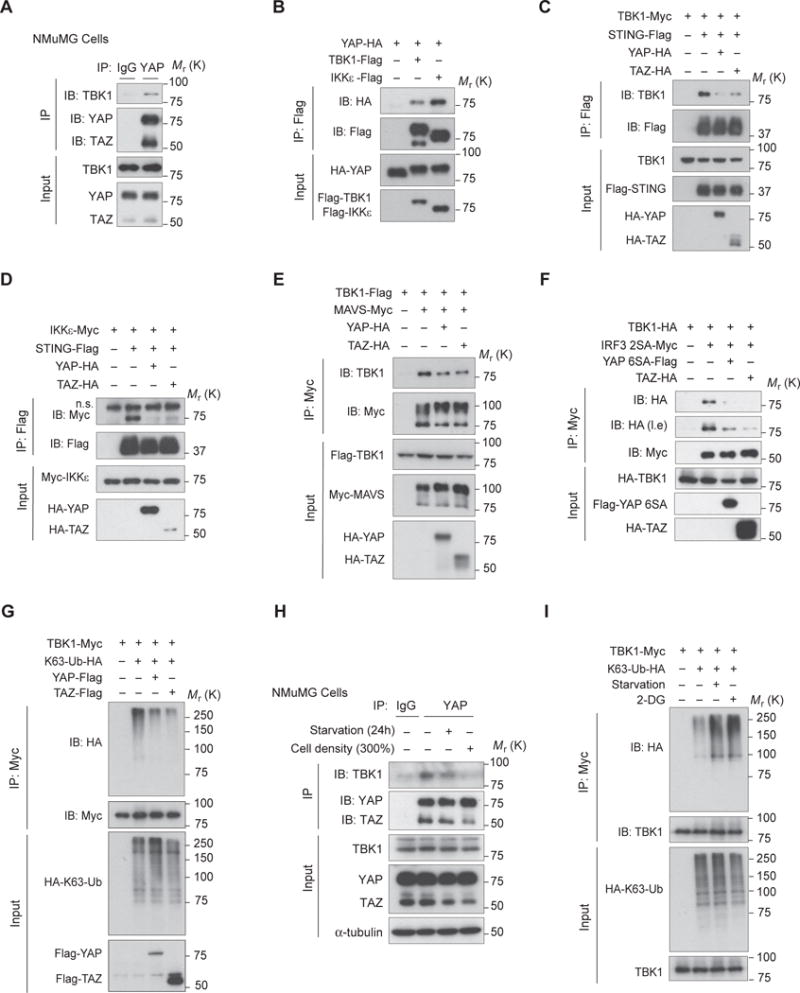
(A), Endogenous complex of YAP/TAZ and TBK1 in NMuMG cells was detected by co-immunoprecipitation using anti-YAP/TAZ antibody and visualized by using anti-TBK1 antibody. (B), Interaction between YAP and TBK1 or IKKε was revealed by co-immunoprecipitation of differentially tagged proteins. (C and D), Cotransfection of YAP or TAZ impaired STING’s recruitment of TBK1 (C) and IKKε (D), revealed by co-immunoprecipitation. (E), Association of MAVS and TBK1 was weakened in the presence of YAP or TAZ, revealed by co-immunoprecipitation. (F), Interaction of TBK1 with its substrate IRF3 was severely dampened in the presence of either YAP 6SA or TAZ, as evaluated by co-immunoprecipitation. Note the IRF3 2SA mutant was used in experiment to strengthen the interaction between TBK1 and IRF3. (G), K63-linked ubiquitination of TBK1, which was detected by coexpression of HA-tagged K63-Ub and immunoblotting, was markedly decreased by YAP or TAZ cotransfection. (H), Endogenous complex of TBK1 and YAP/TAZ was weakened under serum starvation or high cell density, revealed by co-immunoprecipitation similar to (A). (I), TBK1 K63-linked ubiquitination was strongly enhanced under nutrient or energy stress that activated Hippo signaling. Unprocessed images of blots are shown in Supplementary Figure 6.
Intriguingly, we found that YAP prevented the association of TBK1/IKKε with their adaptors STING (Fig 4C and 4D) and MAVS (Fig. 4E). Interaction between TBK1 and its substrate IRF3 was also severely compromised in the presence of YAP/TAZ (Fig. 4F). TAZ and the transcriptionally inactive YAP 6SA similarly disrupted TBK1/IKKε interaction with STING, MAVS, or IRF3 (Fig. 4C–F). These observations strongly suggest that YAP/TAZ prevent TBK1/IKKε kinases from forming a signaling complex with adaptors and substrates.
TRAFs-mediated TBK1 K63-linked ubiquitination is critical for TBK1 activation50. We observed that coexpression of YAP or TAZ reduced TBK1 K63 ubiquitination (Fig. 4G). Consistent with the enhanced antiviral response, a weaker endogenous complex between TBK1 and YAP/TAZ was observed under nutrient or cell-cell contact stresses (Fig. 4H), as well as more a robust TBK1 K63 ubiquitination (Fig. 4I). 2-DG treatment also led to a stronger TBK1 K63 ubiquitination (Fig. 4I), although the effect of 2-DG might be complicated as it suppresses glycolysis and alters inflammatory response51. These observations suggest that YAP/TAZ impair TBK1 K63 ubiquitination and TBK1 signaling complex.
YAP directly inhibits TBK1 activity through the transactivation domain
To dissect the domain(s) of YAP/TAZ required for TBK1 suppression, we generated serial YAP truncations and confirmed their expressions (Fig. 5A). Revealed by the IFNβ reporter assay, we found that the C-terminal transactivation domain of YAP (a.a. 291–488) was necessary and sufficient to abolish IRF3 transactivation (Fig. 5B). Similar to the full-length YAP, transactivation domain of YAP alone was able to interact with TBK1, abrogate TBK1 activation (Fig. 5C and 5D), and block the interaction between TBK1 and IRF3 (Fig. 5E). These observations suggest that YAP transactivation domain is responsible for TBK1 inhibition.
Figure 5. YAP abolishes TBK1 activity through its C-terminal transactivation domain.
