

Abstract
The RASopathy neurofibromatosis 1 is an autosomal dominant hereditary cancer syndrome that represents a major risk for the development of malignancies, particularly malignant peripheral nerve sheath tumors (MPNSTs). MPNSTs are unique sarcomas that originate from the peripheral nerve and represent the only primary cancer of the peripheral nervous system. To date, surgery is the only treatment modality proven to have survival benefit for MPNSTs and even when maximal surgery is feasible, these tumors are rarely curable, despite the use of chemotherapy and radiation. In this review, we discuss the current state-of-the-art treatments for MPNSTs, latest therapeutic developments, and critical aspects of the underlying molecular and pathophysiology that appear promising for therapeutic developments in the future. In particular, we discuss the specific elements of cancer in the peripheral nerve and how that may impel development of unique therapies for this form of sarcoma.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-017-0518-y) contains supplementary material, which is available to authorized users.
Keywords: Malignant peripheral nerve sheath tumor, Malignant transformation, Sarcoma, Neurofibromatosis, Treatment, Chemoprevention
Introduction to Neurofibromatosis 1
The RASopathy neurofibromatosis 1 (NF1) is an autosomal dominant hereditary cancer syndrome that affects ~1 in 3000 individuals [1]. The diagnosis of NF1 is based on National Institutes of Health clinical consensus criteria [2, 3], with the main clinical features including multiple café-au-lait macules, axillary freckling, Lisch nodules, optic pathway gliomas, peripheral nerve sheath tumors, multiple forms of cognitive dysfunction, and a higher risk of cancers such as pheochromocytoma and breast cancer in young women. NF1 is caused by various DNA alterations, including point mutations, deletions, insertions, microdeletions, and splicing mutations of the NF1 tumor suppressor gene at 17q11.2, which encodes the GTPase activating protein neurofibromin that catalyzes the inactivation of Ras by accelerating guanosine triphosphate hydrolysis to guanosine diphosphate [4, 5]. In affected individuals, truncation or loss of neurofibromin results in activated Ras with subsequent activation of the Raf–mitogen-activated protein kinase (MEK)–extracellular signal-regulated kinase (ERK) cascade. The Ras hyperactivation supports the frequent development of multiple benign tumors, especially neurofibromas, and, less frequently, cancers.
NF1 is a highly variable disease and considerable differences in clinical symptoms are noted within the same family [1]. Notably, phenotype–genotype correlation studies have suggested that the type of mutation in the NF1 gene does not solely account for the observed phenotype, with the exception of patients carrying chromosomal deletions (microdeletions) [6]. Although only 5% to 10% of patients with NF1 harbor these microdeletions, affected individuals account for an overproportional morbidity and mortality because the loss of the entire NF1 locus and large parts of adjacent sequences predisposes patients to a more severe clinical phenotype characterized by a higher burden of cutaneous neurofibromas, earlier onset of benign neurofibromas, and a higher incidence of malignancies, particularly malignant peripheral nerve sheath tumors (MPNSTs) [7, 8]. Hence, a high index of suspicion and close medical surveillance program is critical to detect a large range of possible complications, including MPNST, even though they occur in only a relatively small percentage of all patients with NF1 [9].
Plexiform Neurofibromas: Nerve Tumor Precursor for MPNST
A hallmark of NF1 is the presence of plexiform neurofibromas (pNFs) and they are present in 50% of individuals with NF1 [10]. pNFs are tumors that arise in the peripheral nerve and involve multiple nerve fascicles. They typically grow along a nerve and its branches and there are frequently collisions of multiple nerve tumors into large masses in plexi such as the brachial or lumbosacral plexi (Fig. 1). Thus, the involvement of multiple fascicles (and indeed whole plexi) makes complete surgical resections without neurologic deficit impossible. Even in lesions whose location and size would allow meaningful surgical debulking, a complete resection is not possible without transection of the nerve. As a result, patients with pNFs are often recommended to undergo surveillance alone and manifestations (pain, neurologic deficits) are managed expectantly. Ultimately, these tumors can grow over time to cause substantial morbidity. Equally importantly, these benign tumors act as precursor lesions and can transform into MPNSTs, with high body tumor burden being a strong risk factor for MPNST development [11]. This is why some experts recommend targeted and whole-body magnetic resonance imaging to screen for subclinical pNFs early in life; however, such screening method has not been incorporated into the NF1 guidelines yet as there are no prospective data about the optimal screening protocol or age for surveillance, and no proven interventions to prevent malignant transformation [12].
Fig. 1.
