

Abstract
Despite their different implementations, comparison of the best threading approaches to the prediction of evolutionary distant protein structures reveals that they tend to succeed or fail on the same protein targets. This is true despite the fact that the structural template library has good templates for all cases. Thus, a key question is why are certain protein structures threadable while others are not. Comparison with threading results on a set of artificial sequences selected for stability further argues that the failure of threading is due to the nature of the protein structures themselves. Using a new contact map based alignment algorithm, we demonstrate that certain folds are highly degenerate in that they can have very similar coarse grained fractions of native contacts aligned and yet differ significantly from the native structure. For threadable proteins, this is not the case. Thus, contemporary threading approaches appear to have reached a plateau, and new approaches to structure prediction are required.
Graphical abstract
INTRODUCTION
One of the more perplexing observations that comes out of the CASP protein structure prediction experiments is that despite the diversity of implementations1–7 (different profiles8–10, dynamic programming11, hidden Markov models12–13, use or absence of predicted contacts14–15 and/or pair potentials16–20), for target proteins lacking closely homologous templates in the Protein Data Bank, PDB21, the best threading based methods tend to recognize the same targets as being threadable or non-threadadble2. Put another way, with few exceptions associated with the actual alignment quality and the rare case where one class of threading algorithms does better than the others, the performance of different threading algorithms is remarkably similar. This pattern goes back to very early CASPs and persists to the present2, 22. Key questions are why is this the case and does it suggest that for proteins lacking close homologues the performance of classical threading approaches has reached a plateau, much as secondary structure predictions schemes did over a decade ago23–28? If it indeed turns out that there are certain classes of protein structures that preclude them from having successful predictions in the absence of closely homologous templates, then this would suggest that for these proteins, alternative non threading based methods of protein structure prediction are required. If so, the further development of classical threading based approaches would likely experience limited success.
In practice, this issue is important, because threading based structure prediction methods have the most general applicability for predicting protein structure29–30, with ~86% of human proteins having at least one domain predicted with acceptable accuracy (with TM-scores ≥ 0.419, 31–32 to native, with a TM-score of 1.0 indicating identical structures and tertiary structures with TM-scores above the 0.4 threshold having p-values at or below 10−5) for virtual ligand screening32–33,34,35, functional analysis36, as well as to assist in the identification of disease associated missense variations37. Complementary to threading based approaches are template free methods38–39; among the most successful are those that use residue covariation to predict tertiary contacts40–44. However, to accurately predict contacts from correlated mutation analysis requires that there be a very large number of protein sequences in a given family. At present, this limits the general applicability of this promising, and yet quite old methodology. Thus, as a practical matter, at present threading based approaches afford the largest coverage of proteins. As such, it is important to understand why they fail, as this might motivate the development of more powerful threading based methods.
The most important factor that influences the performance of a given threading algorithm is the comprehensiveness of the protein structure template library. The performance of a given threading algorithm can drastically change depending on whether or not closely homologous templates are included in the template library. While in principle the same set of PDB structures are available to all investigators at the time of a given CASP, in practice, the template libraries used by different labs are often somewhat different. In general, this cannot be a major effect, since essentially all of the best threading algorithms perform similarly1–2, 4–5. However, to be absolutely certain that different state of the art threading algorithms perform similarly requires that the template library used by the different algorithms be exactly the same. In that regard, for those cases where threading fails, it is possible that the template library lacks sufficiently close structures that its failure is simply due to the absence of suitable structural templates. This would imply that the PDB library is not complete, a result in contradiction with a significant body of work45–46, but one which has to be investigated. As a corollary, there might be suitable templates in the PDB, but the substructure that aligns to the target contains so many gaps that threading based methods cannot work- the cost of introducing many gaps in the threading alignment is too high. Another possible explanation is that certain proteins are so evolutionary distant from their templates that threading based methods, which are inherently driven by profile similarity, fail. If so, it should be reflected in the set of sequences used to construct the sequence profiles rather than the structure itself. This issue can be directly addressed by computationally generating sequences that in the context of a given potential have low energy in the structure of interest 47–48, and examining if there are any common features between structures that are threadable when native and such artificial sequences are used. If in the majority of cases virtually the same sets of structures are threadable or non-threadable, this would imply that there is (are) some features of the structure responsible for its threadability. In this context, we could use a purely structure based algorithm to detect which structural features of the fold are responsible for the success or failure of threading. In the simplest case, one could just align secondary structural elements; however, different folds can have closely matching, if not absolutely identical, secondary structures. On the other hand, Cα-based contacts could potentially differentiate between protein structures. This might be true when a significant fraction of the contacts in the structure are non-local; whereas if most contacts are local, one is really comparing local secondary structures rather than global folds. One also has to bear in mind that for distantly related pairs of proteins, even if one had native contacts in hand, due to differences in the structures, 100% of their contacts need not overlap. In that regard, when the number of secondary structures is small, viz. the folds are of low complexity, one might also expect significant contact overlap, and yet their global folds might differ appreciably.
