

Abstract
Here, we review three sets of key proteins and their corresponding downstream pathways that have been linked to extending lifespan and promoting health span in a wide range of organisms. In particular, we review the biology of the sirtuin family of proteins, the insulin/insulin-like growth factor (IGF) signaling (IIS) pathway, and the mechanistic target of rapamycin (mTOR). Using insights derived from simple model organisms, mice, and humans we discuss how these proteins and pathways may potentially alter the rate of aging. We further describe how knowledge of these pathways may lead to the rational design of small molecules that modulate aging and hence alter the propensity for a host of age-related diseases.
Keywords: aging, insulin-like growth factor (IGF), mammalian target of rapamycin (mTOR), mouse genetics, sirtuin, lifespan, aging, IGF-1, sirtuins
Introduction
Understanding what the key molecular regulators of human lifespan are represents one of the most important and unresolved questions in biology. Age represents the major risk factor for a host of diseases including cancer, atherosclerosis, and neurodegeneration. As such, if understanding aging led to biological insights that allowed aging to become a modifiable disease risk factor, it raises the specter of simultaneously combating a host of debilitating conditions that now are largely untreatable. Although bioethicists might object to the unintended societal implications, agents that slow the aging process are increasingly viewed as feasible (1). Although such agents might extend lifespan, most view the delay of age-related morbidities as the ultimate goal. In that sense, there remains the hopeful notion that extending lifespan through targeted lifestyle changes or potentially therapeutic small molecules might result in an actual “compression of morbidity”(2). This concept posits that extending lifespan can actually function to reduce the overall time an individual suffers from chronic and debilitating morbidities. Analysis of non-randomized, longitudinal human studies seems to potentially support this hypothesis (3).
Here, we review the key set of proteins in three evolutionary conserved pathways that have been linked to regulation of lifespan. These proteins are the sirtuin family of NAD-dependent enzymes, the various components of the insulin/IGF2 pathway, and the mechanistic target of rapamycin (mTOR) kinase and its downstream effectors. We attempt to understand how these pathways might regulate lifespan and how they are interconnected with each other. Moreover, we describe the initial efforts to develop pharmacological approaches that mimic the well-described life-extending genetic perturbations in these pathways, and that therefore hold promise as potential anti-aging molecules.
Sirtuins as regulators of lifespan
The sirtuins are a family of NAD-dependent enzymes that catalyze post-translational modification of both histone and non-histone proteins (4). Although the first enzymatic activity of this family of proteins was the NAD-dependent deacetylation of target proteins, it is now clear that sirtuin family members can catalyze a growing list of more general deacylation reactions including demalonylation, desuccinylation, and deproprionylation (5). The initial studies of sirtuin biology centered on Sir2, a Saccharomyces cerevisiae family member that was initially implicated in transcriptional silencing. The link of these enzymes to aging became evident when it was noted that simple overexpression of Sir2 was sufficient to extend replicative lifespan in S. cerevisiae (6). Moreover, the life-extending benefit of caloric restriction in yeast was demonstrated to require Sir2 (7). Several potential mechanisms were implicated in the life extension observed by augmenting Sir2 activity including suppressing the formation of extrachromosomal rDNA circles (6), a known cause of yeast aging, as well as promoting asymmetric distribution of damaged proteins between mother and daughter cells (8). In higher organisms, other potential mechanisms for the life-extending effects of sirtuins include changes in mitochondrial function and biogenesis, suppression of inflammation, and regulation of genomic stability (9).
Overexpression of Sir2 orthologs in both Caenorhabditis elegans and Drosophila melanogaster can also extend lifespan, although the precise magnitude of this effect is the subject of debate (10). Extension of these observations to mammalian species has been attempted with mixed results. Part of the difficulty is that mammals have seven sirtuins (SIRT1–7) that exist in various compartments including the predominantly nuclear forms (SIRT1, SIRT6, and SIRT7), as well as the mitochondrial family members (SIRT3, SIRT4, and SIRT5) (11). The closest parallels to the effects of Sir2 overexpression in yeast are perhaps best recapitulated with SIRT6-overexpressing transgenic mice, where male, but not female mice, have an approximate 15% extension in median lifespan (12). Another example is seen in mice overexpressing SIRT1 specifically in the hypothalamus, which also results in a modest increase in median lifespan (e.g. 16% in females and 9% in males) (13). In contrast, other mouse sirtuin overexpression models do not demonstrate longer lifespans, although there are indications that these interventions can augment health span (11). For example, in mice, the beneficial effect of SIRT1 overexpression includes protection from heart failure and cardiovascular disease, reduction in certain forms of cancer, decreased propensity for developing metabolic syndrome, and protection from various neurodegenerative diseases (14). Similarly, SIRT6 overexpression also appears to improve health span (15). Complementing these gain-of-function models involving overexpression, detailed characterization of sirtuin knock-out models has also implicated this family of proteins in age-related pathologies. For instance, the absence of SIRT3 appears to predispose mice to early onset of a wide variety of pathologies associated with aging (16). Similarly, the connection between caloric restriction and sirtuin function first established in yeast was reinforced by the observation that in mice, the ability of caloric restriction to prevent age-related hearing loss required SIRT3 expression (17).
Genetic manipulation of sirtuin activity has been instrumental in identifying this family of proteins in various age-related processes, and as such, the development of sirtuin-activating compounds (STACs) represents a practical and logical extension of these observations (Fig. 1). The first sirtuin-activating small molecule identified was resveratrol, a polyphenol found in the skin of red grapes (18). In some experimental paradigms, resveratrol has been demonstrated to extend the lifespan of yeast, worms, and flies; however, these effects are not universally observed (19). In mammals, although resveratrol does not by itself increase lifespan, it does protect against certain age-related pathologies, particularly the metabolic deficits associated with normal aging or diet-induced obesity. For instance, cardiovascular protective effects appear to be associated with resveratrol administration in high-fat-fed non-human primates (20). Similarly, in obese human subjects, resveratrol appears to confer at least some measurable short-term benefits (21). Part of the difficulty in the interpretation of even these positive studies is the wide range of putative non-sirtuin targets that resveratrol may have. As such, although interesting, data generated with newer, presumably more selective sirtuin activators are potentially more revealing. In that regard, two STACs (SRT1720 and SRT2104) both appear to prolong lifespan and improve health span of mice fed a standard diet, although the effects on mean lifespan were modest (i.e. <10%) (22, 23). Administration of these agents also appears to protect against certain age-related conditions (24). Besides direct activation of sirtuins, considerable attention has been focused recently on manipulating levels of intracellular NAD+ as a means of modulating sirtuin activity. These approaches were spurred on by the observation that augmenting flux through the NAD+ salvage pathways in yeast could extend lifespan (25), and that administering the NAD+ precursor nicotinamide riboside to mice could replete NAD+ stores and thereby improve mitochondrial and stem cell function, as well as providing a modest 5% extension in lifespan (26). Similarly, NAD+ supplementation appears to extend the lifespan in certain mouse premature aging models (27). Interestingly, early clinical trials have already begun using nicotinamide riboside supplementation in humans (28).