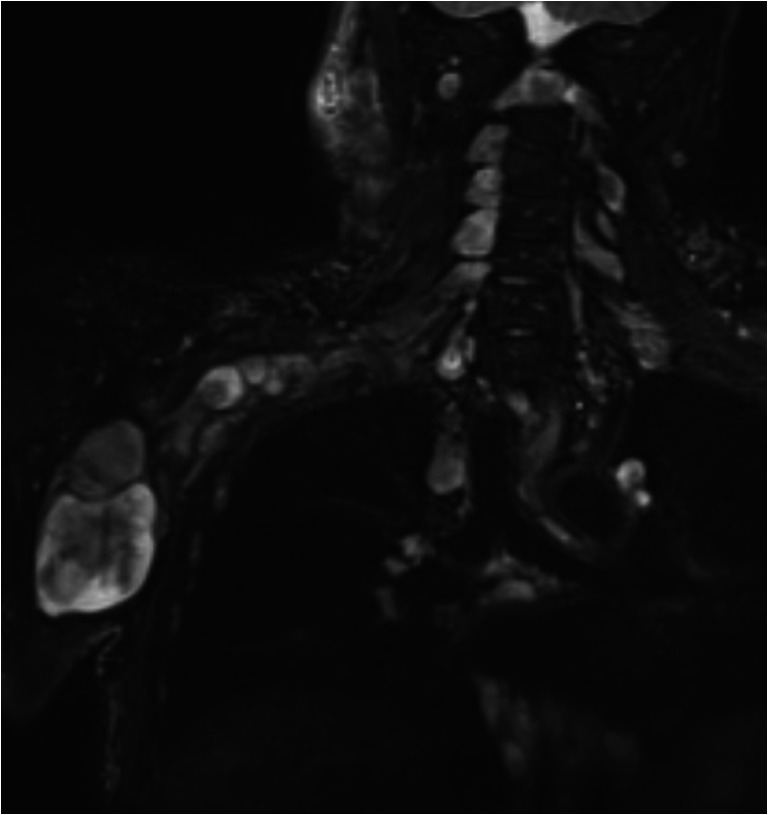
Plexiform neurofibroma involving the right brachial plexus. Multiple nerve sheath tumors (neurofibromas) at each level of the cervical spine and extending through the brachial plexus
In people with known pNF, imaging characteristics that may suggest impending malignant transformation include nodular-appearing lesions with an apparent diffusion coefficient value <1 on magnetic resonance imaging; lesions larger than ≥ 3 cm in diameter; signal heterogeneity; ill-defined margins; presence of edema; or lesions that show avid uptake of fluorodeoxyglucose on positron emission tomography (Fig. 2) [13–15]. When biopsied, these lesions are often classified as atypical neurofibromas (aNFs), a premalignant lesion that exhibits some features of low-grade MPNST, such as higher mitotic activity or neoangiogenesis, and accumulation of molecular abnormalities [16].
Fig. 2.

Imaging features of various peripheral nerve sheath tumors in the left leg of a person with neurofibromatosis 1 (NF1). (A) T2-weighted image with fat saturation sequence showing increased signal, the anterior lesion with poorly defined boundaries (white arrowhead), and the posterior with well-demarcated boundaries (green arrowhead). (B) Apparent diffusion coefficient (ADC) imaging shows low signal and a quantified minimum ADC of only 0.2 × 10–3/mm2/s in the anterior lesion (white arrowhead) compared with the posterior lesion with a normal minimum ADC value of 2.1 × 10–3/mm2/s (green arrowhead). (C) Fluorodeoxyglucose (FDG) positron emission tomography of the anterior lesion demonstrates an increased SUVmax of 6.4 (white arrowhead), while the posterior lesion shows no significant FDG uptake (green arrowhead)
Genetic Evolution of MPNSTs
About 8% to 13% of pNFs across all NF1 patients undergo cancerous transformation into MPNSTs [7, 17], in contrast to 16% to 26% in patients with microdeletions [8]. While > 80% of MPNSTs evolve from pre-existing pNFs, a small proportion can arise de novo. The last few years have provided a wealth of new knowledge on the biology and pathophysiology of NF1-associated MPNST that unveiled important mechanisms in the transition of pNF to MPNST. Similar to other cancer predisposition syndromes, accumulating evidence suggests that for carcinogenesis loss of NF1 alone is not sufficient but requires a multistep process in which accumulated genetic and epigenetic alternations affect the regulation of multiple cellular processes, including growth factor signaling, metabolism, and apoptosis. Although the detailed mechanisms of the malignant transformation remain unknown, biallelic-inactivating mutations must occur in NF1 in Schwann cells as the initial tumor-forming event, leading to either complete loss of function or substantial reduction of functional neurofibromin and subsequent hyperactivation of Ras and multiple downstream Ras effector pathways, including activation of phosphatidylinositol 3-kinase, protein kinase B, mammalian target of rapamycin (mTOR), Raf, MEK and ERK (Fig. 3) [18]. The inactivating NF1 mutations in Schwann cell alleles result in benign tumor formation, and these tumors have increased cell proliferation and hyperplasia, as seen in pNFs and aNFs, but no features of malignancy. Deletion of cell-cycle regulator CDKN2A/B is the next step in the progression toward cancer and this is present in the majority of aNFs but absent in pNFs [19]. Lastly, for the formation of a malignant cancer (MPNST) additional critical genetic and epigenetic events must appear. Those alterations include loss-of-function mutations in the tumor suppressor TP53, which among its many functions activates various growth factors and blocks the activation of HIF-1α leading to subsequent overexpression of vascular endothelial growth factor (VEGF) receptors, as well as loss-of-function of the histone methyltransferase polycomb repressive complex 2 (PRC2), including its core components EED or SUZ12 [19].
Fig. 3.