The goal of this paper is to understand why certain classes of proteins are non-threadable, while others are threadable. We examine each of possible underlying causes described above and explore whether the non-threadability of distantly homologous proteins is predominantly a feature of the target protein’s structure or is due to differences in the evolutionary distance of the target from the closest template structures. By focusing on the structural properties of these proteins, we demonstrate that the difficulty threading experiences is due to the inherent features of threadable vs. non-threadable protein structures. Finally, we discuss promising existing and to be developed approaches that can extend protein structure prediction to enable it to consistently break through the glass ceiling of threading based protein structure prediction methods.
METHODS
Template and target libraries, threading algorithms and assessment metrics
A representative PDB template library that is an extension of the PROSPECTOR_4 template library 49 clustered at the level of 35% sequence identity was prepared. This library, LIST.templates, consists of 16,747 proteins ranging in length from 40 to 1962 residues. The representative set of target proteins, LIST.targets, consists of 3221 proteins that share no more than 35% pairwise sequence identity and range in length from 40 to 200 residues. Both libraries as well as list of threadable and non-threadable proteins are available at http://cssb2.biology.gatech.edu/threading/download.html.
To assess whether the majority of templates are recognized by different threading algorithms, we employed HHpred50–51, SP3 9 and PROSPECTOR_449; for details of these algorithms, we refer the reader to the original papers. These three algorithms were chosen because they use different alignment methods and types of scoring functions. For each threading algorithm, the same threading library, LIST.templates, was employed as well as the sequence dataset used to build the appropriate sequence profiles. This will allow for direct evaluation of the conjecture that there is (are) some property(ies) of the target structure that render them threadable or non-threadable. In that regard, a metric of structure comparison between the target structure and threading alignment to the template protein is required. To assess a given threading alignment, we employ the TM-score 52 generated using the latest version fr-TM-align 53, an improved, more sensitive version of the original TM-align protein structure alignment algorithm31. As previously, we define a protein as being threadable if its TM-score is ≥ 0.4.
Generation of low energy artificial protein sequences for the target and template structures
For each PDB target and template protein, following the procedure described in47, low energy sequences for the given structure were generated. The potential consists of burial, secondary structure and pair statistical potentials. For a given randomly selected overall amino acid composition whose average matches that in the PDB, the sequence is shuffled until a low energy sequence is found. Then, the process is repeated until 60 such low energy sequences are generated for each target and template protein structure. Note that the average pairwise sequence identity is about 11% for the set of low energy sequences that match the given structure. We then use the resulting sequences to generate the sequence profile for use in threading by PROSPECTOR_4 49.
C-Align: A Cα-based contact map alignment algorithm
In what follows, we describe C-align, a newly developed Cα-based contact map based alignment algorithm. It aligns a pair of structures based on the overlap of the set of contacts defined between residues i and j whose distance between pairs of Cα atoms are less than d = 8.5 Å, C(i, j)8.5, and d =14 Å, C(i, j)14 respectively. Here,
| (1) |
where dijis the distance between Cαs i and j. We need to consider both short and long range contacts, i.e. contacts between residues that are < 6 (short range) and ≥ 6 (long range) residues apart respectively. In what follows, lower (upper) case residue indices, e.g. i (J), refer to the ith (Jth) residue in the target (template) protein respectively. Let nS(i)d and nL(i)d be the number of such short and long range contacts for residue i in the target structure.
| (2) |
with β = S or L and Md(l, i) the identity of the lth contacting residue with residue i.
Then, the first pass scoring function to generate the initial alignment given for a given γ is given by
| (3) |
Note that as γ increases, the scoring penalty for contact number mismatch grows as well. In practice, three independent runs will be done, where γ = (2,4,6), respectively. Dynamic programming is implemented in the identical manner as in PROSPECTOR_449.
Following the first pass for a given γ, we then perform a series of 30 successive iterations. The score of a given alignment for the Kth iteration is given as follows: Let aln(J)K−1 be the alignment of template residue J to target residue i generated by the K-1st alignment. Then, the number of correctly aligned contacts with a distance cutoff d for residue i=aln(J) is given by
| (4) |
where δεζ is the Kronecker delta and NT is the number of template residues, and T is the residue index. The score associated with aligning residue i with residue J is given by
| (5a) |
where
| (5b) |
The best of the successive set of alignments is chosen from the maximum value of the fraction of native contacts aligned in the template structure and is calculated for an M residue target protein following the Kth iteration by
| (6) |
In practice, fN is obtained by iterating over a range of 11 gap opening and gap extension penalties, where the gap opening penalties range from [−1,−2] and the gap extension penalties range from [0.–0.9], and are decremented by −0.1 per iteration. In addition to generating the alignment over the entire template molecule, we consider alignments to template substructures. We first consider the template substructure from residues 6 to NT − 5. The next substructure is from 11 to NT − 10. The process is iterated up to a total of up to 10 times, each time successively decreasing the substructures’ length by 10 residues, provided that the template is at least 10% longer than the target protein. The goal is to try and capture good substructure alignments to the template even when the global contact map overlap is quite poor. This was found to be necessary especially when the target and templates differ significantly in length. While in principle a perfect global search algorithm should find the best alignment, given that we employ an iterative dynamic programming algorithm, sometimes the alignment gets trapped in deep local minima. This procedure helps alleviate this problem.