Figure 1.

Environmental or pharmacological strategies to slow aging. Drugs such as rapamycin and STACs, and lifestyle interventions such as fasting or caloric restriction, alter the activity of the IIS, mTOR, and sirtuin pathways. This alteration induces a complex web of functional alterations that individually or collectively might slow the aging process.
The insulin/insulin-like growth factor signaling (IIS) pathway
The IIS pathway is a hormonally regulated cell-signaling pathway that includes insulin and insulin-like peptides, their cognate cell surface transmembrane receptors, substrates of these receptors, and downstream effectors. There is considerable genetic and biochemical evidence linking this pathway to aging across a wide spectrum of species. The first line of evidence came from the identification in C. elegans of loss-of-function mutations in age-1 and daf-2, gene products that function in the worm as the sole phosphatidylinositol-3-OH kinase (PI3K) and insulin-like receptor, respectively, and whose manipulation can result in a doubling of lifespan (29–32). Subsequent evidence demonstrated that these two gene products regulate lifespan within a single pathway (33). Further analysis revealed that the Forkhead box O (FOXO) transcription factor Daf-16 functions as an important transcriptional target of the IIS pathway in C. elegans (30, 34). The lifespans of other model organisms such as flies were also soon shown to be regulated by similar genetic alterations that modestly reduce IIS signaling (35).
Although there is considerable evidence that this pathway is relevant to mammalian aging, it is important to note that there are major differences between IIS signaling in model organisms and mammalian species. For instance, worms have nearly 40 different insulin-like peptides that could potentially modulate longevity (36). Moreover, although collectively insulin and insulin-like peptides regulate development, body size, and cellular growth, in higher organisms, insulin signaling is predominantly involved in nutrient regulation, whereas IGF-1 regulates growth. In contrast, these function are less distinctly separable in model organisms (37).
In mammals, levels of IGF-1, produced predominantly in the liver, are regulated by growth hormone (GH), a factor secreted by the pituitary. Consistent with the divergence of the somatotropic axis over evolution, there is no clear GH equivalent in yeast, flies, or worms. Mice that have a reduction in GH signaling due to a central defect in secretion or a genetic alteration in peripheral GH receptor function are in general long-lived (38). Examples include Snell mice (PIT-1 mutants; lifespan increase can approach 50% in female mice), Ames mice (PROP-1 mutants), lit/lit mice (GH-releasing hormone receptor mutants), Ghr−/− mice (GH receptor deletion), and GH receptor knock-out mice (39). Moreover, mouse genetic models that lead to too much GH secretion appear to have a reduced lifespan (40).
Mice with reduced GH secretion have, as expected, reduced circulating levels of IGF-1. That this reduction in circulating IGF-1 contributes to the observed longevity of GH mutants is supported by the observation that although Igf-1 null mice have severely reduced survival (41), conditional deletion of hepatic Igf-1, at 4 weeks of age, results in a 16% increase in median lifespan of female mice (42). A similar deletion in male mice, however, did not alter lifespan. This sexual dimorphic response, which is not well understood, is also seen in long-lived insulin receptor substrate 1 (IRS-1) knock-out mice (43). Similarly, in one study, loss of one allele of the Igf-1 receptor (IGF-1R) was shown to increase lifespan by 33% in females, with a non-significant trend toward increased lifespan in male mice (44). The precise magnitude of effects seen in these haploinsufficient IGF-1R+/− mice appears, however, to heavily depend on the strain of mice being studied (44). In addition to reducing the level of circulating IGF-1 or reducing the expression of IGF-1R, mouse models that reduce tissue levels of IGF-1 by genetic perturbations that alter the degradation of IGF-1-binding proteins can also markedly extend mean lifespan (45). The relevance of these observations to human aging is underscored by genetic evidence that certain loss-of-function mutations in IGF-1R are enriched in a well-characterized cohort of centenarians (46).
How does a reduction in insulin or IGF-1 signaling result in an increase in lifespan? In worms, as mentioned, this pathway involves both daf-2 and daf-16, two genes that got their names as known regulators of dauer, the stress-resistant, alternative developmental state worms can adopt when faced with harsh environmental stress. In C. elegans, the transcription factor DAF-16 coordinately regulates the expression of several hundred genes that are broadly involved in stress resistance, immune function, and metabolism (47, 48). In this context, long-lived worms that have alterations in the IIS pathway appear to have a broad resistance to environmental stresses including oxidative, osmotic, ultraviolet, heat, and hypoxic stress (49). These functions appear to be broadly conserved in higher species. For instance, although DAF-16 regulates antioxidant levels in worms (47, 48), a similar regulation is also seen by the mammalian FOXO homologs (50, 51).