Schematic overview of underlying pathophysiology and targeted therapies in neurofibromatosis 1 (NF1)-associated malignant peripheral nerve sheath tumors (MPNSTs). MPNSTs arise from plexiform neurofibromas (pNFs) via a stepwise malignant transformation process that includes the loss of NF1 (gray box), deletion of CDKN2A, and loss of TP53 and PRC2. The hyperactivation of the Ras pathway and acquired epigenetic alterations provide therapeutic targets for various molecule inhibitors (yellow boxes) GPCR = G-protein coupled receptor; RTK = receptor tyrosine kinase; HSP = heat shock protein; MEK = mitogen-activated protein kinase; ERK = extracelluar regulated kinase; PI3K = phosphatidylinositol 3-kinase; AKT = protein kinase B; mTOR = mammalian target of rapamycin; AURKA = Aurora kinase A; BET = bromodomain and extraterminal; PRC2 = polycomb repressive complex 2; aNF = atypical neurofibroma; pNF = plexiform neurofibroma
In particular, SUZ12 is also lost in patients with microdeletions, which, as stated earlier, are associated with an increased risk for MPNST and poor survival. The loss of PRC2 components results in complete loss of trimethylation at lysine 27 of histone H3 (H3K27me3) and transcriptional activation of multiple PRC2-repressed homeobox master regulators, as well as their pathways, thereby potentiating the effects of NF1 mutations by amplifying Ras-driven transcription [20]. Those alterations in the context of TP53 loss provide a mitogenic advantage and promote malignant tumor formation by upregulating various proliferative stimuli, such as epidermal growth factor receptor (EGFR), neuregulin-1 co-receptor erbB2, c-Kit, platelet-derived growth factor-α, and c-Met [21].
Current Standard of Treatment and Outcomes in NF1-Associated MPNST
MPNSTs are the most common malignancy and the leading cause of death in individuals with NF1, particularly for those younger than 40 years of age [22]. It is the only primary cancer of the peripheral nervous system; however, peripheral nerves can also be involved in other cancers by compression or infiltration from metastasis and lymphomas. Histologically, MPNSTs are comprised of spindle cells reminiscent of Schwann cells that can be distinguished in high-grade MPNST and low-grade MPNST based on cellularity, mitotic figures, and presence of necrosis [23]. As MPNSTs often arise within peripheral nerves, considerable tissue heterogeneity exists which often complicates the diagnostic process.
MPNSTs are staged and treated as malignant soft tissue sarcomas. These tumors require a multidisciplinary team of surgical, medical, and radiation oncologists; radiologists; and pathologists. Tumor grade, location, extent of surgical resection, and presence or absence of metastases are the key factors influencing therapeutic decision-making. The only known definitive curative treatment for MPNSTs is complete surgical resection with wide negative margins, which may not be feasible in a number of patients, owing to variables such as tumor size, location, and metastases [17, 22]. In patients with high-grade lesions > 5 cm in size or where the attainment of wide excision margins by surgery alone is difficult, preoperative radiation therapy (RT) is often considered to improve local control, although its neoadjuvant role is not defined and a clear survival improvement has not been demonstrated [24–26]. RT is standard in non-NF1-associated MPNSTs, but controversial in the setting of NF1 because there is no proven survival advantage and ongoing concerns about an increased risk for malignant conversion of neighboring benign tumors after exposure to RT [26, 27]. Likewise, the role of chemotherapy remains unclear. A number of neurofibromatosis and sarcoma centers in the US utilize chemotherapy (ifosfamide, doxorubicin), pre- and/or postoperatively, in their treatment protocols with disease stabilization for the majority of patients and partial responses in a small fraction of patients [28]. Regardless, there is little prospective NF1-specific data about outcomes with this approach and these therapies have high risks of associated toxicity. Hence, there is currently no consensus about the role of chemotherapy in the setting of newly diagnosed NF1-associated MPNST. Despite maximal efforts, MPNST frequently recur [22], and for patients with recurrent, unresectable, or metastatic disease no treatment standards are available and enrollment in clinical trials should be considered. Overall, only 20% of patients with NF1-associated MPNST survive longer than 5 years, reflecting the urgent need for new and more effective therapeutics for this cancer.
Investigational Drugs in Clinical Trials
Although efforts are needed and ongoing to standardize and optimize existing therapies, we have possibly reached the limits of what can be accomplished with traditional cytotoxic agents. Advancements in understanding signaling mechanisms and molecular networks have identified new nodes of vulnerabilities. However, the rarity of these tumors and their heterogeneity has limited the number of trials available.
Owing to the lack of clinically effective anti-Ras drugs, translational research has focused on critical downstream effectors of Ras. In MPNSTs, loss of NF1 has been shown to activate downstream effectors of the receptor tyrosine kinase (RTK) pathway, including mitogen-activated protein kinases (MAPK) and phosphatidylinositol 3-kinase/protein kinase B/mTOR signaling pathways [29]. RTKs comprise a family of surface receptors that, under physiologic conditions, are intermittently activated by ligand binding to regulate changes in cell proliferation and differentiation [30]. However, in oncologic processes such as MPNSTs, these kinases are constitutively activated and support tumor propagation, including cell proliferation, survival, and therapeutic resistance [30].