As a preliminary validation step, C-align was applied to generate the contact map alignment of the 3221 target proteins. 95.2% have a TM-score≥ 0.95, with 98% of the targets having greater than 95% of their native contacts recovered. We next compared the resulting best TM-score obtained from using C-align to that of the best structural alignment, SA, obtained from fr-TM-align on templates whose sequence identity to the target is ≤ 30%; for this comparison, the best SA templates are used in both cases. The correlation coefficient for their TM-scores is 0.93, with the full set of pairs of TM-scores shown in Figure 1. As expected, in almost all cases, the TM-score obtained from the best structural alignment ≥ the TM-score obtained from aligning contact maps, with the greatest discrepancy between the two methods found in the neighborhood of TM-scores of <0.40; see below for the discussion of these are non-threadable proteins. In this regime, the best contact map alignment need not produce the best TM-score, with the discrepancy generally decreasing as the TM-score increases. Nevertheless, these results show that C-align can generate good quality alignments. It will be used in what follows to help dissect what are the features of those proteins where threading fails.
Figure 1.

Comparison of the best TM-score obtained using C-align to the best structural alignment obtained from fr-TM-align on templates whose sequence identity to the target is ≤ 30%.
Finally, in what follows, we will need to calculate the contact order of a given target from:
| (7) |
RESULTS
Do different threading algorithms perform similarly on the same target protein?
The first question that needs to be addressed is whether or not the failure of a given threading algorithm is due to the nature of the target structure or reflects differences among the various implementations of the different threading approaches. The latter is often the implicit expectation. However, as shown in Figure 2, this is not the case.
Figure 2.
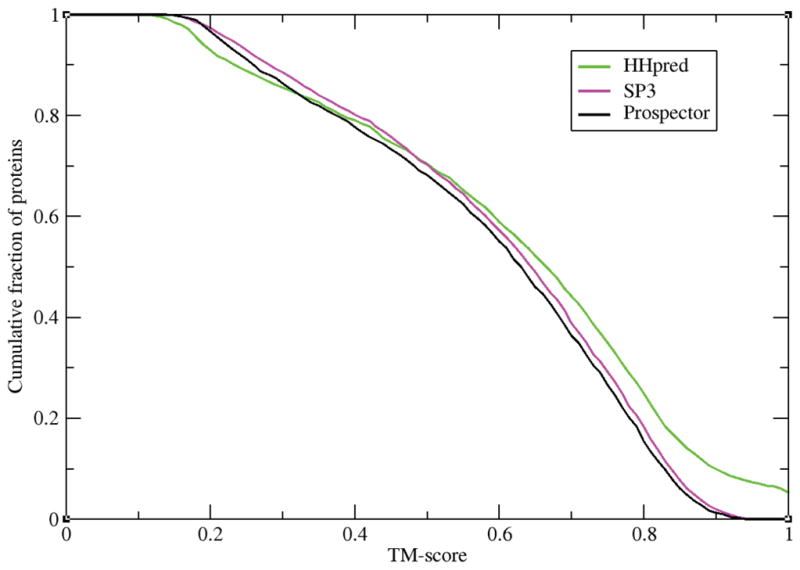
For the best of the top 5 ranked template alignments, the cumulative fraction of proteins whose TM-score ≥ TM-score on the x-axis.
In the TM-score range <0.5, SP3 performs the best, while HHpred generates better high quality alignments in the >0.5, high TM-score range. But with regards to the ability to identify suitable templates, the performance of all three approaches, despite the details of implementation, is remarkably similar. This conclusion is further reinforced in Figure 3 where for proteins that are threadable (the TM-score of the best of top 5 ranked templates ≥ 0.4) and non-threadable (TM-score<0.4), we plot the rank of the very best template (whose threading TM-score is maximal) obtained by the three different threading approaches. In this case, the performance of HHpred and PROSPECTOR_4 are virtually identical, with SP3 selected templates having slightly higher rank, with lower rank being better. Of course, it is possible that each of the three algorithms on average performs better on mostly different protein targets, and yet the average behavior is the same. That this is not the case at all is clearly demonstrated in Table 1.
Figure 3.

For both threadable and non-threadable protein targets, the cumulative fraction of templates whose rank ≤ value indicated on the x-axis.
Table 1.
Comparison of threadable and non-threadable targets in HHpred, SP3 and PROSPECTOR_4..
| Algorithm | HHpred | SP3 | PROSPECTOR_4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TH-TH | TH-NTH | NTH-TH | NTH-NTH | TH-TH | TH-NTH | NTH-TH | NTH-NTH | TH-TH | TH-NTH | NTH-TH | NTH-NTH | |
| HHpred | 2544 | 0 | 0 | 677 | 2471 | 73 | 113 | 563 | 2355 | 189 | 148 | 528 |
| SP3 | 2471 | 113 | 73 | 563 | 2584 | 0 | 0 | 637 | 2392 | 192 | 111 | 526 |
| Prospector_4 | 2355 | 189 | 189 | 528 | 2392 | 111 | 192 | 526 | 2503 | 0 | 0 | 718 |
X-Y, with X and Y= TH or NTH means that the given target is threadable, TH, or non-threadable, NTH in the first, (X) and second (Y) members of the pair of compared threading algorithms.
Clearly based on Table 1, in no more than 6% of the cases are the same proteins classified as threadable by one algorithm and non-threadable by the other. Indeed, 2323 proteins are assigned to be threadable by all three methods. Put another way, which proteins are threadable or not depends mainly on the target protein itself and not on the threading algorithm which is employed. In what follows, we wish to elucidate why this is the case.