In addition to an increase in overall stress resistance, long-lived IIS mutants appear to possess other attributes that might explain their increased longevity (Fig. 1). As mentioned, these mammalian models are often characterized by a reduction in circulating IGF-1. In humans, there is a strong correlation between increased circulating IGF-1 levels and the development of a wide range of malignancies (52). Fascinatingly, patients with Laron syndrome, an autosomal recessive disorder caused by mutation in the growth hormone receptor, have extremely low levels of circulating IGF-1, resulting in short stature but also an extremely low incidence of cancer (53). These patients also appear to be resistant to diabetes (53), and increased metabolic fitness might also contribute to the increase in lifespan seen with IIS pathway mutants. Similarly, GH mutant mice have altered mitochondrial function, changes in oxygen consumption, and an increased reliance on fatty acid oxidation that may be beneficial and contribute to the observed changes in lifespan (54). Similarly, alterations in the IIS pathway affect mTOR signaling, which, as discussed below, can have a major influence on lifespan. As such, it is difficult to know exactly what mechanism accounts for the increased lifespan in mammalian IIS mutants as these animals appear to have a number of alterations including increased stress resistance, decreased cancer incidence, altered metabolism, and reduced mTOR signaling that may be beneficial alone or in combination. One strategy to sort out these various possibilities would be to try to identify in mice the tissue(s) in which the reduction of IIS signaling is critical for mediating lifespan extension. Unfortunately, although various conditional GH receptor mutants have been made in a wide array of tissues (e.g. adipose tissue, liver, skeletal muscle, etc.), to date, none of these models have been able to recapitulate the life extension observed in the total body knockouts (54).
Mechanistic target of rapamycin
The mTOR kinase is a serine/threonine protein kinase belonging to the phosphoinositide 3-kinase-related family that is highly conserved among eukaryotes and that can be inhibited by the immunosuppressive drug rapamycin. In mammals, mTOR exists in two well-characterized complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). These complexes can be differentiated by the their unique interacting partners, with the mTORC1 complex containing Raptor and PRAS40, while mTORC2 complex contains Rictor, mSin1, and Protor-1/2 (55). Both complexes share certain components including mLST8/GβL and DEPTOR. mTORC1 has been known as a metabolic sensor for nutrients, growth factors, energy, and stress. These upstream signals act to tune the activity of mTORC1. Downstream effectors of mTORC1 are myriad (Fig. 2) and include regulation of ribosomal biogenesis, autophagy, protein translation, lipid synthesis, mitochondrial metabolism, pyrimidine synthesis, and most recently, modulation of the senescence-associated secretory phenotype (56, 57). Although not a direct regulator of transcription, mTORC1 does regulate a host of key transcription factors including sterol regulatory element-binding protein (SREBP) 1 (lipogenesis), peroxisome proliferator-activated receptor γ (PPARγ) (adipogenesis), transcription factor EB (TFEB) (autophagy), HIF-1α (metabolism), and NF-κB (inflammation) via regulation of interleukin 1α (55). In contrast, the less well-characterized mTORC2 complex, which is also less sensitive to the acute effects of rapamycin, has been linked to the modulation of metabolism, cytoskeleton dynamics, regulation of cell polarity, and control of cell survival (56). Generally speaking, mTOR acts as an energy sensor such that in response to abundant nutrients (e.g. amino acids) or growth factor stimulation, mTOR is activated and thereby enhances anabolic processes (e.g. translation, ribosomal biogenesis) and inhibits catabolic processes (e.g. autophagy) to promote cellular growth and cell proliferation (56, 59). Conversely, in the setting of limited nutrients (e.g. caloric restriction), mTOR is inhibited and the opposite set of events transpires.
Figure 2.
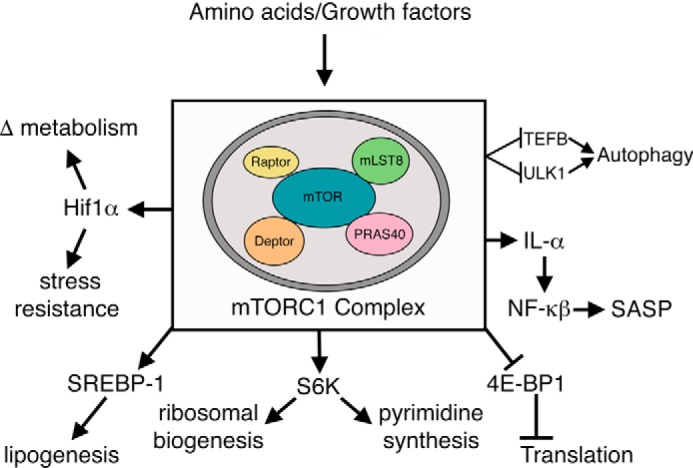
mTORC1 and its downstream effectors. The molecular components of the mTORC1 complex as well as some of the downstream effectors are shown. See text for further details.
The first indication that the mTOR pathway was an important regulator of lifespan came from analysis of simple organisms such as worms and flies where inhibition of this pathway was demonstrated to significantly increase longevity (60, 61). At least in yeast, it would appear that the beneficial effects of caloric restriction were not observed in organisms that were long-lived due to mTOR inhibition (62). This would imply that the biological effects of caloric restriction are mediated through this pathway, at least in this experimental paradigm. Interestingly, in flies, inhibiting the mTOR downstream effector S6K, involved in the regulation of protein translation, was sufficient to extend lifespan (61). Similar relationships between S6K and lifespan have also been seen in mice (63). Other mouse models include animals lacking one allele of both mTOR and mLST8 that have reduced mTORC1 activity, and where there is an approximate 15% increase in median survival of female mice (64). Similarly, in a mouse model of hypomorphic mTOR expression, due to targeting a neomycin cassette in an intron of the mTOR locus, mice live roughly 20% longer than control littermates and have slower age-dependent decline in some, but not all, tissues and organs (65).
Interest in the mTOR pathway was significantly bolstered by the observation that the mTOR inhibitor rapamycin could extend the lifespan of genetically heterogeneous mice by about 10% (66). This initial rapamycin study involved treating the animals beginning at 20 months of age, which represents a rather late intervention. Nonetheless, these results have been confirmed with early intervention schedules (67, 68). Moreover, the effects of rapamycin on lifespan extension are not strain-specific as positive effects have been observed in a number of different mouse strains (e.g. C57BL/6J, C57BL/6J R, 129/Sv, and FVB/N HER-2/neu mice).