A number of RTKs are closely linked to the tumorigenesis of MPNST, such as EGFR, platelet-derived growth factor receptor (PDGFR)-α and PDGFR-β, vascular endothelial growth factor receptor (VEGFR), c-Kit, and c-Met [29, 31–33]. Multiple studies have implicated EGFR as a key oncogenic target in MPNST that is overexpressed in more than two-thirds of patients and associated with high-grade, p53 positivity and invasiveness [21, 29, 34–38]. Despite a sound rationale and the fact that MPNST derived from patients and NF1 +/–/TP53 +/– mice can be stimulated by epidermal growth factor and inhibited by EGFR inhibitors [36–38], a phase II trial with EGFR inhibitor erlotinib, the first targeted agent used in a NF1-associated MPNST, largely failed [39]. Other inhibitors have targeted PDGFR, which also shows an increased expression of both proteins in MPNSTs and carries prognostic relevance [29, 40, 41]. In particular, the function of PDGFR-α is associated with mitogenic signaling through the Ras/MAPK pathway and promotes angiogenesis, tumor proliferation, and invasion [32, 42, 43]. Preclinical studies revealed that inhibition of PDGFR effectively suppressed MPNST cell invasion and cell growth [32, 44]. However, the monotherapeutic use of multikinase inhibitors with anti-PDGFR activity, such as imatinib mesylate, which blocks PDGFR, VEGFR, and c-Kit [45], the dual VEGFR/PDGFR inhibitor sorafenib [46], and dasatinib, which targets PDGFR and c-Kit in addition to BCR-ABL and the Src family [47] did not show a meaningful treatment response in phase II trials when tested in patients with refractory MPNSTs.
The failure of RTK pathway inhibitors following the strong molecular evidence for RTK pathway targeting has been disappointing, especially when considering the treatment successes in other malignancies. There are several potential explanations for these treatment failures, most important are perhaps the aggressive nature and fast progression of advanced, metastatic MPNSTs; the lack of sufficient Ras suppression and development of intrinsic resistance with both parallel signaling pathways and bypass feedback loops. Importantly, several oncogenic targets may also play a critical role for healthy nerve development and repair, including multiple growth factors and MAPKs such as Erk and c-Jun N-terminal kinase [48]. However, to date there has been little investigation about the interaction between normal nerve signaling and propagation of cancer in the peripheral nerve.
Recognizing those limitations, newer trials have focused on combination treatments to increase efficacy and delay the development of resistance. For example, a phase II trial with bevacizumab, a monoclonal antibody against the VEGF-α ligand, plus RAD001, a first-generation mTOR inhibitor (SARC016), in recurrent MPNST has recently completed and data are expected this year.
One of the most promising non-RTK inhibitors currently undergoing clinical investigations is heat shock protein (Hsp)90 inhibitor ganetespib. Hsp90 stabilizes certain proteins and reduces oxidative stress to favor cancer cell survival, and treatment with Hsp90 inhibitor ganetespib in combination with mTOR inhibitor sirolimus led to dramatic tumor shrinkage in an NF1 +/–/TP53 +/– murine model [49]. A combined phase I/II trial in refractory MPNSTs is currently ongoing (NCT02008877).
Preclinical Drug Development
Although we know that NF1 –/– Schwann cells are a key requirement for tumor formation and the ultimate development of MPNSTs, it is not clear that any specific neuronal or Schwann cell-specific elements are contributing to tumor maintenance or growth. Hence, efforts to develop novel therapeutics have been focused on oncogenic pathways. The identification of new compounds or targets in the treatment of MPNSTs has mainly been driven by large-scale compound screening libraries, targeting of nodes in oncogenic pathways and perhaps in the future may utilize synthetic lethality screenings, that is, the identification of genes for which the loss of function would be lethal in the presence of hyperactivated Ras and NF1 loss.
The use of MEK inhibitors provides a very strong rationale for the treatment of NF1-associated neoplasms based on the hyperactivation of Raf/MEK/ERK. Thus, it is not surprising that the highly selective pharmacological MEK inhibitor PD0325901 reduced tumor growth and prolonged survival in a NF1 mouse model [NF1(fl/fl);Dhh-Cre] and in NF1 patient-derived MPNST cell xenografts [50]. Although early clinical trials of MEK inhibitors in patients with NF1 with pNFs have resulted in very encouraging results [51], MEK inhibitors have not yet advanced into clinical trial investigations for MPNSTs. However, work is underway to bring combination therapies with MEK inhibitors to clinical investigation for NF1-associated MPNST.
Furthermore, current research supports a therapeutic role for c-Met and its ligand hepatocyte growth factor, which when overexpressed in MPNSTs induces cell motility, invasion, angiogenesis, and VEGF expression. Knockdown of Met and the use of specific c-Met inhibitor XL184 markedly decreased human MPNST xenograft growth in mice [31]. c-Met inhibitors have not been clinically investigated in MPNSTs yet; however, they are in active investigation for pNFs (NCT02101736).
Epigenetic aberrations play a crucial role in tumorigenesis of a variety of cancers, including MPNSTs. Recent evidence by De Raedt et al. [20] indicates that loss of PRC2 complex is a key oncogenic driver in NF1-associated MPNSTs that can be therapeutically targeted by BRD4 inhibitors. BRD4 is a BET (bromodomain and extraterminal) family member that contains 2 bromodomains, which regulate expression of mitotic genes required for cell-cycle progression. Preclinical animal experiments showed that single use of BRD4 inhibitors exerted modest cytostatic effects, while the combination with a MEK inhibitor resulted in significant tumor regressions, with tumor shrinkages up to 67% of mice [20]. This observation was confirmed by a previous study from Patel et al. [52], who also reported that BRD4 inhibition in MPNSTs profoundly suppressed both growth and tumorigenesis. Additional exciting preclinical results were seen with inhibition of Aurora kinase A using selective inhibitor MLN8237, which stabilized tumor volume and significantly increased survival of mice with MPNST xenografts [53].