Are there always good structures (TM-score ≥ 0.4) in the threading template library?
The most straightforward explanation for the fact that all three threading algorithms behave essentially the same is that for the non-threadable targets there are no good structural templates in the template library, whereas for threadable proteins there are. That this is not the explanation is convincingly demonstrated in Figure 4, where we plot the cumulative fraction of proteins whose best template TM-score obtained from the structural alignment algorithm fr-TM-align53 versus best TM-score is presented. We also include the corresponding cumulative fraction for the best TM-score obtained from threading. While it is true that threadable templates have on average considerably higher TM-scores, in no case is there a target for which the best template structural alignment TM-score is not ≥0.4. To preclude the possibility that the best templates have a considerable number of gaps in the alignment (i.e. are discontinuous alignments in very large proteins with lots of gaps), we also plot the corresponding distribution restricted to templates < 301 residues. Again, the same behavior is clearly seen. What is also evident is that the difference in TM-scores of proteins from their best structural alignment is about 0.3 TM-score units for non-threadable proteins and at most 0.1 TM-score units for threadable proteins. Thus, while non-threadable targets have templates that are structurally more distant than their templates which are of poorer quality than threading targets, nevertheless there are certainly adequate templates in the PDB library to yield far better results than those obtained from threading. For the best templates, the very best templates have lower (higher) ranks when threadable (non-threadable) proteins are used (see Figure 3) (lower ranks are better). Thus, more complex explanations of the cause of consensus non-threadable targets are required.
Figure 4.

For both threadable and non-threadable protein targets, the cumulative fraction of templates whose best TM-score obtained by the structural alignment algorithm fr-TM-align, Best SA, and from PROSPECTOR_4, Best Threading. Threadable and non-threadable targets are indicated in black and red respectively.
Comparison of threading results for artificial and native protein sequences
For distantly related targets and templates, another possible origin of non-threadable proteins is that they are evolutionary more distant from their templates than threadable ones and/or they have too few sequences to construct sensitive sequence profiles. To examine this issue, we generated a set of 60 sequences with protein like, amino acid composition per target protein selected on the basis that they have a low, knowledge based force field47 energy in the target structure. The average pairwise sequence identity of the sequences that adopt a given target fold is 11.9%, with a standard deviation of 1.1%. These targets were then threaded against a set of 12,532 templates (the subset of structures in LIST.templates<300 residues in length) whose sequence profiles are also generated from 60 artificial sequences/template. For each template protein structure, their mean pairwise sequence identity is 11.3%, with a standard deviation of 1.3%. PROSPECTOR_4 was then employed to thread these artificial targets to the artificial template structures. The mean sequence identity of the target/best threading template is 13.8%. 2141 have a TM-score to native in the best of top 5 threading templates ≥ 0.4; viz. are threadable. Of these, 2027 are also threadable when native sequences are used. Similarly, for the non threadable targets, 603 are in common when artificial and native protein sequences are used.
Interestingly, as shown in Figure 5, the correlation coefficient between the TM-score of the best template obtained using native sequences to that using artificial sequences is 0.82. While there are clearly some differences as to which proteins are threadable and which are not, overall, the performance when artificial and wild type sequences are used is similar. Note that all artificial sequences when threaded to their native structure recover the native alignment; the issue is what happens when distantly related template structures are used. This observation was previously used in an attempt to help improve threading by using artificial template protein sequences in one incarnation of threading54, but as seen from the figure, the improvement is uneven.
Figure 5.

TM-score of the best of top 5 ranked PROSPECTOR_4 threading template alignments obtained for artificial target and template protein sequences versus that when native protein sequences are used. Black (red) circles indicate (non) threadable proteins using native target and template sequences as input.
Figure 5 also clearly shows that in reality threading need not in fact detect evolutionarily related sequences as the majority of artificial sequences generated by the criterion of protein stability are also threadable. Yet, the two sets of sequences behave in a similar fashion as to which protein structures are threadable and which are not. The potentials used to generate the artificial sequences could provide a clue as to what in reality is driving selection/threadability. We first consider the case of threading by matching secondary structure alone. When this is done, only 1283/3221 targets are threadable; this reflects the fact that often the template with the best match to the native protein’s secondary structure does not have the same global fold as the target. When we then use secondary structure plus pair potential terms to drive the threading alignment, then 2066/3221 targets are threadable. This suggests, not surprisingly, that the selection of sequences is strongly influenced by tertiary interactions, a rather reasonable result. But, for this method to work, pairwise residue contacts have to be preserved at an appropriate level between the target and template structures. Based on this conjecture, we now examine what happens when contact map alignment is done and whether this will provide deeper insight into what constitutes a threadable versus a non-threadable protein target.