Although rapamycin is believed to work primarily through inhibition of mTORC1 signaling, many of the deleterious side effects appear to be mediated through the ability of long-term rapamycin administration to also inhibit mTORC2 signaling. For instance, the harmful effects of chronic rapamycin therapy with regard to glucose tolerance appear to be mediated by mTORC2 signaling in the liver and are separable from the positive pro-longevity effects of the drug (64). This would suggest that agents that solely inhibit mTORC1 might maintain the observed increase in lifespan and potentially avoid some of the negative side effects (e.g. glucose intolerance, delayed wound healing, immunosuppression) that may be detrimental in an elderly cohort. In the absence of this selective agent, there have been attempts to shorten the duration or intensity of rapamycin treatment to lessen the chance of inhibiting mTORC2. In that regard, it is encouraging that transient or intermittent rapamycin appears to be effective (69, 70). Although data from healthy humans are limited, one study using the mTOR inhibitor RAD001 demonstrated that a 6-week exposure to this agent in patients over 65 actually improved responses to subsequent influenza vaccination, suggesting that the elderly immunosenescent phenotype might be at least partially amenable to short-term mTOR inhibition (71).
Similar to IIS signaling, the precise explanation as to how reduced mTOR signaling affects lifespan remains unclear. One hypothesis is that the benefit occurs through reduction of global mRNA translation and protein synthesis, which may reduce the burden and energetic demands associated with protein folding, repair, and degradation, thus maintaining better overall protein homeostasis (Fig. 1). These effects are believed to be largely mediated by two downstream effectors of mTOR, namely S6K and eukaryotic translation initiation factor 4E-binding protein (4E-BP). As mentioned above, deletion of S6K1 in mice results in increased lifespan by about 20% in female mice (63); however, these longevity effects appear to occur without any measurable effect on protein translation (72). Similarly, although chronic rapamycin treatment is capable of extending life in mice, this drug regimen also appears to have little effect on the level of tissue protein translation (73). There is, however, some evidence that 4E-BP might mediate the beneficial lifespan effects of mTOR inhibition. For instance, in flies, caloric restriction up-regulates the Drosophila equivalent of 4E-BP (d4E-BP), and augmenting d4E-BP is sufficient to extend lifespan in this organism (61). Moreover, in mice, increasing 4E-BP in skeletal muscle prevents certain deleterious age-related metabolic changes, although the opposite changes occur when 4E-BP is activated in adipocytes (74).
Activation of autophagy is another potential explanation of how mTORC1 inhibition promotes longevity. It is believed that because the general capacity for autophagic degradation declines with age, there is an accumulation of cellular damage such as protein aggregates and dysfunctional mitochondria. In C. elegans, the lifespan extensions observed by either caloric restriction or mTOR inhibition require an intact autophagic machinery (75). In that context, in mice, overexpressing the essential autophagy gene product ATG5 is sufficient to extend lifespan by 17% (76). Finally, there are several other properties of mTOR activity that link to the aging process. First, mTOR inhibition improves stem cell self-renewal both in the hematopoietic system as well as in the intestine (77). In addition, as mentioned, mTOR inhibition can suppress the secretion of inflammatory cytokines by senescent cells, the so-called senescence-associated secretory phenotype (SASP) that is believed to contribute to age-related pathologies (57, 78). Similar to the IIS pathway, the inhibition of the mTOR pathway is also linked to the ability of the cell or tissue to withstand a variety of stresses including energetic stress, oxidative stress, and hypoxia (79). Finally, mTOR has important effects on mitochondrial number (biogenesis and mitophagy) and function, although the precise role of mTOR with regard to mitochondrial activity appears to differ in different tissues (80).
Interaction between longevity pathways
Although evidence suggests that the sirtuins, mTOR, and the IIS pathway can individually modulate lifespan, it is important to understand that these three signaling networks are not autonomous or unconnected from each other. For instance, all three of these pathways respond to nutrient availability. Although in general, mTOR activity declines and sirtuin activity increases when tissues are depleted of nutrients, recent examples have suggested that this paradigm may have important exceptions (81). One major node of interconnection between these various pathways is through the energy-sensing, AMP-activated protein kinase (AMPK). For instance, under starved conditions, AMPK is activated, and this activation alters intracellular metabolism, culminating in an increase in NAD+ levels, with a concomitant increase in SIRT1 activity (82). Once activated, sirtuins can directly deacetylate and thereby regulate FOXO transcriptional activity (82, 83), thus linking SIRT1 to the important downstream effector of the IIS pathway. The interaction between AMPK and the sirtuins appears bidirectional as SIRT1 can also deacetylate liver kinase B1 (LKB1), an upstream activator of AMPK (84). In turn, AMPK can directly phosphorylate FOXO proteins both in model organisms such as worms, as well as in mammals (85). Finally, AMPK directly regulates mTOR activity, working both at the level of the upstream regulator TSC2 (86), as well as at the level of the mTORC1 component raptor (87). Thus, these examples suggest a network of complex interactions between sirtuins, mTOR, and IIS signaling (Fig. 3).
Figure 3.

Interaction between sirtuins, mTOR, and IIS signaling. Various points of interaction between these three signaling pathways have been described, some of which are depicted here. See text for details. P, phosphorylation site; Ac, acetylation site.
In addition to these biochemical interactions, these pathways can also interact on a more global, physiological level. One prime example is in the regulation of autophagy. For instance, insulin signaling inhibits autophagy through two mechanisms. First, by activating PI3K/AKT signaling, insulin stimulates mTOR, which in turn phosphorylates ATG1 (yeast) or ULK1/2 (mammals) to shut off autophagosome formation (88). AKT activation can also negatively regulate FOXO transcriptional activity, and in turn, FOXO transcriptional activity appears to regulate the expression of a host of autophagic genes (89). As mentioned above, sirtuins can also regulate FOXO activity through deacetylation, and hence by extension, this family of proteins can also regulate autophagic flux. This SIRT1/FOXO pathway has been shown to be critical for the starvation-induced cardiac autophagy response (90). In addition to regulating FOXO-dependent transcriptional pathways, SIRT1 can also deacetylate essential autophagy genes and hence directly modulate autophagic flux (91). Overall, this modulation of autophagy by IIS, mTOR, and sirtuins appears important for the longevity response. For instance, in the worm, autophagy is required for the lifespan extension resulting from reduced IIS signaling (92), inhibition of mTOR (75, 93), or sirtuin overexpression (94). Similar relationships are seen in other model organisms (95). Thus, once again, it would seem that the three longevity pathways discussed (e.g. sirtuins, IIS, and mTOR) converge to regulate autophagic flux through multiple interdependent effectors. It should, however, be noted that in C. elegans, double mutants in both the IIS and the mTOR pathway appear to live 5-fold longer than controls, which is a larger life extension than would be anticipated from the additive effect of combining each single mutant (96). This synergy appears to be a result of a positive feedback loop involving AMPK and FOXO proteins and suggests that the connectivity of these pathways is considerably more dynamic and complex than can be conveyed in simple linear flow diagrams. Finally, it is important to note that the pathways discussed are not the only regulators of lifespan. For instance, substantial evidence in both model organisms and mammals suggests a role for mitochondria in aging (97). Although these mitochondrial pathways may intersect with the elements discussed above (e.g. NAD levels, AMPK, etc.), they can also exert influence on aging through alternative means including activation of distinct signaling pathways such as the mitochondrial unfolded protein response (97).