It has been well established that the tumor environment plays a critical role in the treatment response and can elicit therapy resistance [54]. In NF1, there is growing evidence that the recruitment of macrophages and mast cells to the environment of pNFs and MPNSTs contributes to the disruption of the nerve axon–Schwann interactions and creates a supportive stroma to facilitate tumor proliferation and invasion via alteration of immune functions, angiogenesis, and the release of cytokines and growth factors [55–57]. There are probably a number of factors that mediate this chemoattraction; however, the exact factors and mechanisms remain largely unexplored. One of the best-described factors is c-Kit ligand/stem cell factor that is secreted by NF1 –/– Schwann cells and results in the migration of c-Kit-expressing mast cells to the tumor where they ultimately stimulate tumor proliferation [55]. In addition, CXCL12, which is highly expressed in NF1-deficient MPNSTs, and colony-stimulating factor-1, which is released by MPNSTs, are 2 other potent macrophage chemoattractants [55, 57, 58]. Correlating with these findings, the genetic and pharmacological disruption of c-Kit with multikinase inhibitors imatinib, pexidartinib, or PLX3397, the latter of which also blocks the colony-stimulating factor-1 receptor, prevented immune cell recruitment and delayed MPNST growth in preclinical NF1 animal models [55, 57, 59]. Unfortunately, these results could not be confirmed in a phase II clinical trial with imatinib [45], but more targeted approaches for c-Kit inhibition are in development and these may have a role in combination with additional agents moving forward, as done with pexidartinib and sirolimus (NCT02584647).
Future preclinical directions may also investigate the role of immunotherapeutics in MPNSTs, which has remained a largely unexplored field in the NF community. Immune checkpoint inhibitors, that is, blocking antibodies of surface receptors expressed on the host’s T cells and tumor, such as cytotoxic T lymphocyte-associated antigen 4, the programmed death-1 (PD-1) receptor, and the PD-L1 ligand have recently gained clinical significance because of their ability to induce a potent antitumor immune response and tumor remissions, particularly in tumors with prominent lymphocyte infiltration and mismatch repair deficiency [60]. Newly published data indicate that MPNSTs exhibit a “non-inflamed phenotype” with low PD-1 and PD-L1 expression levels [61]. However, although the expression of PD-L1 on the surface of tumor cells or immune cells is an important marker to predict response, it is not a definitive marker [60]. Thus, a combination approach using effector T-cell activation with other drugs could still be beneficial for patients with MPNST and does warrant further investigations.
Furthermore, direct intratumoral therapies delivered by implanted biomaterials, convection-enhanced delivery, or replication-competent biological agents such as oncolytic viruses and bacteria are also becoming more advanced and may be useful in a subset of patients, with localized, nonresectable, nonmetastatic MPNSTs. For example, an oncolytic measles virus encoding thyroidal sodium iodide symporter is currently being investigated in a phase I trial in MPNSTs (NCT02700230). Based on reports, measles viruses attach to the cancer cell membrane, kill cancer cells, and cause disease regression [62]. In addition, it is able to take up iodine and thus may be used to visualize infected cancer cells [63].
Aside from viruses, anaerobic bacteria have also been used to treat cancer and are currently in preclinical testing for MPNSTs, owing to their hypoxic nature. One particular promising strain is Clostridium novyi, which is a highly motile spore-forming anaerobe that is exquisitely sensitive to oxygen [64, 65]. For therapeutic purposes, this strain was rendered nonpathogenic by eliminating a residential phage carrying α-toxin and the resulting clone was named C. novyi-NT [64]. Once administered as a single dose, C. novyi-NT spores geminate locally within tumors and precisely spread throughout the tumor and its microsatellites, causing hemorrhagic necrosis, tumor cell lysis, and tumor destruction [66, 67], presumably by direct cell lysis and sensitization of the immune system, although the mechanisms are poorly understood. A variety of syngeneic and xenograft experimental tumors across multiple animal species have successfully been treated with C. novyi-NT spores, including syngeneic and xenograft orthotopic glioblastomas in rats [64–68]. Notably, C. novyi-NT spores have already been evaluated in companion dogs with spontaneous peripheral nerve sheath tumors, and clinical responses were seen in > 50% of the animals, with 28.5% of animals having complete responses [66]. A phase I human trial with C. novyi-NT spores is currently ongoing for patients with recurrent sarcoma (NCT01924689).
The Role of Chemoprevention in MPNST and NF1
As current MPNST-targeted therapies are largely ineffective, a multistep genetic transformation has been proposed to underlie MPNST evolution, and a high-risk population among the NF1 patients (e.g., patients who carry a microdeletion) can be easily identified, early chemoprevention has gained the attention of some research groups. The success of chemoprevention, that is, the use of drugs to reduce the risk of the development of cancer, has been impressively demonstrated in multiple epithelial malignancies, particularly breast, prostate, and colorectal cancers with the use of selective estrogen receptor modulators (e.g., tamoxifen and raloxifen), 5α-reductase inhibitors (e.g., finasteride), and cyclooxygenase-2 (COX-2) inhibitors, a type of nonsteroidal anti-inflammatory drug, which inhibited the appearance of colorectal polyps in various familial colorectal cancer predisposing syndromes [69].