Analysis of threadability in contact map driven alignments
In what follows, we employ the native contacts of the target sequence to generate the best contact map alignment to the template. This is being done deliberately as we are seeking to understand why certain protein structures are threadable, whereas others are not. If one cannot generate native like alignments even when all the contacts are native, then the performance when predicted contacts are used (which is the actually situation), will certainly be worse. Consider the cases of the top 5 templates selected from threading versus the best structural alignments. For threading selected templates, C-align generates predictions whose best of top 5 ranked alignments has a TM-score ≥ 0.4 for 2614 targets versus 2949 targets when the best non trivial (sequence identity <0.3) structural alignment, SA, template identified from fr-TM-align is used. Figure 6 shows the correlation between the C-align generated best TM-score and the PROSPECTOR_4 best TM-score for the same set of threading templates. Clearly, the two sets are highly correlated with a correlation coefficient of 0.92. Once again, independent of the particular method used to generate the alignment, the quality of the results depends on the particular target structure chosen. The question then is what makes a given structure threadable or not. We next turn to the analysis of this key question.
Figure 6.

TM-score of the best of top 5 ranked PROSPECTOR_4 threading template alignments obtained from contact map based alignment vs. that when threading is used. Black (red) circles indicate (non-) threadable proteins using native target and template sequences as input.
Let us first examine the performance of C-align using the top five templates selected not from threading but from the best structural alignment. Despite the fact that fr-TM-align generates structures whose TM-score ≥ 0.4 in every case, C-align still yields 247 non-threadable targets. In contrast, if the PROSPECTOR_4 selected templates are used, then there are a total of 581 non-threadable targets despite the fact that the target structure’s native contacts are used to drive the alignment. In Figure 7, we present the distribution of the fraction of aligned native contacts obtained in each case. For templates obtained from the best structural alignments, the p-value of Student’s t-test for threadable and non-threadable proteins is 0.96. This is the same p-value obtained for that between threadable and non-threadable PROSPECTOR_4 generated templates. Thus, the difference in the distributions of the fraction of aligned native contacts for each of the two respective distributions is not statistically significant. Put another way, for evolutionarily distant proteins, due to structural differences, the aligned native contact fractions behave similarly. For threadable proteins, these templates bear a significant global structural similarity to native, whereas for non-threadable ones they don’t. When structural alignment selected templates are used, for the 247 non-threadable targets, there is an alternative high scoring alignment generated such that the resulting aligned coordinates have a TM-score <0.4 to the native fold (see black vs. red curves). One can even have very high contact map overlap (see fc>0.9). There are non-threadable targets where all native contacts are aligned; e.g., 1bbt4 whose TM-score=0.22. These proteins tend to be small and have very open, non globular protein conformations. In fact, 26% (65/247) of these non-threadable proteins are ≤ 60 residues in length. In contrast, for templates identified on the basis of their structural alignment to native, about 8% of threadable proteins are in this size range. Clearly, smaller proteins contain a smaller number of contacts and secondary structure elements that can be aligned, and the possibility of finding a globally dissimilar structure that satisfies these constraints is large. These small proteins need to be accounted for in any threading alignment algorithm. For threading identified templates, the fraction of threadable proteins ≤ 60 residues in length drops to 6% of the total number of threadable proteins, and the number of such small, non-threadable targets essentially doubles to 131/581, with a somewhat reduced fraction of 23%. Of course, this a just part of the story, but the qualitative effect remains unchanged.
Figure 7.

Distribution of the fraction of targets versus the fraction of aligned native contacts generated by C-align for the best of top 5 templates identified using the fr-TM-align structural alignment algorithm and by PROSPECTOR_4.
While it is certainly true on the basis of Figure 7 that on average there are more templates whose fN exceeds 0.6 (95%) in threadable than non-threadable templates(88%) structural alignment, SA, identified templates; nevertheless there is considerable overlap. Similarly, for PROSPECTOR_4 identified templates, we can be confidently assign a protein as being non-threadable when fN <0.6 (<93% of threadable targets exceed this value, whereas 49% of non-threadable targets do). However, since the two fN distributions overlap significantly, contact overlap criteria cannot be reliable used to differentiate threadable from non-threadable proteins. In non-threadable cases, at a given fN even when the same template as that giving the best structural alignment is used, a poor alignment will result. Other folds, that tend to be larger, suffer from less fN redundancy, and as such are more likely to be threadable. Thus, whether or not a protein is threadable simply depends on whether its fold is such that high fN scoring templates adopt the native fold or not. For non-threadable proteins, given differences in the structure of the target and the template, there are nonnative folds with very similar fN values, whereas for threadable proteins the best scoring folds have a similar structure to the target.
DISCUSSION AND CONCLUSION
The qualitatively similar performance of different threading algorithms on the same set of threading targets strongly suggests that there are common features of the proteins that are threadable (those having good quality alignment to native, taken here as a TM-score ≥0.4) when there are no closely related proteins in the template library. This issue is relevant in that threading based structure prediction methods seem to have hit a plateau, and it is important to understand why this is the case. Does it reflect a fundamental limitation of the approach or is it just a matter of the structural completeness of the template library? Of course, as the number of protein structures and sequences continues to grow, eventually, there will be no distantly related threading templates, and one would expect threading to work in general. In the interim, however, when a significant fraction of an arbitrary exome has over half of its sequences in the distant homology regime to the PDB, this is critical issue if structure prediction is be able to assist in the annotation of protein function35.