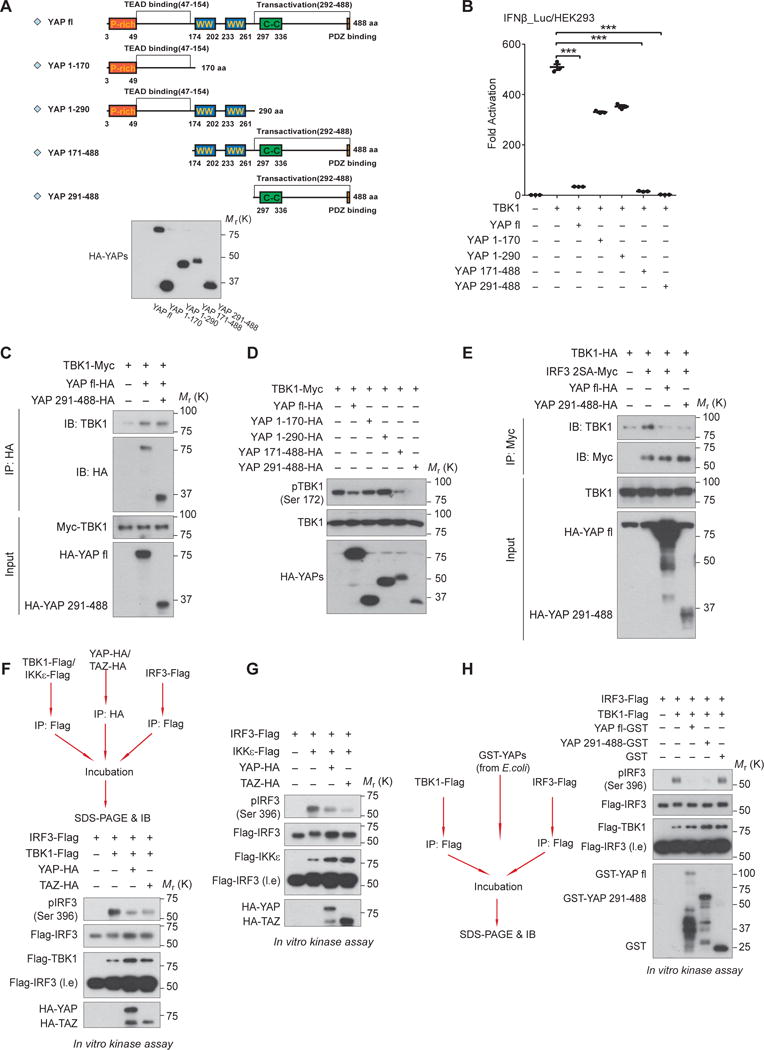
(A and B), Serial truncations of YAP were generated as depicted (A, upper panel) and their expressions were verified by immunoblotting (A, lower panel). Effects of the full-length or YAP truncations on antiviral signaling were measured by IRF3 transactivation, which revealed a marked inhibition by YAP transactivation domain (a.a. 291 – 488) (B). n=3 independent experiments. Mean ± SEM. ***P<0.001, by ANOVA test and Bonferroni correction. (C and D), Similar to the full-length protein, YAP transactivation domain was sufficient to interact with TBK1 (C) and to block TBK1 activation (D), assessed by co-immunoprecipitation and by immunoblotting of TBK1 Ser172 phosphorylation, respectively. (E), Interaction of TBK1 and IRF3 was severely interrupted in the presence of YAP transactivation domain, revealed by co-immunoprecipitation. (F and G), Addition of either YAP or TAZ separately purified from HEK293T cells in the in vitro kinase assays suppressed the catalytic activity of TBK1 (F) or IKKε (G) on the substrate IRF3. (H), Likewise, addition of GST-tagged full-length or transactivation domain of YAP that was expressed and purified from E.coli blocked TBK1-mediated IRF3 phosphorylation, in an in vitro kinase assay. Unprocessed images of blots are shown in Supplementary Figure 6. Statistics source data are provided in Supplementary Table 1.
Intriguingly, we observed a direct modification of TBK1 on either full-length YAP or its transactivation domain (sFig. 3A and 3B). We also found that the addition of either YAP or TAZ purified from HEK293T cells abrogated most TBK1- or IKKε-mediated IRF3 phosphorylation in vitro (Fig. 5F and 5G). To further verify this observation, we expressed and purified full-length YAP or its transactivation domain from E. coli, and found that full-length YAP as well as its transactivation domain were both sufficient to block TBK1 catalytic activity in the in vitro kinase assay (Fig. 5H). These observations suggest that YAP may directly abolish the catalytic activity of TBK1 by its transactivation domain, probably due to the interference of TBK1-substrate interaction.
YAP/TAZ-mediated TBK1 suppression is relieved by Lats1/2 kinases
We unexpectedly observed that endogenous YAP/TAZ proteins that reside in the nucleus in resting cells were significantly more cytoplasmic in response to VSV infection (Fig. 6A), which was verified by the nuclear/cytoplasmic fractionation (sFig. 6A), suggesting a dynamic and reciprocal regulation between Hippo signaling and antiviral response. Although the underlying mechanism exporting YAP/TAZ upon virus infection is currently unknown, we noticed that coexpression of either MAVS or TBK1 led to a marked redistribution of transfected YAP or YAP 6SA to the cytoplasm (Fig. 6B). The obviously overlapping distribution of TBK1 with YAP or YAP 6SA also suggested their interaction in the cytoplasm (Fig. 6B).
Figure 6. Lats1/2 relieve YAP/TAZ’s association and inhibition of TBK1.