A substantial body of work supports the evidence that chronic inflammation may contribute to a variety of cancers by inducing proneoplastic mutations, resistance to apoptosis, and environmental changes such as stimulation of angiogenesis [70]. COX-2 overexpression, which is triggered by inflammation, was detected in 2/3 of NF1-related MPNSTs and has been linked to carcinogenesis and angiogenesis [71], whereas the inhibition of COX-2 resulted in significant antitumor responses in various NF1 patient-derived MPNST cell lines [72, 73]. Likewise, manipulations to the tumor environment by inhibiting recruitment of immune cells via BRD4 inhibitors is an exciting strategy that is currently actively pursued and could potentially delay or prevent the malignant transformation of MPNSTs. This is particularly important in patients with NF1 microdeletions, where SUZ12 is known to be co-deleted with NF1. However, whether these strategies truly affect the clinical course of pNFs or MPNSTs remains to be determined in the future.
Conclusion
MPNST is a very rare primary cancer of the peripheral nervous system that occurs with increased incidence in the setting of NF1 and carries a poor prognosis. Although no treatment standard has been established yet, active work is underway to determine the optimal “standard” approach for these tumors, including defining the role of traditional cytotoxic chemotherapies, radiation, and radical surgical approaches, such as amputation, and with the consideration that both the cancer and the treatments directed against the cancer may cause irreversible nerve injury. Even with the most aggressive standard interventions, there is no guarantee of long-term survival, and these cancers will likely recur.
However, there is also reason for guarded optimism in the field. Remarkable advancements have been made in furthering our understanding of the molecular and pathophysiologic underpinnings of MPNSTs that identified novel oncogenic targets and inspired new therapeutic approaches in multiple directions. Indeed, there are more NF1-specific MPNST clinical therapeutics in development at the moment than there ever have been previously.
Many challenges to the successful development of promising therapeutic approaches remain. Firstly, these tumors arise from the same mesenchymal origin of normal nerve elements, as well as some of the most resistant cancer’s known. Secondly, there is a very clear role of the microenvironment in both benign and malignant nerve sheath tumors that may result in a constant flux of factors contributing to tumor heterogeneity and resistance and may highlight the need of personalized medicine. Finally, as these are rare tumors, more efficient clinical trial designs are critical. This paired with our rapidly emerging understanding of the tumor pathophysiology (including the discovery that allows consideration of therapeutics that prevent MPNSTs) and entirely novel therapeutic approaches raises hope that meaningful therapeutics for MPNST are forthcoming in the near future.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Required Author Forms Disclosure forms provided by the authors are available with the online version of this article. (PDF 1224 kb)
Acknowledgements
V.S. is supported by the Francis S Collins Scholars Program of the Neurofibromatosis Therapeutic Acceleration Program (NTAP) and the Children's Tumor Foundation (CTF). R.-Y.B. is supported by National Cancer Institute grant 1R03CA178118-01A1 and CTF. J.B. is supported by the Neurofibromatosis Therapeutic Acceleration Program (NTAP) and the Bloomberg and Gottesman Foundations.
References
- 1.Carey JC, Baty BJ, Johnson JP, Morrison T, Skolnick M, Kivlin J. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45–56. doi: 10.1111/j.1749-6632.1986.tb48061.x. [DOI] [PubMed] [Google Scholar]
- 2.NIH. National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13–15, 1987. Neurofibromatosis 1988; 1: 172-178. [PubMed]
- 3.Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57. [PubMed] [Google Scholar]
- 4.Abramowicz A, Gos M. Neurofibromin in neurofibromatosis type 1—mutations in NF1gene as a cause of disease. Dev Period Med. 2014;18:297–306. [PubMed] [Google Scholar]
- 5.Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15:290–301. doi: 10.1038/nrc3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabbagh A, Pasmant E, Laurendeau I, et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum Mol Genet. 2009;18:2768–2778. doi: 10.1093/hmg/ddp212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pasmant E, Sabbagh A, Spurlock G, et al. NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat. 2010;31:E1506–E1518. doi: 10.1002/humu.21271. [DOI] [PubMed] [Google Scholar]
- 8.De Raedt T, Brems H, Wolkenstein P, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003;72:1288–1292. doi: 10.1086/374821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huson SHR. The neurofibromatoses: a pathogenic and clinical overview. London: Chapman and Hall; 1994. [Google Scholar]
- 10.Mautner VF, Asuagbor FA, Dombi E, et al. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro Oncol. 2008;10:593–598. doi: 10.1215/15228517-2008-011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen R, Jett K, Harris GJ, Cai W, Friedman JM, Mautner VF. Benign whole body tumor volume is a risk factor for malignant peripheral nerve sheath tumors in neurofibromatosis type 1. J Neurooncol. 2014;116:307–313. doi: 10.1007/s11060-013-1293-1. [DOI] [PubMed] [Google Scholar]
- 12.Porter DE, Prasad V, Foster L, Dall GF, Birch R, Grimer RJ. Survival in malignant peripheral nerve sheath tumours: a comparison between sporadic and neurofibromatosis type 1-associated tumours. Sarcoma. 2009;2009:756395. doi: 10.1155/2009/756395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194:1568–1574. doi: 10.2214/AJR.09.2724. [DOI] [PubMed] [Google Scholar]