Thus, there is a clear need to identify why contemporary threading methods fail on the certain targets. Here, we showed that even for non-threadable proteins, there are structurally related template proteins in the PDB. We also eliminated that trivial explanation that non-threadable targets are more evolutionarily distant from their templates than threadable ones by showing that the behavior of artificially generated sequences selected for their stability in the structure of interest are highly correlated with that of native proteins. Since these artificial sequences are not related to each other by evolution and have the same sequence diversity, this suggests that it is mainly the properties of the protein’s structure that make it threadable or not. Furthermore, even when a pair of proteins are matched by threading, one cannot conclude that they are evolutionarily related, as the artificial proteins are clearly unrelated by evolution and yet they match as well.
Motivated by the results from the artificial sequences, we sought a structure based solution to the question of threadability. Clearly, aligning secondary structure is not informative, as there are many different folds with essentially the same secondary structure pattern and length of secondary structure elements. We then focused on contact map alignment and found that the results in most consonance with direct target-template structure alignments involve the use of contact maps between Cα atoms defined at the 8.5 Å and 14 Å cutoff level. This generates alignments whose TM-scores are highly correlated with those obtained from threading. What is clear is that the distribution of the fraction of aligned native contacts are very similar in both threadable and non-threadable templates. Put succinctly, there are very high scoring alignments in non-threadable templates that result from differences in the target and template structures. When the native structure is used, almost all targets have a contact alignment based TM-score ≥ 0.95. When structural alignment selected templates whose sequence identity to the target protein <30% are used, then for the majority of cases, good alignments are generated, but for 7.6%, poor quality alignments are generated. Yet, in many cases, these have a high fraction of native contacts. A significant number of these non threadable folds are small proteins of low fold complexity, but this is not always so. Rather, non-threadable proteins simply reflect the fact that the average fraction of these native Cα contacts do not uniquely specify the native state. That is, it is a too coarse grained measure. While in the majority of cases, it is the native fold which is recovered; in a minority (15%), there is an alternative alignment which has a higher contact fraction that that of the native alignment and threading fails. Despite our attempts to tease out a single cause as to why some folds are non-threadable, we were unable to find it. Rather, non threadable protein structures often have a synergistic combination of effects: small proteins with a small number of secondary structural elements, low fold complexity, low contact order and sometimes just the fact that a non native fold has better contact map overlap than native like folds.
What then are possible avenues of improvement when secondary structure and native contact overlap are insufficient to select the native fold? This reflects the implicit assumption of any threading approach that when an appropriate comparison metric is high, the more likely it is that native like features are recovered. It is an open question whether side chain contacts can be routinely predicted by residue covariation40,41 with sufficient accuracy for enough targets that the coverage of non-threadable targets can be improved. However, perhaps similar problems as encountered by threading will be encountered. As demonstrated here, even when all nonlocal contacts are satisfied, alternative non-native alignments can be generated for certain structures. More generally, we could turn the question around. Rather than asking what is the property of non threadable target structures, we could ask (in the twilight zone of sequence identity) if there are certain templates that when matched by threading are almost always indicative that the target protein is non-threadable. This can provide another means of assessing the reliability of threading in advance and possible sets of alternative folds consistent with the identified, non-threadable template. For targets mapped to these non-threadable templates, template free methods will need to be applied as threading based approaches are not reliable. In this way, the manifold of non-threadable templates could be exploited to provide additional information. Alternatively, perhaps methods that could predict higher order patterns of contacts or global contact maps might sometimes be of help in the regime where there is roughly 50–60% overlap and yet the folds differ. What is clear, however, is that traditional approaches to threading have more or less realized their maximum potential, and different methods need to be developed to deal with the 15–20% of targets that remain outside the reliable regime of current structure prediction methods. This remains a crucial problem that needs to be addressed.
Acknowledgments
This project was funded in part by 1R35GM118039 of the Division of General Medical Sciences of the NIH.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. JS conceived of the idea and performed the majority of the calculations and analysis. HZ performed the threading calculations using the HHpred and SP3 threading algorithms.