Conclusion and future directions
The identification of evolutionary conserved pathways that appear to regulate lifespan suggests that aging may, in the near future, become amenable to pharmacological intervention (Table 1). Indeed, for over a decade, the NIA, National Institutes of Health has funded an Interventions Testing Program (ITP) in both mice and C. elegans to help identify potential life-extending small molecules (98). For the case of mTOR inhibition, there are already some small-scale human trials that have obtained hints of efficacy (71). Similarly, a large human trial that seeks to activate AMPK using the commonly prescribed medication metformin is being contemplated (99). It is likely that if AMPK provides any benefit, it does so by modulating one or more of the pathways we have described.
Table 1.
A summary of some of the small molecules or approved and/or investigational drugs that may be beneficial to slow the aging process
| Molecular target | Drug/Small molecule | Mode of action |
|---|---|---|
| Sirtuins | Resveratrol | ? Direct/indirect STAC |
| SRT1720/2104 | Direct sirtuin activator | |
| Nicotinamide riboside | NAD precursor | |
| Apigenin | Inhibits CD38, enzyme that degrades NAD | |
| mTOR | Rapamycin/Sirolimus/ Everolimus | Rapamycin and rapalogs/mTORC1 inhibitors |
| AZD8055/INK128 | Direct mTOR kinase inhibitors | |
| IIS/GH | Pegvisomant | GH receptor antagonist |
| Trametinib | Ras inhibitor resulting in decreased IIS signaling | |
| AMPK and others | Metformin | Potential AMPK activator |
| Ruxolitinib | JAK inhibitor/↓ inflammation | |
| Spermidine | Natural product ↑ autophagy | |
| Quescetin/Dasatinib | Senolytics |
Remarkably, the average human lifespan has nearly doubled over the last century (58). This change is largely ascribed to improvements in infant mortality, vaccinations, and other public health measures. The benefits of such interventions are generally believed to have been largely maximized in the developed world. The challenge of the next century is to test whether direct intervention on the aging process can result in a similar beneficial effect on both lifespan and health span.
Acknowledgment
We are grateful to Ilsa Rovira for help with this manuscript.
This work was supported by National Institutes of Health Intramural Funds and a grant from the Leducq Transatlantic Network Consortium (to T. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IGF
- insulin-like growth factor
- mTOR
- mechanistic target of rapamycin
- IIS
- insulin/insulin-like growth factor signaling
- STAC
- sirtuin-activating compound
- GH
- growth hormone
- S6K
- S6 kinase
- 4E-BP
- 4E-binding protein
- AMPK
- AMP-activated protein kinase.
References
- 1. Newman J. C., Milman S., Hashmi S. K., Austad S. N., Kirkland J. L., Halter J. B., and Barzilai N. (2016) Strategies and challenges in clinical trials targeting human aging. J. Gerontol. A Biol. Sci. Med. Sci. 71, 1424–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fries J. F. (1980) Aging, natural death, and the compression of morbidity. N. Engl. J. Med. 303, 130–135 [DOI] [PubMed] [Google Scholar]
- 3. Fries J. F., Bruce B., and Chakravarty E. (2011) Compression of morbidity 1980–2011: a focused review of paradigms and progress. J. Aging Res. 2011, 261702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haigis M. C., and Guarente L. P. (2006) Mammalian sirtuins: emerging roles in physiology, aging, and calorie restriction. Genes Dev. 20, 2913–2921 [DOI] [PubMed] [Google Scholar]
- 5. Bonkowski M. S., and Sinclair D. A. (2016) Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 17, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaeberlein M., McVey M., and Guarente L. (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin S. J., Defossez P. A., and Guarente L. (2000) Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128 [DOI] [PubMed] [Google Scholar]
- 8. Aguilaniu H., Gustafsson L., Rigoulet M., and Nyström T. (2003) Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753 [DOI] [PubMed] [Google Scholar]
- 9. Wątroba M., and Szukiewicz D. (2016) The role of sirtuins in aging and age-related diseases. Adv. Med. Sci. 61, 52–62 [DOI] [PubMed] [Google Scholar]
- 10. Dang W. (2014) The controversial world of sirtuins. Drug. Discov. Today Technol. 12, e9-e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Giblin W., Skinner M. E., and Lombard D. B. (2014) Sirtuins: guardians of mammalian healthspan. Trends Genet. 30, 271–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanfi Y., Naiman S., Amir G., Peshti V., Zinman G., Nahum L., Bar-Joseph Z., and Cohen H. Y. (2012) The sirtuin SIRT6 regulates lifespan in male mice. Nature 483, 218–221 [DOI] [PubMed] [Google Scholar]
- 13. Satoh A., Brace C. S., Rensing N., Cliften P., Wozniak D. F., Herzog E. D., Yamada K. A., and Imai S. (2013) Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 18, 416–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herskovits A. Z., and Guarente L. (2014) SIRT1 in neurodevelopment and brain senescence. Neuron 81, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roichman A., Kanfi Y., Glazz R., Naiman S., Amit U., Landa N., Tinman S., Stein I., Pikarsky E., Leor J., and Cohen H. Y. (2016) SIRT6 overexpression improves various aspects of mouse healthspan. J. Gerontol. A Biol. Sci. Med. Sci. 10.1093/gerona/glw152 [DOI] [PubMed] [Google Scholar]
- 16. McDonnell E., Peterson B. S., Bomze H. M., and Hirschey M. D. (2015) SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. Metab. 26, 486–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Someya S., Yu W., Hallows W. C., Xu J., Vann J. M., Leeuwenburgh C., Tanokura M., Denu J. M., and Prolla T. A. (2010) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Howitz K. T., Bitterman K. J., Cohen H. Y., Lamming D. W., Lavu S., Wood J. G., Zipkin R. E., Chung P., Kisielewski A., Zhang L. L., Scherer B., and Sinclair D. A. (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 [DOI] [PubMed] [Google Scholar]