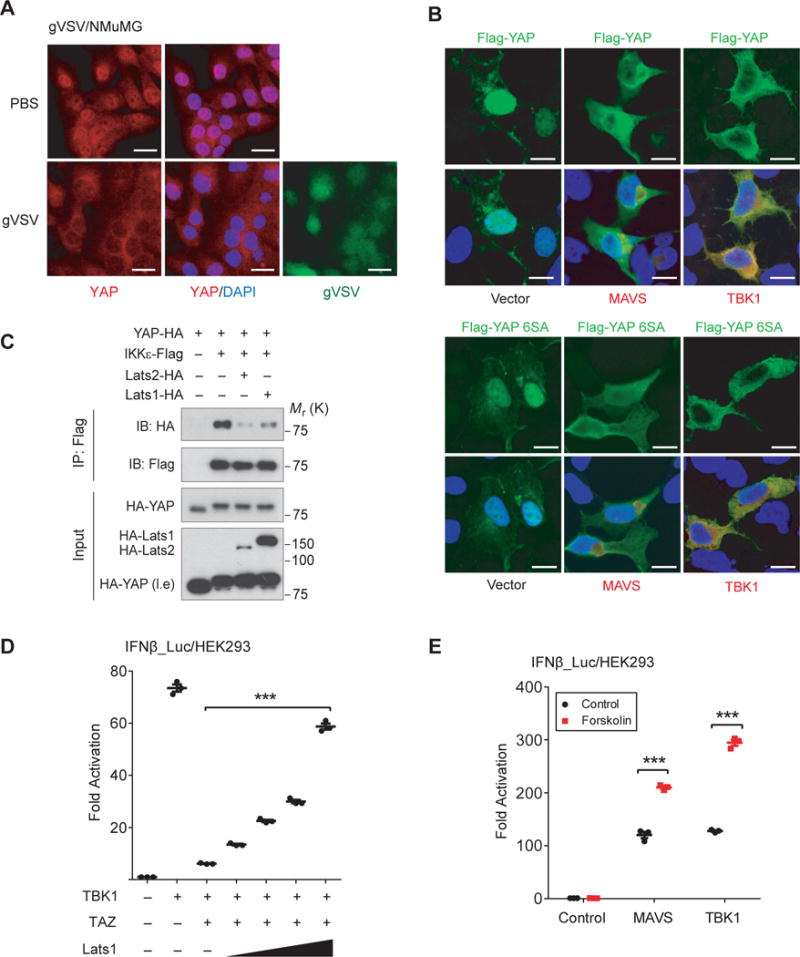
(A), Immunofluorescence assay revealed that endogenous YAP/TAZ, which resided richly in the nucleus, was partially exported to the cytoplasm in response to VSV infection. Scale bars, 20 μm. (B), YAP wild-type or 6SA mutant, which resided mostly in the nucleus, was exported into the cytoplasm when coexpressed with MAVS or TBK1, revealing by immunofluorescence. The overlap of YAPs and TBK1 in the cytoplasm was evidently under confocal microscopy. Scale bars, 10 μm. (C), YAP-IKKε complex was dissociated in the presence of Lats1/2, assessed by co-immunoprecipitation. (D), Expression of Lats1 relieved TAZ-mediated suppression of TBK1 in a dose-dependent manner, revealed by IRF3 responsiveness. n=3 independent experiments. Mean ± SEM. ***P<0.001, by ANOVA test and Bonferroni correction. (E), IRF3 responsiveness, which was stimulated by MAVS or TBK1 coexpression, was boosted in the presence of Forskolin, which is known to activate Hippo pathway. n=3 independent experiments. Mean ± SEM. ***P<0.001, by ANOVA test and Bonferroni correction. Unprocessed images of blots are shown in Supplementary Figure 6. Statistics source data are provided in Supplementary Table 1.
We next assessed the IKKε-YAP interaction by co-immunoprecipitation and found that both Lats1 and Lats2 weakened IKKε-YAP interaction (Fig. 6C). We also observed that Lats1 relieved the suppressing effect of TAZ on TBK1, evaluated by IRF3 transactivation (Fig. 6D). However, individual point mutations of five known Lats1/2-phosphorylated YAP residues into Aspartate showed little effect on YAP-mediated TBK1 suppression (sFig. 4B) and YAP Ser127 phosphorylation mimetic also interacted with TBK1 (sFig. 4C). On the other hand, activation of Hippo signaling by Forskolin52 also boosted IRF3 transactivation (Fig. 6E). These observations suggest that association of YAP/TAZ to the TBK1/IKKε complex and the inhibition effects are regulated.
YAP and TAZ control the host antiviral defense in cells and zebrafish
We subsequently investigated the physiological significance of YAP/TAZ-mediated TBK1 regulation in antiviral immunity. Stable NMuMG cells for Dox-inducible YAP 6SA expression were generated and verified (Fig. 7A and 7B, right panels). When YAP 6SA was induced, we observed an enhanced replication level of GFP-tagged VSV or the DNA virus HSV-1, shown by microscopy of GFP+ (virus replicating) cells and by anti-GFP immunoblotting (Fig. 7A and 7B). Application of the TBK1 inhibitor BX795 alleviated the effect of YAP 6SA induction (sFig. 5A), suggesting that the inhibitory effect of YAP 6SA is mostly through TBK1/IKKε. Expression of MAVS activates antiviral defense and endows host cells for viral resistance (10 and Fig. 7C), whereas coexpression of YAP, TAZ, or YAP 6SA impaired MAVS-driven viral resistance and restored VSV replication (Fig. 7C). These data demonstrate the biological function of YAP/TAZ in antiviral host defense and the “unexpected” function of the transcriptionally inactive YAP.
Figure 7. YAP/TAZ control host antiviral defense in cells.
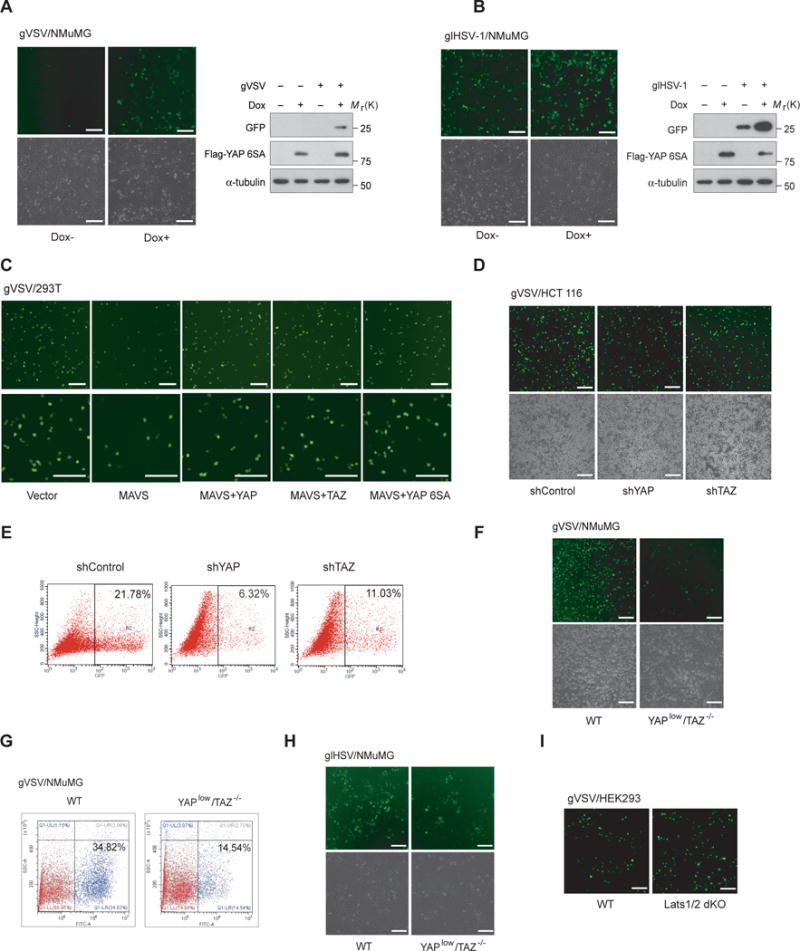
(A and B), Dox-inducible expression of YAP 6SA in NMuMG cells was verified by immunoblotting (right panels). Cellular resistance to GFP tagged RNA virus VSV (A) or DNA virus HSV-1 (B) was assessed by microscopy of viral replication (GFP+) cells (left panels) or by GFP immunoblotting (right panels), both revealed an impaired cellular viral resistance under YAP 6SA induction. Scale bars, 100 μm. (C), HEK293T cells transfected with MAVS were infected by gVSV, in the absence or presence of YAP wild-type, 6SA, or TAZ. The restored number of virus-replication (GFP+) cells indicated that YAPs or TAZ impeded the antiviral function of MAVS. Scale bars, 100 μm. (D and E), HCT 116 cells with shRNA-mediated YAP or TAZ knockdown exhibited a reduced