References
- 1.Kryshtafovych A, Fidelis K, Moult J. Casp9 Results Compared to Those of Previous Casp Experiments. Proteins. 2011;79(Suppl 10):196–207. doi: 10.1002/prot.23182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y. [accessed October 10, 2016];Automated Assessment of Protein Structure Prediction in Casp11. http://zhanglab.ccmb.med.umich.edu/casp11/
- 3.Joo K, et al. Template Based Protein Structure Modeling by Global Optimization in Casp11. Proteins. 2016;84(Suppl 1):221–32. doi: 10.1002/prot.24917. [DOI] [PubMed] [Google Scholar]
- 4.Yang J, Zhang W, He B, Walker SE, Zhang H, Govindarajoo B, Virtanen J, Xue Z, Shen HB, Zhang Y. Template-Based Protein Structure Prediction in Casp11 and Retrospect of I-Tasser in the Last Decade. Proteins. 2016;84(Suppl 1):233–46. doi: 10.1002/prot.24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Modi V, Xu Q, Adhikari S, Dunbrack RL., Jr Assessment of Template-Based Modeling of Protein Structure in Casp11. Proteins. 2016;84(Suppl 1):200–20. doi: 10.1002/prot.25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Modi V, Dunbrack RL., Jr Assessment of Refinement of Template-Based Models in Casp11. Proteins. 2016;84(Suppl 1):260–81. doi: 10.1002/prot.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang S, Li W, Liu S, Xu J. Raptorx-Property: A Web Server for Protein Structure Property Prediction. Nucleic Acids Res. 2016;44:W430–5. doi: 10.1093/nar/gkw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panchenko AR, Marchler-Bauer A, HBS Combination of Threading Potentials and Sequence Profiles Improves Fold Recognition. J Mol Biol. 2000;296:1319–1331. doi: 10.1006/jmbi.2000.3541. [DOI] [PubMed] [Google Scholar]
- 9.Zhou H, Zhou Y. Sparks 2 and Sp3 Servers in Casp6. Proteins. 2005;61(Suppl 7):152–6. doi: 10.1002/prot.20732. [DOI] [PubMed] [Google Scholar]
- 10.Wu S, Zhang Y. Muster: Improving Protein Sequence Profile-Profile Alignments by Using Multiple Sources of Structure Information. Proteins. 2008;72:547–56. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marti-Renom MA, Madhusudhan MS, Sali A. Alignment of Protein Sequences by Their Profiles. Protein Sci. 2004;13:1071–87. doi: 10.1110/ps.03379804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soding J, Biegert A, Lupas AN. The Hhpred Interactive Server for Protein Homology Detection and Structure Prediction. Nucleic Acids Res. 2005;33:W244–8. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meier A, Soding J. Automatic Prediction of Protein 3d Structures by Probabilistic Multi-Template Homology Modeling. PLoS Comput Biol. 2015;11:e1004343. doi: 10.1371/journal.pcbi.1004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, Li M, Kim D, Xu Y. Raptor: Optimal Protein Threading by Linear Programming. J Bioinformatics Comp Bio. 2003;1:95–117. doi: 10.1142/s0219720003000186. [DOI] [PubMed] [Google Scholar]
- 15.Xu J, Peng J, Zhao F. Template-Based and Free Modeling by Raptor++ in Casp8. Proteins. 2009;77(Suppl 9):133–7. doi: 10.1002/prot.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skolnick J, Kihara D, Zhang Y. Development and Large Scale Benchmark Testing of the Prospector_3 Threading Algorithm. Proteins. 2004;56:502–18. doi: 10.1002/prot.20106. [DOI] [PubMed] [Google Scholar]
- 17.Wu S, Zhang Y. Lomets: A Local Meta-Threading-Server for Protein Structure Prediction. Nucleic Acids Res. 2007;35:3375–82. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S, Zhang Y. A Comprehensive Assessment of Sequence-Based and Template-Based Methods for Protein Contact Prediction. Bioinformatics. 2008;24:924–31. doi: 10.1093/bioinformatics/btn069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou H, Skolnick J. Improving Threading Algorithms for Remote Homology Modeling by Combining Fragment and Template Comparisons. Proteins. 2010;78:2041–8. doi: 10.1002/prot.22717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Zhang W, He B, Walker SE, Zhang H, Govindarajoo B, Virtanen J, Xue Z, Shen HB, Zhang Y. Template-Based Protein Structure Prediction in Casp11 and Retrospect of I-Tasser in the Last Decade. Proteins. 2015 doi: 10.1002/prot.24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berman H, Henrick K, Nakamura H, Markley JL. The Worldwide Protein Data Bank (Wwpdb): Ensuring a Single, Uniform Archive of Pdb Data. Nucleic Acids Res. 2007;35:D301–3. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y. [accessed October 10, 2016];Automated Assessment of Predicted Models by Casp7 Servers. 2006 http://Zhang.Bioinformatics.Ku.Edu/Casp7/Index_All.Html.
- 23.Rost B, Sander C, Schneider R. Phd--an Automatic Mail Server for Protein Secondary Structure Prediction. Comput Appl Biosci. 1994;10:53–60. doi: 10.1093/bioinformatics/10.1.53. [DOI] [PubMed] [Google Scholar]
- 24.Jones DT. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 25.Aydin Z, Altunbasak Y, Borodovsky M. Protein Secondary Structure Prediction for a Single-Sequence Using Hidden Semi-Markov Models. BMC Bioinformatics. 2006;7:178. doi: 10.1186/1471-2105-7-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen MN, Rajapakse JC. Prediction of Protein Secondary Structure with Two-Stage Multi-Class Svms. Int J Data Min Bioinform. 2007;1:248–69. doi: 10.1504/ijdmb.2007.011612. [DOI] [PubMed] [Google Scholar]
- 27.Zhou T, Shu N, Hovmoller S. A Novel Method for Accurate One-Dimensional Protein Structure Prediction Based on Fragment Matching. Bioinformatics. 2010;26:470–7. doi: 10.1093/bioinformatics/btp679. [DOI] [PubMed] [Google Scholar]