- 19. Gambini J., Inglés M., Olaso G., Lopez-Grueso R., Bonet-Costa V., Gimeno-Mallench L., Mas-Bargues C., Abdelaziz K. M., Gomez-Cabrera M. C., Vina J., and Borras C. (2015) Properties of resveratrol: in vitro and in vivo studies about metabolism, bioavailability, and biological effects in animal models and humans. Oxid. Med. Cell. Longev. 2015, 837042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mattison J. A., Wang M., Bernier M., Zhang J., Park S. S., Maudsley S., An S. S., Santhanam L., Martin B., Faulkner S., Morrell C., Baur J. A., Peshkin L., Sosnowska D., Csiszar A., et al. (2014) Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab. 20, 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Timmers S., Konings E., Bilet L., Houtkooper R. H., van de Weijer T., Goossens G. H., Hoeks J., van der Krieken S., Ryu D., Kersten S., Moonen-Kornips E., Hesselink M. K., Kunz I., Schrauwen-Hinderling V. B., Blaak E. E., et al. (2011) Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 14, 612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mitchell S. J., Martin-Montalvo A., Mercken E. M., Palacios H. H., Ward T. M., Abulwerdi G., Minor R. K., Vlasuk G. P., Ellis J. L., Sinclair D. A., Dawson J., Allison D. B., Zhang Y., Becker K. G., Bernier M., and de Cabo R. (2014) The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 6, 836–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mercken E. M., Mitchell S. J., Martin-Montalvo A., Minor R. K., Almeida M., Gomes A. P., Scheibye-Knudsen M., Palacios H. H., Licata J. J., Zhang Y., Becker K. G., Khraiwesh H., González-Reyes J. A., Villalba J. M., Baur J. A., et al. (2014) SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 13, 787–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Milne J. C., Lambert P. D., Schenk S., Carney D. P., Smith J. J., Gagne D. J., Jin L., Boss O., Perni R. B., Vu C. B., Bemis J. E., Xie R., Disch J. S., Ng P. Y., Nunes J. J., et al. (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., and Sinclair D. A. (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang H., Ryu D., Wu Y., Gariani K., Wang X., Luan P., D'Amico D., Ropelle E. R., Lutolf M. P., Aebersold R., Schoonjans K., Menzies K. J., and Auwerx J. (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443 [DOI] [PubMed] [Google Scholar]
- 27. Scheibye-Knudsen M., Mitchell S. J., Fang E. F., Iyama T., Ward T., Wang J., Dunn C. A., Singh N., Veith S., Hasan-Olive M. M., Mangerich A., Wilson M. A., Mattson M. P., Bergersen L. H., Cogger V. C., et al. (2014) A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in Cockayne syndrome. Cell Metab. 20, 840–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trammell S. A., Schmidt M. S., Weidemann B. J., Redpath P., Jaksch F., Dellinger R. W., Li Z., Abel E. D., Migaud M. E., and Brenner C. (2016) Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 7, 12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Friedman D. B., and Johnson T. E. (1988) A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 118, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kenyon C., Chang J., Gensch E., Rudner A., and Tabtiang R. (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464 [DOI] [PubMed] [Google Scholar]
- 31. Morris J. Z., Tissenbaum H. A., and Ruvkun G. (1996) A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382, 536–539 [DOI] [PubMed] [Google Scholar]
- 32. Kimura K. D., Tissenbaum H. A., Liu Y., and Ruvkun G. (1997) daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–946 [DOI] [PubMed] [Google Scholar]
- 33. Dorman J. B., Albinder B., Shroyer T., and Kenyon C. (1995) The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics 141, 1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gottlieb S., and Ruvkun G. (1994) daf-2, daf-16 and daf-23: genetically interacting genes controlling Dauer formation in Caenorhabditis elegans. Genetics 137, 107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kenyon C. (2011) The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philos. Trans. R Soc. Lond. B Biol. Sci. 366, 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pierce S. B., Costa M., Wisotzkey R., Devadhar S., Homburger S. A., Buchman A. R., Ferguson K. C., Heller J., Platt D. M., Pasquinelli A. A., Liu L. X., Doberstein S. K., and Ruvkun G. (2001) Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 15, 672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bitto A., Wang A. M., Bennett C. F., and Kaeberlein M. (2015) Biochemical genetic pathways that modulate aging in multiple species. Cold Spring Harb. Perspect. Med. 5, a025114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bartke A., Sun L. Y., and Longo V. (2013) Somatotropic signaling: trade-offs between growth, reproductive development, and longevity. Physiol. Rev. 93, 571–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown-Borg H. M., Borg K. E., Meliska C. J., and Bartke A. (1996) Dwarf mice and the ageing process. Nature 384, 33. [DOI] [PubMed] [Google Scholar]
- 40. Bartke A. (2003) Can growth hormone (GH) accelerate aging? Evidence from GH-transgenic mice. Neuroendocrinology 78, 210–216 [DOI] [PubMed] [Google Scholar]
- 41. Liu J. P., Baker J., Perkins A. S., Robertson E. J., and Efstratiadis A. (1993) Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59–72 [PubMed] [Google Scholar]
- 42. Svensson J., Sjögren K., Fäldt J., Andersson N., Isaksson O., Jansson J. O., and Ohlsson C. (2011) Liver-derived IGF-I regulates mean life span in mice. PLoS ONE 6, e22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Selman C., Lingard S., Choudhury A. I., Batterham R. L., Claret M., Clements M., Ramadani F., Okkenhaug K., Schuster E., Blanc E., Piper M. D., Al-Qassab H., Speakman J. R., Carmignac D., et al. (2008) Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 22, 807–818 [DOI] [PubMed] [Google Scholar]
- 44. Holzenberger M., Dupont J., Ducos B., Leneuve P., Géloën A., Even P. C., Cervera P., and Le Bouc Y. (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421, 182–187 [DOI] [PubMed] [Google Scholar]