level of virus replication (GFP+), revealed by microscopy (D) or FACS analysis (E). Scale bars, 100 μm. (F and G), Boosted antiviral resistance to gVSV was revealed in YAPlow/TAZ−/− NMuMG cells, evidenced by microscopy (F) or FACS analysis (G). Scale bars, 100 μm. (H), Enhanced cellular resistance to HSV-1 infection was observed in YAPlow/TAZ−/− NMuMG cells, determined by microscopy of virus replication (GFP+) cells. Scale bars, 100 μm. (I), Impaired viral resistance to gVSV infection was observed in Lats1/2 dKO cells by microscopy. Scale bars, 100 μm. Unprocessed images of blots are shown in Supplementary Figure 6.
In contrast, shRNA-mediated depletion of YAP or TAZ decreased the active replication of VSV in HCT 116 cells (Fig. 7D and 7E), and CRISPR/Cas9-mediated knockout/knockdown of TAZ/YAP in NMuMG cells similarly led to a marked enhancement of antiviral defense, revealed by microscopy or FACS analysis of VSV replication (Fig. 7F and 7G). Replication of the DNA virus HSV-1 was similarly suppressed in YAPlow/TAZ−/− cells (Fig. 7H). Conversely, dKO of Lats1/2 down-regulated antiviral signaling (sFig. 5B) and boosted VSV replication (Fig. 7I). These observations together suggest a negative biological regulation of YAP/TAZ and a positive regulation of Lats1/2 on cellular antiviral defense.
We then investigated YAP’s function in antiviral defense in whole animals, by using a system previously developed in zebrafish19–20. Human YAP 6SA or GFP was ectopically expressed in zebrafish embryos by mRNA microinjection at the one-cell stage, followed by gVSV infection at 48 hpf. As shown in Fig. 8A and previous reports19–20, zebrafish embryos underwent a severe VSV infection and started to die around 24–30 hpi. Expression of YAP 6SA sensitized embryos to VSV infection as evidenced by a significant increase in death rate upon virus attack (Fig. 8B), as well as suppressed antiviral responses, revealed using qRT-PCR to assess mRNA expressions of zebrafish IFNs and ISGs (Fig. 8C). These observations suggest a biological and cross-species function of YAP/TAZ in suppressing of the antiviral defense in zebrafish.
Figure 8. YAP attenuates cytosolic nucleic acid sensing and antiviral defense in zebrafish.
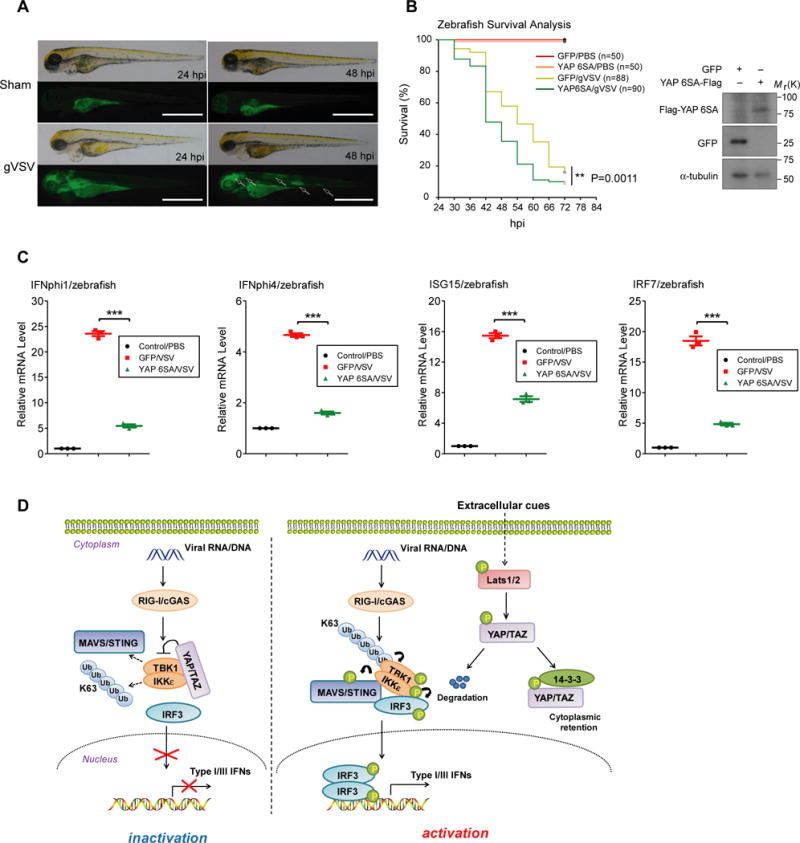
(A), gVSV was microinjected into the yolk of zebrafish embryos (48 hpf) to elicit a robust virus infection state, occurred mainly in brain, muscle and gut tissues of fishes, which eventually led to embryonic death at 24–30 hpi. Scale bars, 1 mm. (B and C), Zebrafish embryos were microinjected at one-cell stage with in vitro transcribed mRNA to gain expression of GFP or YAP 6SA, verified by immunoblottings (B, right panel). A vulnerable phenotype of YAP 6SA expressing embryos was observed upon VSV challenge (B, left panel). n=278 zebrafish. **P=0.0011, by log rank test. In parallel experiments (C), zebrafish embryos at 24 hpi were subjected to qRT-PCR analysis to determine the expression of zebrafish IFNs and ISGs mRNA, revealed a phenotype of suppressed antiviral responses in zebrafish expressing YAP 6SA. n=3 independent experiments using 25 embryos for each group. Mean ± SEM. ***P<0.001 by ANOVA test and Bonferroni correction. (D), Model for the Hippo-YAP regulation of cytosolic nucleic acid sensing and antiviral defense. YAP/TAZ associate with TBK1/IKKε kinases to prevent their K63 ubiquitination and adaptor/substrate association, thus restricting TBK1/IKKε activation in response to cytosolic nucleic acid sensing. Activation of Hippo signaling through extracellular clues, such as nutrient stress or cell-cell contact, activates Lats1/2 kinases that lead to YAP/TAZ phosphorylation and degradation, thereby relieving their association and inhibition of TBK1/IKKε. Unprocessed images of blots are shown in Supplementary Figure 6. Statistics source data are provided in Supplementary Table 1.