- 28.Nasrul Islam M, Iqbal S, Katebi AR, Tamjidul Hoque M. A Balanced Secondary Structure Predictor. J Theor Biol. 2016;389:60–71. doi: 10.1016/j.jtbi.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y. I-Tasser Server for Protein 3d Structure Prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H, Skolnick J. Template-Based Protein Structure Modeling Using Tasser(Vmt) Proteins. 2011 doi: 10.1002/prot.23183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Skolnick J. Tm-Align: A Protein Structure Alignment Algorithm Based on the Tm-Score. Nucleic Acids Res. 2005;33:2302–9. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou H, Skolnick J. Findsite(Comb): A Threading/Structure-Based, Proteomic-Scale Virtual Ligand Screening Approach. J Chem Inf Model. 2013;53:230–40. doi: 10.1021/ci300510n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wass MN, Kelley LA, Sternberg MJ. 3dligandsite: Predicting Ligand-Binding Sites Using Similar Structures. Nucleic Acids Res. 2010;38:W469–73. doi: 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Srinivasan B, Zhou H, Kubanek J, Skolnick J. Experimental Validation of Findsite(Comb) Virtual Ligand Screening Results for Eight Proteins Yields Novel Nanomolar and Micromolar Binders. J Cheminform. 2014;6:16. doi: 10.1186/1758-2946-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou H, Gao M, Skolnick J. Comprehensive Prediction of Drug-Protein Interactions and Side Effects for the Human Proteome. Sci Rep. 2015;5:11090. doi: 10.1038/srep11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brylinski M, Skolnick J. A Threading-Based Method (Findsite) for Ligand-Binding Site Prediction and Functional Annotation. Proc Natl Acad Sci U S A. 2008;105:129–34. doi: 10.1073/pnas.0707684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou H, Gao M, Skolnick J. Entprise: An Algorithm for Predicting Human Disease-Associated Amino Acid Substitutions from Sequence Entropy and Predicted Protein Structures. PLoS One. 2016;11:e0150965. doi: 10.1371/journal.pone.0150965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou H, Skolnick J. Ab Initio Protein Structure Prediction Using Chunk-Tasser. Biophys J. 2007;93:1510–8. doi: 10.1529/biophysj.107.109959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu D, Zhang J, Roy A, Zhang Y. Automated Protein Structure Modeling in Casp9 by I-Tasser Pipeline Combined with Quark-Based Ab Initio Folding and Fg-Md-Based Structure Refinement. Proteins. 2011;79(Suppl 10):147–60. doi: 10.1002/prot.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gobel U, Sander C, Schneider R, Valencia A. Correlated Mutations and Residue Contacts in Proteins. Proteins. 1994;18:309–17. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- 41.Skolnick J, Kolinski A, Ortiz AR. Monsster: A Method for Folding Globular Proteins with a Small Number of Distance Restraints. J Mol Biol. 1997;265:217–41. doi: 10.1006/jmbi.1996.0720. [DOI] [PubMed] [Google Scholar]
- 42.Kinch LN, Li W, Monastyrskyy B, Kryshtafovych A, Grishin NV. Evaluation of Free Modeling Targets in Casp11 and Roll. Proteins. 2015 doi: 10.1002/prot.24973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ovchinnikov S, Kinch L, Park H, Liao Y, Pei J, Kim DE, Kamisetty H, Grishin NV, Baker D. Large-Scale Determination of Previously Unsolved Protein Structures Using Evolutionary Information. Elife. 2015;4:e09248. doi: 10.7554/eLife.09248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinch LN, Li W, Monastyrskyy B, Kryshtafovych A, Grishin NV. Assessment of Casp11 Contact-Assisted Predictions. Proteins. 2016 doi: 10.1002/prot.25020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Skolnick J. The Protein Structure Prediction Problem Could Be Solved Using the Current Pdb Library. Proc Natl Acad Sci U S A. 2005;102:1029–34. doi: 10.1073/pnas.0407152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skolnick J, Zhou H, Brylinski M. Further Evidence for the Likely Completeness of the Library of Solved Single Domain Protein Structures. The journal of physical chemistry B. 2012 doi: 10.1021/jp211052j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skolnick J, Gao M. Interplay of Physics and Evolution in the Likely Origin of Protein Biochemical Function. Proc Natl Acad Sci U S A. 2013;110:9344–9. doi: 10.1073/pnas.1300011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skolnick J, Gao M, Zhou H. How Special Is the Biochemical Function of Native Proteins? F1000Res. 2016;5 doi: 10.12688/f1000research.7374.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SY, Skolnick J. Tasser_Wt: A Protein Structure Prediction Algorithm with Accurate Predicted Contact Restraints for Difficult Protein Targets. Biophys J. 2010;99:3066–75. doi: 10.1016/j.bpj.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soding J, Biegert A, Lupas AN. The Hhpred Interactive Server for Protein Homology Detection and Structure Prediction. Nucleic Acids Res. 2005;33:W244–8. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hildebrand A, Remmert M, Biegert A, Söding J. Fast and Accurate Automatic Structure Prediction with Hhpred. Proteins. 2009;77:128–32. doi: 10.1002/prot.22499. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Skolnick J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins. 2004;57:702–10. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 53.Pandit SB, Skolnick J. Fr-Tm-Align: A New Protein Structural Alignment Method Based on Fragment Alignments and the Tm-Score. BMC Bioinformatics. 2008;9:531. doi: 10.1186/1471-2105-9-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee SY, Skolnick J. Benchmarking of Tasser_2. 0: An Improved Protein Structure Prediction Algorithm with More Accurate Predicted Contact Restraints. Biophys J. 2008;95:1956–64. doi: 10.1529/biophysj.108.129759. [DOI] [PMC free article] [PubMed] [Google Scholar]