- 45. Conover C. A. (2013) Role of PAPP-A in aging and age-related disease. Exp. Gerontol. 48, 612–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suh Y., Atzmon G., Cho M. O., Hwang D., Liu B., Leahy D. J., Barzilai N., and Cohen P. (2008) Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. U.S.A. 105, 3438–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee S. S., Kennedy S., Tolonen A. C., and Ruvkun G. (2003) DAF-16 target genes that control C. elegans life-span and metabolism. Science 300, 644–647 [DOI] [PubMed] [Google Scholar]
- 48. Murphy C. T., McCarroll S. A., Bargmann C. I., Fraser A., Kamath R. S., Ahringer J., Li H., and Kenyon C. (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283 [DOI] [PubMed] [Google Scholar]
- 49. Altintas O., Park S., and Lee S. J. (2016) The role of insulin/IGF-1 signaling in the longevity of model invertebrates, C. elegans and D. melanogaster. BMB Rep. 49, 81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nemoto S., and Finkel T. (2002) Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 295, 2450–2452 [DOI] [PubMed] [Google Scholar]
- 51. Kops G. J., Dansen T. B., Polderman P. E., Saarloos I., Wirtz K. W., Coffer P. J., Huang T. T., Bos J. L., Medema R. H., and Burgering B. M. (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321 [DOI] [PubMed] [Google Scholar]
- 52. Milman S., Huffman D. M., and Barzilai N. (2016) The somatotropic axis in human aging: framework for the current state of knowledge and future research. Cell Metab. 23, 980–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guevara-Aguirre J., Balasubramanian P., Guevara-Aguirre M., Wei M., Madia F., Cheng C. W., Hwang D., Martin-Montalvo A., Saavedra J., Ingles S., de Cabo R., Cohen P., and Longo V. D. (2011) Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 3, 70ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bartke A., List E. O., and Kopchick J. J. (2016) The somatotropic axis and aging: benefits of endocrine defects. Growth Horm. IGF Res. 27, 41–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kennedy B. K., and Lamming D. W. (2016) The Mechanistic target of rapamycin: the grand ConducTOR of metabolism and aging. Cell Metab. 23, 990–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shimobayashi M., and Hall M. N. (2014) Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 15, 155–162 [DOI] [PubMed] [Google Scholar]
- 57. Laberge R. M., Sun Y., Orjalo A. V., Patil C. K., Freund A., Zhou L., Curran S. C., Davalos A. R., Wilson-Edell K. A., Liu S., Limbad C., Demaria M., Li P., Hubbard G. B., Ikeno Y., et al. (2015) MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 17, 1049–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dong X., Milholland B., and Vijg J. (2016) Evidence for a limit to human lifespan. Nature 538, 257–259 [DOI] [PubMed] [Google Scholar]
- 59. Laplante M., and Sabatini D. M. (2012) mTOR signaling. Cold Spring Harb. Perspect. Biol. 4, a011593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vellai T., Takacs-Vellai K., Zhang Y., Kovacs A. L., Orosz L., and Müller F. (2003) Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426, 620. [DOI] [PubMed] [Google Scholar]
- 61. Kapahi P., Zid B. M., Harper T., Koslover D., Sapin V., and Benzer S. (2004) Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 14, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kaeberlein M., Powers R. W. 3rd, Steffen K. K., Westman E. A., Hu D., Dang N., Kerr E. O., Kirkland K. T., Fields S., and Kennedy B. K. (2005) Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196 [DOI] [PubMed] [Google Scholar]
- 63. Selman C., Tullet J. M., Wieser D., Irvine E., Lingard S. J., Choudhury A. I., Claret M., Al-Qassab H., Carmignac D., Ramadani F., Woods A., Robinson I. C., Schuster E., Batterham R. L., Kozma S. C., et al. (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lamming D. W., Ye L., Katajisto P., Goncalves M. D., Saitoh M., Stevens D. M., Davis J. G., Salmon A. B., Richardson A., Ahima R. S., Guertin D. A., Sabatini D. M., and Baur J. A. (2012) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu J. J., Liu J., Chen E. B., Wang J. J., Cao L., Narayan N., Fergusson M. M., Rovira I. I., Allen M., Springer D. A., Lago C. U., Zhang S., DuBois W., Ward T., deCabo R., et al. (2013) Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 4, 913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Harrison D. E., Strong R., Sharp Z. D., Nelson J. F., Astle C. M., Flurkey K., Nadon N. L., Wilkinson J. E., Frenkel K., Carter C. S., Pahor M., Javors M. A., Fernandez E., and Miller R. A. (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miller R. A., Harrison D. E., Astle C. M., Baur J. A., Boyd A. R., de Cabo R., Fernandez E., Flurkey K., Javors M. A., Nelson J. F., Orihuela C. J., Pletcher S., Sharp Z. D., Sinclair D., Starnes J. W., et al. (2011) Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol A. Biol. Sci. Med. Sci. 66, 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Miller R. A., Harrison D. E., Astle C. M., Fernandez E., Flurkey K., Han M., Javors M. A., Li X., Nadon N. L., Nelson J. F., Pletcher S., Salmon A. B., Sharp Z. D., Van Roekel S., Winkleman L., and Strong R. (2014) Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 13, 468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bitto A., Ito T. K., Pineda V. V., LeTexier N. J., Huang H. Z., Sutlief E., Tung H., Vizzini N., Chen B., Smith K., Meza D., Yajima M., Beyer R. P., Kerr K. F., Davis D. J., et al. (2016) Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 5, e16351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Arriola Apelo S. I., Pumper C. P., Baar E. L., Cummings N. E., and Lamming D. W. (2016) Intermittent administration of rapamycin extends the life span of female C57BL/6J mice. J. Gerontol. A Biol. Sci. Med. Sci. 71, 876–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mannick J. B., Del Giudice G., Lattanzi M., Valiante N. M., Praestgaard J., Huang B., Lonetto M. A., Maecker H. T., Kovarik J., Carson S., Glass D. J., and Klickstein L. B. (2014) mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 6, 268ra179. [DOI] [PubMed] [Google Scholar]