- 14.Ahlawat S, Blakeley J, Montgomery E, Subramaniam RM, Belzberg A, Fayad LM. Schwannoma in neurofibromatosis type 1: a pitfall for detecting malignancy by metabolic imaging. Skeletal Radiol. 2013;42:1317–1322. doi: 10.1007/s00256-013-1626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meany H, Dombi E, Reynolds J, et al. 18-fluorodeoxyglucose-positron emission tomography (FDG-PET) evaluation of nodular lesions in patients with neurofibromatosis type 1 and plexiform neurofibromas (PN) or malignant peripheral nerve sheath tumors (MPNST) Pediatr Blood Cancer. 2013;60:59–64. doi: 10.1002/pbc.24212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beert E, Brems H, Daniels B, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–1032. doi: 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- 17.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–314. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubin JB, Gutmann DH. Neurofibromatosis type 1—a model for nervous system tumour formation? Nat Rev Cancer. 2005;5:557–564. doi: 10.1038/nrc1653. [DOI] [PubMed] [Google Scholar]
- 19.Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–251. doi: 10.1038/nature13561. [DOI] [PubMed] [Google Scholar]
- 21.Carroll SL. The challenge of cancer genomics in rare nervous system neoplasms: malignant peripheral nerve sheath tumors as a paradigm for cross-species comparative oncogenomics. Am J Pathol. 2016;186:464–477. doi: 10.1016/j.ajpath.2015.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249:1014–1022. doi: 10.1097/SLA.0b013e3181a77e9a. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295–319. doi: 10.1007/s00401-012-0954-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stucky CC, Johnson KN, Gray RJ, et al. Malignant peripheral nerve sheath tumors (MPNST): the Mayo Clinic experience. Ann Surg Oncol. 2012;19:878–885. doi: 10.1245/s10434-011-1978-7. [DOI] [PubMed] [Google Scholar]
- 25.Kaushal A, Citrin D. The role of radiation therapy in the management of sarcomas. Surg Clin North Am. 2008;88:629–646. doi: 10.1016/j.suc.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kahn J, Gillespie A, Tsokos M, et al. Radiation therapy in management of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Front Oncol. 2014;4:324. doi: 10.3389/fonc.2014.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watson KL, Al Sannaa GA, Kivlin CM, et al. Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J Neurosurg. 2017;126:319–329. doi: 10.3171/2015.12.JNS152443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Widemann B, et al. SARC006: Phase II trial of chemotherapy in sporadic and neurofibromatosis type 1 (NF1)-associated high-grade malignant peripheral nerve sheath tumors (MPNSTs). J Clin Oncol 2013; 31.
- 29.Perrone F, Da Riva L, Orsenigo M, et al. PDGFRA, PDGFRB, EGFR, and downstream signaling activation in malignant peripheral nerve sheath tumor. Neuro Oncol. 2009;11:725–736. doi: 10.1215/15228517-2009-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 31.Torres KE, Zhu QS, Bill K, et al. Activated MET is a molecular prognosticator and potential therapeutic target for malignant peripheral nerve sheath tumors. Clin Cancer Res. 2011;17:3943–3955. doi: 10.1158/1078-0432.CCR-11-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holtkamp N, Okuducu AF, Mucha J, et al. Mutation and expression of PDGFRA and KIT in malignant peripheral nerve sheath tumors, and its implications for imatinib sensitivity. Carcinogenesis. 2006;27:664–671. doi: 10.1093/carcin/bgi273. [DOI] [PubMed] [Google Scholar]
- 33.Rahrmann EP, Watson AL, Keng VW, et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat Genet. 2013;45:756–766. doi: 10.1038/ng.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu J, Patmore DM, Jousma E, et al. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene. 2014;33:173–180. doi: 10.1038/onc.2012.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holtkamp N, Malzer E, Zietsch J, et al. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008;10:946–957. doi: 10.1215/15228517-2008-053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ling BC, Wu J, Miller SJ, et al. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell. 2005;7:65–75. doi: 10.1016/j.ccr.2004.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeClue JE, Heffelfinger S, Benvenuto G, et al. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J Clin Invest. 2000;105:1233–1241. doi: 10.1172/JCI7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tabone-Eglinger S, Bahleda R, Cote JF, et al. Frequent EGFR positivity and overexpression in high-grade areas of human MPNSTs. Sarcoma. 2008;2008:849156. doi: 10.1155/2008/849156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albritton K, et al. Phase II trial of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumor (MPNST). J Clin Oncol 2006; 24.
- 40.Holtkamp N, Mautner VF, Friedrich RE, et al. Differentially expressed genes in neurofibromatosis 1-associated neurofibromas and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2004;107:159–168. doi: 10.1007/s00401-003-0797-8. [DOI] [PubMed] [Google Scholar]
- 41.Kilvaer TK, Smeland E, Valkov A, et al. The VEGF- and PDGF-family of angiogenic markers have prognostic impact in soft tissue sarcomas arising in the extremities and trunk. BMC Clin Pathol. 2014;14:5. doi: 10.1186/1472-6890-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao R, Brakenhielm E, Li X, et al. Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-alphaalpha and -alphabeta receptors. FASEB J. 2002;16:1575–1583. doi: 10.1096/fj.02-0319com. [DOI] [PubMed] [Google Scholar]
- 43.Ki DH, He S, Rodig S, Look AT. Overexpression of PDGFRA cooperates with loss of NF1 and p53 to accelerate the molecular pathogenesis of malignant peripheral nerve sheath tumors. Oncogene 2016 Aug 1 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 44.Aoki M, Nabeshima K, Koga K, et al. Imatinib mesylate inhibits cell invasion of malignant peripheral nerve sheath tumor induced by platelet-derived growth factor-BB. Lab Invest. 2007;87:767–779. doi: 10.1038/labinvest.3700591. [DOI] [PubMed] [Google Scholar]
- 45.Chugh R, Wathen JK, Maki RG, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J Clin Oncol. 2009;27:3148–3153. doi: 10.1200/JCO.2008.20.5054. [DOI] [PubMed] [Google Scholar]
- 46.Maki RG, D'Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133–3140. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schuetze S, et al. Results of a Sarcoma Alliance for Research through Collaboration (SARC) phase II trial of Dasatinib in previously treated, highgrade, advanced sarcoma. J Clin Oncol 2010; 28.