DISCUSSION
Host antiviral sensing and defense are strictly controlled by intrinsic molecules53–54. Still, little is known regarding their regulation by extracellular signals. Here we show that cellular nutrient/density status, through the Hippo-YAP pathway, regulates antiviral host defense (Fig. 8D). Our study reveals that intrinsic activity of the Hippo pathway can integrate and coordinate the outcome of innate host defense. Given that the Hippo pathway mediates signals from cell-cell contact, mechanical stress, matrix stiffness, and long-range hormonal signals55–56, this finding illustrates the possibility for the regulation of innate antiviral immunity by a variety of extracellular cues.
The observation that YAP/TAZ-mediated TBK1 regulation controls a magnitude of host cells for sensing dangerous signals, such as heterogeneous RNA or DNA, adds a further dimension for the function of the Hippo pathway. This additional layer of regulation could be an adaptive host mechanism to ensure the removal of pathogenic factors but add protection to avoid excessive responses which jeopardize cell survival57, or to evade potential autoimmune damages from the exposure of self nucleic acids in the cytosol21–22. Although how YAP/TAZ are regulated by particular conditions, such as GPCR regulation, energy stress, and serum starvation, has been well defined43–46,54–55, their regulation by intracellular conditions or extracellular cues is still not fully understood. Considering the general role of YAP/TAZ in promoting cell proliferation and inhibiting apoptosis58–59, it is not surprising that YAP/TAZ also control cytosolic nucleic acid sensing, which often leads to cell death60–61. We believe the direct inhibition of TBK1/IKKε by YAP/TAZ provides a mechanism to neglect the danger signal and to ensure cell survival and proliferation when favorable growth conditions are available. This inhibition may also contribute to regulation of apoptosis and the tumorigenic role of TBK1/IKKε27,62–63, which awaits further investigation. TBK1 is involved in maturation of autophagy and bacteria defense25,64, but little is known if autophagy, which is also triggered by serum starvation, regulate TBK1 activation. The dependence of Lats1/2 and YAP/TAZ reveals that Hippo signaling, rather than autophagy, is important to mediate antiviral regulation by nutrient/physical stresses. Our current data illustrate the essential focus of Hippo signaling and YAP/TAZ in cytosolic RNA/DNA sensing, which is also well supported by physiological data obtained from cell culture and zebrafish. The fact that cells and zebrafish expressing YAP 6SA are sensitized to RNA/DNA virus infection provides us direct evidence for the physiological involvement of YAP/TAZ in antiviral defense, independent of their transcriptional activity. Since the host defense imbalance is a main cause of autoimmune diseases21–22, it is worthy to examine if YAP/TAZ and the Hippo pathway are involved in these situations.
Pathogenic nucleic acids are sensed in the cytosol by RIG-I-like receptors and/or cGAS1,6. TBK1 is central for this cytosolic RNA/DNA sensing, acting as a downstream signal mediator of mitochondria-conjugated MAVS or ER-associated STING to transduce the recognition signal to the transcriptional factors IRF3/IRF7, to induce the expression of antiviral cytokines and a variety of ISGs54. MAVS self-associates and polymerizes on mitochondria to set the platform for functional signal complexes14, while STING-mediated TBK1 activation is thought to be executed in the microsome65–66. Our data of YAP/TAZ re-localization during virus infection and their formation of endogenous complex with TBK1 suggest that YAP/TAZ are regulatory components for these antiviral signaling complexes. The presence of YAP/TAZ prevents the K63 ubiquitination of TBK1, which is critical for TBK1 activation15,50,67–68. We did not dissect the possible causation for this regulation, but noticed that the interaction between TBK1 and adaptors MAVS or STING, or with the substrate IRF3, is interfered by YAP/TAZ. Our data also showed that YAP/TAZ inhibit TBK1 kinase activity in vitro, probably through direct association with TBK1 to cover its catalytic center or to compete with IRF3 as a substrate.
Conversely, nutrient starvation or cell-cell contact activates Lats1/2, which removes YAP/TAZ’s inhibition on TBK1 and sensitizes host cells for danger signals. Distinct from our previous finding of Mst1 in antiviral immunity that is independent of Lats1/2 and Hippo signaling20, the regulation by cellular nutrient/physical stress requires Lats1/2 kinase. Intriguingly, YAP with the Ser94 to Alanine mutation, which disrupts the YAP-TEAD complex formation49, retains the same inhibitory effect. Purified full-length or transactivation domain of YAP also directly blocks the kinase activities of TBK1/IKKε, suggesting an alternate function mode of YAP/TAZ by direct protein-protein interaction, rather than through its transcriptional coactivators potential. We noticed that Lats1/2 can effectively dissociate YAP from TBK1/IKKε and relieve TBK1 inhibition, indicating that YAP/TAZ-mediated TBK1 inhibition is controlled, although the exact mechanism requires further investigation.
In conclusion, our study provides an unusual function and signal integration of the Hippo pathway to TBK1 activation through an unexpected mechanism. Our model indicates that the level and activity of the Hippo components can serve a determinant to regulate the magnitude of host antiviral responses. Consistent with this notion, our research suggests that pharmacological manipulation of these signal mediators may offer potential therapeutic benefits for antiviral prevention.