- 72. Mieulet V., Roceri M., Espeillac C., Sotiropoulos A., Ohanna M., Oorschot V., Klumperman J., Sandri M., and Pende M. (2007) S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover. Am. J. Physiol. Cell Physiol. 293, C712–C722 [DOI] [PubMed] [Google Scholar]
- 73. Garelick M. G., Mackay V. L., Yanagida A., Academia E. C., Schreiber K. H., Ladiges W. C., and Kennedy B. K. (2013) Chronic rapamycin treatment or lack of S6K1 does not reduce ribosome activity in vivo. Cell Cycle 12, 2493–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tsai S., Sitzmann J. M., Dastidar S. G., Rodriguez A. A., Vu S. L., McDonald C. E., Academia E. C., O'Leary M. N., Ashe T. D., La Spada A. R., and Kennedy B. K. (2015) Muscle-specific 4E-BP1 signaling activation improves metabolic parameters during aging and obesity. J. Clin. Invest. 125, 2952–2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hansen M., Chandra A., Mitic L. L., Onken B., Driscoll M., and Kenyon C. (2008) A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 4, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pyo J. O., Yoo S. M., Ahn H. H., Nah J., Hong S. H., Kam T. I., Jung S., and Jung Y. K. (2013) Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun 4, 2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yilmaz Ö. H., Katajisto P., Lamming D. W., Gültekin Y., Bauer-Rowe K. E., Sengupta S., Birsoy K., Dursun A., Yilmaz V. O., Selig M., Nielsen G. P., Mino-Kenudson M., Zukerberg L. R., Bhan A. K., Deshpande V., and Sabatini D. M. (2012) mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 486, 490–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Herranz N., Gallage S., Mellone M., Wuestefeld T., Klotz S., Hanley C. J., Raguz S., Acosta J. C., Innes A. J., Banito A., Georgilis A., Montoya A., Wolter K., Dharmalingam G., Faull P., et al. (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 17, 1205–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Reiling J. H., and Sabatini D. M. (2006) Stress and mTORture signaling. Oncogene 25, 6373–6383 [DOI] [PubMed] [Google Scholar]
- 80. Albert V., and Hall M. N. (2015) mTOR signaling in cellular and organismal energetics. Curr. Opin. Cell Biol. 33, 55–66 [DOI] [PubMed] [Google Scholar]
- 81. Igarashi M., and Guarente L. (2016) mTORC1 and SIRT1 cooperate to foster expansion of gut adult stem cells during calorie restriction. Cell 166, 436–450 [DOI] [PubMed] [Google Scholar]
- 82. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., and Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Brunet A., Sweeney L. B., Sturgill J. F., Chua K. F., Greer P. L., Lin Y., Tran H., Ross S. E., Mostoslavsky R., Cohen H. Y., Hu L. S., Cheng H. L., Jedrychowski M. P., Gygi S. P., Sinclair D. A., et al. (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 84. Lan F., Cacicedo J. M., Ruderman N., and Ido Y. (2008) SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1: possible role in AMP-activated protein kinase activation. J. Biol. Chem. 283, 27628–27635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Greer E. L., Oskoui P. R., Banko M. R., Maniar J. M., Gygi M. P., Gygi S. P., and Brunet A. (2007) The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 282, 30107–30119 [DOI] [PubMed] [Google Scholar]
- 86. Inoki K., Ouyang H., Zhu T., Lindvall C., Wang Y., Zhang X., Yang Q., Bennett C., Harada Y., Stankunas K., Wang C. Y., He X., MacDougald O. A., You M., Williams B. O., and Guan K. L. (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126, 955–968 [DOI] [PubMed] [Google Scholar]
- 87. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., and Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kim J., Kundu M., Viollet B., and Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S. J., Di Lisi R., Sandri C., Zhao J., Goldberg A. L., Schiaffino S., and Sandri M. (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471 [DOI] [PubMed] [Google Scholar]
- 90. Hariharan N., Maejima Y., Nakae J., Paik J., Depinho R. A., and Sadoshima J. (2010) Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 107, 1470–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee I. H., Cao L., Mostoslavsky R., Lombard D. B., Liu J., Bruns N. E., Tsokos M., Alt F. W., and Finkel T. (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U.S.A. 105, 3374–3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Meléndez A., Tallóczy Z., Seaman M., Eskelinen E. L., Hall D. H., and Levine B. (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391 [DOI] [PubMed] [Google Scholar]
- 93. Tóth M. L., Sigmond T., Borsos E., Barna J., Erdélyi P., Takács-Vellai K., Orosz L., Kovács A. L., Csikós G., Sass M., and Vellai T. (2008) Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4, 330–338 [DOI] [PubMed] [Google Scholar]
- 94. Morselli E., Maiuri M. C., Markaki M., Megalou E., Pasparaki A., Palikaras K., Criollo A., Galluzzi L., Malik S. A., Vitale I., Michaud M., Madeo F., Tavernarakis N., and Kroemer G. (2010) Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gelino S., and Hansen M. (2012) Autophagy: an emerging anti-aging mechanism. J. Clin. Exp. Pathol. Suppl. 4, 006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chen D., Li P. W., Goldstein B. A., Cai W., Thomas E. L., Chen F., Hubbard A. E., Melov S., and Kapahi P. (2013) Germline signaling mediates the synergistically prolonged longevity produced by double mutations in daf-2 and rsks-1 in C. elegans. Cell Rep. 5, 1600–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sun N., Youle R. J., and Finkel T. (2016) The mitochondrial basis of aging. Molecular cell 61, 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Miller R. A., Harrison D. E., Astle C. M., Floyd R. A., Flurkey K., Hensley K. L., Javors M. A., Leeuwenburgh C., Nelson J. F., Ongini E., Nadon N. L., Warner H. R., and Strong R. (2007) An Aging Interventions Testing Program: study design and interim report. Aging Cell 6, 565–575 [DOI] [PubMed] [Google Scholar]
- 99. Barzilai N., Crandall J. P., Kritchevsky S. B., and Espeland M. A. (2016) Metformin as a tool to target aging. Cell Metab. 23, 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]