- 48.Abe N, Cavalli V. Nerve injury signaling. Curr Opin Neurobiol. 2008;18:276–283. doi: 10.1016/j.conb.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Raedt T, Walton Z, Yecies JL, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–413. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jessen WJ, Miller SJ, Jousma E, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340–347. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher-Smith LE, Rizvi TA, Wu J, Ershler R, Wolters P, Therrien J, Glod J, Belasco JB, Schorry E, Brofferio A, Starosta AJ, Gillespie A, Doyle AL, Ratner N, Widemann BC. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med. 2016 Dec 29;375(26):2550-2560. [DOI] [PMC free article] [PubMed]
- 52.Patel AJ, Liao CP, Chen Z, Liu C, Wang Y, Le LQ. BET bromodomain inhibition triggers apoptosis of NF1-associated malignant peripheral nerve sheath tumors through Bim induction. Cell Rep. 2014;6:81–92. doi: 10.1016/j.celrep.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel AV, Eaves D, Jessen WJ, et al. Ras-driven transcriptome analysis identifies aurora kinase A as a potential malignant peripheral nerve sheath tumor therapeutic target. Clin Cancer Res. 2012;18:5020–5030. doi: 10.1158/1078-0432.CCR-12-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang FC, Ingram DA, Chen S, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/– and c-kit-dependent bone marrow. Cell. 2008;135:437–448. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang FC, Ingram DA, Chen S, et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/– mast cells. J Clin Invest. 2003;112:1851–1861. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prada CE, Jousma E, Rizvi TA, et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol. 2013;125:159–168. doi: 10.1007/s00401-012-1056-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mo W, Chen J, Patel A, et al. CXCR4/CXCL12 mediate autocrine cell- cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell. 2013;152:1077–1090. doi: 10.1016/j.cell.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patwardhan PP, Surriga O, Beckman MJ, et al. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res. 2014;20:3146–3158. doi: 10.1158/1078-0432.CCR-13-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shurell E, Singh AS, Crompton JG, et al. Characterizing the immune microenvironment of malignant peripheral nerve sheath tumor by PD-L1 expression and presence of CD8+ tumor infiltrating lymphocytes. Oncotarget. 2016;7:64300–64308. doi: 10.18632/oncotarget.11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Russell SJ, Federspiel MJ, Peng KW, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc. 2014;89:926–933. doi: 10.1016/j.mayocp.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grunwald GK, Vetter A, Klutz K, et al. Systemic image-guided liver cancer radiovirotherapy using dendrimer-coated adenovirus encoding the sodium iodide symporter as theranostic gene. J Nucl Med. 2013;54:1450–1457. doi: 10.2967/jnumed.112.115493. [DOI] [PubMed] [Google Scholar]
- 64.Dang LH, Bettegowda C, Huso DL, Kinzler KW, Vogelstein B. Combination bacteriolytic therapy for the treatment of experimental tumors. Proc Natl Acad Sci U S A. 2001;98:15155–15160. doi: 10.1073/pnas.251543698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diaz LA, Jr, Cheong I, Foss CA, et al. Pharmacologic and toxicologic evaluation of C. novyi-NT spores. Toxicol Sci. 2005;88:562–575. doi: 10.1093/toxsci/kfi316. [DOI] [PubMed] [Google Scholar]
- 66.Roberts NJ, Zhang L, Janku F, et al. Intratumoral injection of Clostridium novyi-NT spores induces antitumor responses. Sci Transl Med 2014; 6: 249ra111. [DOI] [PMC free article] [PubMed]
- 67.Staedtke V, Bai RY, Sun W, et al. Clostridium novyi-NT can cause regression of orthotopically implanted glioblastomas in rats. Oncotarget. 2015;6:5536–5546. doi: 10.18632/oncotarget.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agrawal N, Bettegowda C, Cheong I, et al. Bacteriolytic therapy can generate a potent immune response against experimental tumors. Proc Natl Acad Sci U S A. 2004;101:15172–15177. doi: 10.1073/pnas.0406242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.William WN, Jr, Heymach JV, Kim ES, Lippman SM. Molecular targets for cancer chemoprevention. Nat Rev Drug Discov. 2009;8:213–225. doi: 10.1038/nrd2663. [DOI] [PubMed] [Google Scholar]
- 70.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol. 2004;31:2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 72.Hakozaki M, Tajino T, Konno S, et al. Overexpression of cyclooxygenase-2 in malignant peripheral nerve sheath tumor and selective cyclooxygenase-2 inhibitor-induced apoptosis by activating caspases in human malignant peripheral nerve sheath tumor cells. PLOS ONE. 2014;9:e88035. doi: 10.1371/journal.pone.0088035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frahm S, Kurtz A, Kluwe L, Farassati F, Friedrich RE, Mautner VF. Sulindac derivatives inhibit cell growth and induce apoptosis in primary cells from malignant peripheral nerve sheath tumors of NF1-patients. Cancer Cell Int. 2004;4:4. doi: 10.1186/1475-2867-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Required Author Forms Disclosure forms provided by the authors are available with the online version of this article. (PDF 1224 kb)