METHODS
Expression plasmids, reagents, and antibodies
Expression plasmids encoding Flag-, Myc-, or HA-tagged wild-type or mutations of human TBK1, IKKε, IRF3, caRIG-I, MAVS, STING, TAZ, YAP, Lats1, Lats2, TRIF, MyD88, K63-Ub, caALK5, and reporters of TCF, Gli1, 4SBE, NF-κB, 5xUAS, IFNβ_Luc and 5xISRE_Luc have been described previously19,69. YAP truncations including YAP a.a. 1–170, 1–290, 171–488, 291–488, and the GST-tagged YAP full-length and a.a. 291–488 were generated by PCR-based cloning performed by pfu DNA polymerase from Stratagene. Detailed information will be provided upon request. All coding sequences were verified by DNA sequencing.
GFP and Luciferase double tagged HSV-1 was gifted from Dr. Jiahuai Han (Xiamen University, Xiamen), GFP tagged VSV was gifted from Dr. Zhijian J. Chen (UT Southwestern Medical Center, Dallas), and Sendai virus (Cantell strain) was from Charles River Laboratories. The pharmacological reagents BX795 (Millipore), 2-DG (Sangon Biotech), Doxycycline (Sangon Biotech), and poly(I:C) (Invivogen) were purchased.
Detailed information of all antibodies applied in immunoblotting, immunoprecipitation and immunofluorescence is provided in the attached Supplementary Table 2.
Cell culture, transfections, and infections
NMuMG, HEK293, HCT 116, HaCaT and THP-1 cells were obtained from ATCC. Peritoneal macrophages were obtained from C57BL/6 male mice at 6–8 weeks age by Brewer thioglycollate medium (Sigma)-induced approach, and MEFs were obtained from E12.5 – E13.5 embryos in pregnant C57BL/6 female mice at 8-week age, and immortalized by infection of viral vector packaging SV40. Care of experimental animals was in accordance with guidelines and approved by laboratory animal committee of Zhejiang University. No cell lines used in this study were found in the database of commonly misidentified cell lines that is maintained by ICLAC and NCBI Biosample. Cell lines were frequently checked in morphology under microscopy and tested for mycoplasma contamination, but were not authenticated. All cell lines, except for THP-1 that was maintained in RPMI 1640 medium, were cultured in DMEM medium with 10% fetal bovine serum (FBS) at 37℃ in 5% CO2 (v/v). The YAP 6SA inducible expressing NMuMG and MEF cells were generated by lentiviral vector containing the inducible Tet-on system followed by open read frame (ORF) of YAP 6SA mutant, and selected by G418 antibiotic at concentration of 1500μg ml−1 for one week. Xtremegene HP (Roche) or Polythylenimine (PEI, Polysciences) transfection reagents were used for plasmid transfection. Transfection of poly(I:C) was performed by using Lipofectamine RNAiMAX (Invitrogen) reagent. Infection of Sendai virus (SeV), Vesicular stomatitis virus (VSV), and Herpes simplex virus-1 (HSV-1) was as previously described19–20. Briefly, viruses with indicated amount (0.5 – 5 moi) were added into the fresh and serum-free medium, and cells were incubated at 37℃ in 5% CO2 (v/v) for 1 hour, shaking mildly every 15 minutes. Virus-containing medium was then replaced by fresh medium containing 10% FBS.
Luciferase reporter assay
HEK293T or HEK293A cells were transfected with indicated reporters (100 ng) bearing an open read frame (ORF) coding Firefly luciferase, along with the pRL-Luc with Renilla luciferase coding as the internal control for transfection, and other expression vectors specified in results section. In brief, cells were cultured for 12 hours post transfection, and stimulated by virus infection or transfection with poly(I:C). After 24 hours of transfection and with indicated treatment, cells were lysed by passive lysis buffer (Promega). Luciferase assays were performed using a dual luciferase assay kit (Promega), quantified with POLARstar Omega (BMG Labtech), and normalized to the internal Renilla luciferase control.
Quantitative RT-PCR assay
The HCT 116 cells or embryos of zebrafish with specified viral infection was lysed and total RNA was extracted using RNAeasy extraction kit (Invitrogen). cDNA was generated by one-step iScript cDNA synthesis kit (Vazyme), and quantitative real-time PCR was performed using the EvaGreen Qpcr MasterMix (Abm) and CFX96 real-time PCR system (Bio-Rad). Relative quantification was expressed as 2-^Ct, where ^Ct is the difference between the main Ct value of triplicates of the sample and that of an endogenous L19 or GAPDH mRNA control. The human or zebrafish primer sequences used can be found in Supplementary Table 3.
Co-immunoprecipitations and immunoblottings
HEK293T or NMuMG cells infected with VSV/SeV, or transfected 36 hours with specified plasmids encoding N-terminal Myc-, Flag-, or HA-tagged YAP, TAZ, TBK1, IKKε, IRF3, caRIG-I, MAVS or STING, were lysed using a modified Myc lysis buffer (MLB)69 (20 mM Tris-Cl, 200 mM NaCl, 10 mM NaF, 1 mM NaV2O4, 1% NP-40, 20 mM β-glycerophosphate, and protease inhibitor, pH 7.5). Cell lysates were then subjected to immunoprecipitation using anti-Flag, anti-Myc, or anti-HA antibodies for transfected proteins, or using anti-YAP/TAZ antibodies for endogenous proteins. After 3–4 washes with MLB, adsorbed proteins in beads was resolved by 2 × SDS loading buffer and analyzed by SDS-PAGE and immunoblotting with indicated antibodies. Cell lysates were also analyzed by SDS-PAGE and immunoblotting to control protein abundance. Nuclear and cytoplasmic extracts were prepared using the NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce), according to the manufacturer’s instructions. Detailed information of all antibodies used in immunoprecipitation assays is provided in Supplementary Table 2.
siRNA or shRNA-mediated RNA interference
Double stranded siRNA (RiboBio) to silence endogenous YAP or TAZ expression in HEK293 cells targeted the human YAP or TAZ mRNA (sequence information is in Supplementary Table 3). Control siRNA (RiboBio) was used to control for possible nonspecific effects of RNA interference. Cells were transfected with siRNA using the Lipofectamine RNAiMAX (Invitrogen) reagent for 48 hours before the further assay, and the reverse transfection method was used to reach optimal efficiency. The shRNA-mediated knockdown of YAP or TAZ in HCT 116 cells was generated by shRNAs as previously described34,37, delivered by the lentiviral vector produced by the Mission shRNA (Sigma Aldrich) plasmids (TRCN information is in Supplementary Table 3), together with pMD2.G and psPAX2 plasmids in 293T cells.
In vitro kinase assay
HEK293T cells were transfected with Flag- or Myc-tagged TBK1 plasmid in the absence or presence of HA-YAPs or HA-TAZ plasmid, or transfected with Flag-IRF3 plasmid, and lysed by the modified MLB lysis buffer after 36 hours of transfection. Immunoprecipitations were performed by using with anti-Flag, anti-Myc, or anti-HA antibodies. With four washes by the MLB and one wash by the kinase assay buffer (20 μM ATP, 20 mM Tris-HCl, 1 mM EGTA, 5 mM MgCl2, 0.02% 2-mercapto-Ethanol, 0.03% Brij-35, and 0.2 mg/mL BSA, PH7.4), immunoprecipitated Flag- or Myc-tagged TBK1, Flag-IRF3, HA-YAPs, or the GST proteins or GST-tagged wild-type YAP or YAP truncation expressed and purified from E.coli, were incubated in the kinase assay buffer at 30℃ for 60 min on THERMO-SHAKER. Reaction was stopped by addition of 2 × SDS loading buffer and subjected to SDS-PAGE and specified immunoblotting. Detailed information of antibodies used in immunoprecipitation assays is provided in Supplementary Table 2.
Immunofluorescence, microscopy, and FACS
To visualize the subcellular localization of endogenous or transfected YAP/TAZ, MAVS, or TBK1, NMuMG or HEK293A cells were infected with gVSV virus at 8 hpi, or transfected with plasmid specified in the results section for 24 hours, fixed in 4% paraformaldehyde, permeablized, blocked in 10% horse serum in PBS for 2 hours, and incubated sequentially with primary antibodies anti-YAP/TAZ or anti-Flag and Alexa-labeled secondary antibodies with extensive washing. Slides were then mounted with Vectorshield and stained with DAPI (Vector Laboratories). Immunofluorescence images were obtained and analyzed using the Nikon Eclipse Ti inverted microscope or by the Zeiss LSM710 confocal microscope. FACS analysis of GFP+ cells was performed at BD FACSCalibur, according to manufacturer’s manual. Detailed information of antibodies used in immunofluorescence assays is provided in Supplementary Table 2.
CRISPR/Cas9-mediated generation of YAPlow/TAZ−/− cells
CRISPR/Cas9 genomic editing for gene deletion was described70. Guide RNA sequences targeting TAZ (5′-GAGGATTAGGATGCGTCAAG-3′) and YAP (5′-CGGGGACTC GGAGACCGACT-3′) exons were cloned into the plasmids px330. Constructs together with puromycin vector pRK7-puromycin in the ratio of 15:1 were transfected into NMuMG by PEI transfection reagent. 24 hours after transfection, cells were selected by puromycin (1.5 μg ml−1) for 72 hours, and single colonies were obtained by serial dilution and amplification. Clones were identified by immunoblotting with anti-YAP/TAZ antibodies, and YAPlow/TAZ−/− clone was used for indicated analyses.
Zebrafish ectopic expression and VSV challenge
A system for challenge of GFP-tagged VSV in zebrafish embryos to rapidly assess the gene function in antiviral defense was previously developed19–20. Zebrafish AB wild-type embryos (male/female) were raised at 28.5℃ in E3 egg water. Forced expression of exogenous genes GFP or YAP 6SA was obtained by microinjection 25 pg of in vitro transcribed mRNA by mMESSAGE mMACHINE SP6 Transcription Kit (Life technology) in the one-cell stage of embryogenesis, that is, the first 20-min. At this stage, exogenous mRNAs distribute most evenly into most cells by cell division and last for 72–96 hours in zebrafish embryos. Injected embryos with normal development were selected and used for the gVSV virus microinjection (1 × 103 pfu/embryo) at the embryo yolk at 48 hpf, and a simple randomization method was used for allocation groups. The infection and death rate of injected embryos were monitored at desired stages. To detect the expression of GFP or YAP 6SA by immunoblotting, tissue samples of zebrafish embryos at 48 hpf were homogenized, lysed in MLB, and subjected to SDS-PAGE and immunoblotting. To detect the expression of zebrafish IFNs and ISGs in response to gVSV infection, tissue samples of zebrafish embryos at 24 hpi were homogenized and lysed, and subjected to RNA extraction and qRT-PCR assays as described in the previous section. Care of experimental animals were in accordance with guidelines and approved by laboratory animal committee of Zhejiang University.
Statistics and reproducibility
Quantitative data are presented as mean ± standard error of mean (SEM) from at least three independent experiments. When appropriate, statistically differences between multiple comparisons were analyzed using the ANOVA test with Bonferroni correction, and survival curve was analyzed using the log rank test, both by Origin 9.1 software. Differences were considered significant at p<0.05 and the P value was precisely specified unless it is smaller than 0.001. All samples if preserved and properly processed were included in the analyses, and no samples or animals were excluded, except for zebrafish with conventional injection damage. No statistical method was used to predetermine sample size, and the experiments except for animal samples were not randomized. Immunoblottings, for which representative experiments are shown in the figures, as well as reporter assay, and qRT-PCR experiments were repeated to a minimum of three independent experiments to ensure reproducibility. The investigators were not blinded to allocation during experiments and outcome assessment.
Data availability
All data supporting the findings of this study are available from the corresponding author on reasonable request. The source data for statistical analyses of Figures 1A, 1C, 1D, 1E, 1G, 2A, 2B, 2C, 2F, 2H, 3A, 3B, 5B, 6D, 6E, 8B and 8C, and Supplementary Figures 1A, 2A, 2B, 2C, 2E, 2F, 4B and 5B are provided in Supplementary Table 1.
Supplementary Material
Acknowledgments
We are grateful to Drs. Zhijian Chen for gVSV virus, Jiahuai Han for glHSV-1 virus, and Zongping Xia, Yaowei Huang, Xiaojian Wang, and Jin Jin for reagents. This research was partly supported by MoST 973 Plan (2015CB553800), NSFC Project (81472665, 91540205 and 31571447), CPSF (581220-X91602), DoD grant (1W81XWH-15-1-0650), NIH (R01GM051586, R35CA196878, and R21CA209007), and Project 985 and the Fundamental Research Funds for the Central Universities to the Life Sciences Institute at Zhejiang University. K.-L.G. co-founded but receives no direct financial support from Vivace Therapeutics. P.X. is a scholar in the National 1000 Young Talents Program.
Footnotes
AUTHOR CONTRIBUTIONS
Q.Z. and F.M. carried out most experiments. S.C., S.W., S.L., R.Z., J.W., and J.Q. contributed with several experiments, S.P., X.L., B.Z., J.L., J.Z., X.-H.F., and K.-L.G. helped with data analyses and discussions. P.X. conceived the study and experimental design and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 2.Hornung V, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 3.Li XD, et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Civril F, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–337. doi: 10.1038/nature12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao P, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orzalli MH, Knipe DM. Cellular sensing of viral DNA and viral evasion mechanisms. Annu Rev Microbiol. 2014;68:477–492. doi: 10.1146/annurev-micro-091313-103409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzgerald KA, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 9.Sharma S, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 10.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan YK, Gack MU. RIG-I-like receptor regulation in virus infection and immunity. Curr Opin Virol. 2015;12:7–14. doi: 10.1016/j.coviro.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 14.Hou F, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu D, et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 2013;3:747–758. doi: 10.1016/j.celrep.2013.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.tenOever BR, et al. Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol. 2004;78:10636–10649. doi: 10.1128/JVI.78.19.10636-10649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tojima Y, et al. NAK is an IkappaB kinase-activating kinase. Nature. 2000;404:778–782. doi: 10.1038/35008109. [DOI] [PubMed] [Google Scholar]
- 18.Solis M, et al. Involvement of TBK1 and IKKepsilon in lipopolysaccharide-induced activation of the interferon response in primary human macrophages. Eur J Immunol. 2007;37:528–539. doi: 10.1002/eji.200636090. [DOI] [PubMed] [Google Scholar]
- 19.Xiang W, et al. PPM1A silences cytosolic RNA sensing and antiviral defense through direct dephosphorylation of MAVS and TBK1. Sci Adv. 2016;2:e1501889. doi: 10.1126/sciadv.1501889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng F, et al. Mst1 shuts off cytosolic antiviral defense through IRF3 phosphorylation. Genes Dev. 2016;30:1086–1100. doi: 10.1101/gad.277533.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao D, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. 2015;112:E5699–5705. doi: 10.1073/pnas.1516465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crampton SP, Bolland S. Spontaneous activation of RNA-sensing pathways in autoimmune disease. Curr Opin Immunol. 2013;25:712–719. doi: 10.1016/j.coi.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999;18:6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baldwin AS. Regulation of cell death and autophagy by IKK and NF-kappaB: critical mechanisms in immune function and cancer. Immunol Rev. 2012;246:327–345. doi: 10.1111/j.1600-065X.2012.01095.x. [DOI] [PubMed] [Google Scholar]
- 25.Pilli M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SE, Hong M, Cho J, Lee J, Kim KM. IKKepsilon and TBK1 expression in gastric cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457–467. doi: 10.1016/s0092-8674(03)00557-9. [DOI] [PubMed] [Google Scholar]
- 29.Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- 30.Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol. 2003;5:921–927. doi: 10.1038/ncb1051. [DOI] [PubMed] [Google Scholar]
- 31.Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol. 2003;5:914–920. doi: 10.1038/ncb1050. [DOI] [PubMed] [Google Scholar]
- 32.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao B, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varelas X, et al. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev Cell. 2010;18:579–591. doi: 10.1016/j.devcel.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Mo JS, Yu FX, Gong R, Brown JH, Guan KL. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs) Genes Dev. 2012;26:2138–2143. doi: 10.1101/gad.197582.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu FX, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–791. doi: 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller E, et al. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol. 2012;19:955–962. doi: 10.1016/j.chembiol.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Bao Y, et al. A cell-based assay to screen stimulators of the Hippo pathway reveals the inhibitory effect of dobutamine on the YAP-dependent gene transcription. J Biochem. 2011;150:199–208. doi: 10.1093/jb/mvr063. [DOI] [PubMed] [Google Scholar]
- 40.Varelas X, et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol. 2008;10:837–848. doi: 10.1038/ncb1748. [DOI] [PubMed] [Google Scholar]
- 41.Camargo FD, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–2060. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 42.Liu B, et al. Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell. 2016;164:406–419. doi: 10.1016/j.cell.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13:877–883. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thaventhiran JE, et al. Activation of the Hippo pathway by CTLA-4 regulates the expression of Blimp-1 in the CD8+ T cell. Proc Natl Acad Sci U S A. 2012;109:E2223–2229. doi: 10.1073/pnas.1209115109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao B, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mo JS, et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17:500–510. doi: 10.1038/ncb3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang W, et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17:490–499. doi: 10.1038/ncb3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen L, et al. Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes Dev. 2010;24:290–300. doi: 10.1101/gad.1865310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, et al. Structural insights into the YAP and TEAD complex. Genes Dev. 2010;24:235–240. doi: 10.1101/gad.1865810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li S, Wang L, Berman M, Kong YY, Dorf ME. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity. 2011;35:426–440. doi: 10.1016/j.immuni.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu FX, et al. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013;27:1223–1232. doi: 10.1101/gad.219402.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu J, Qian C, Cao X. Post-Translational Modification Control of Innate Immunity. Immunity. 2016;45:15–30. doi: 10.1016/j.immuni.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 54.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 55.Low BC, et al. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014;588:2663–2670. doi: 10.1016/j.febslet.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 56.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1–17. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porritt RA, Hertzog PJ. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015;36:150–160. doi: 10.1016/j.it.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 58.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 60.Barber GN. Host defense, viruses and apoptosis. Cell Death Differ. 2001;8:113–126. doi: 10.1038/sj.cdd.4400823. [DOI] [PubMed] [Google Scholar]
- 61.Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell. 2014;54:273–280. doi: 10.1016/j.molcel.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ou YH, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–470. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xie X, et al. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci U S A. 2011;108:6474–6479. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 67.Larabi A, et al. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep. 2013;3:734–746. doi: 10.1016/j.celrep.2013.01.034. [DOI] [PubMed] [Google Scholar]
- 68.Shu C, et al. Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure. 2013;21:1137–1148. doi: 10.1016/j.str.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu P, et al. Innate antiviral host defense attenuates TGF-beta function through IRF3-mediated suppression of Smad signaling. Mol Cell. 2014;56:723–737. doi: 10.1016/j.molcel.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available from the corresponding author on reasonable request. The source data for statistical analyses of Figures 1A, 1C, 1D, 1E, 1G, 2A, 2B, 2C, 2F, 2H, 3A, 3B, 5B, 6D, 6E, 8B and 8C, and Supplementary Figures 1A, 2A, 2B, 2C, 2E, 2F, 4B and 5B are provided in Supplementary Table 1.