

Abstract
The 53BP1-dependent end-joining pathway plays a critical role in double strand break repair and is uniquely responsible for cellular sensitivity to poly(ADP-ribose) polymerase inhibitors (PARPi) in BRCA1-deficient cancers. We and others have investigated the downstream effectors of 53BP1, including replication timing regulatory factor 1 (RIF1) and Pax transactivation domain-interacting protein (PTIP), in the past few years to elucidate how loss of the 53BP1-dependent repair pathway results in PARPi resistance in BRCA1 patients. However, questions regarding the upstream regulation of the 53BP1 pathway remain unanswered. In this study, we identified the Tudor-interacting repair regulator (TIRR) that specifically associates with the ionizing radiation-induced foci formation region of 53BP1. 53BP1 and TIRR form a stable complex, which is required for their expression. Moreover, the 53BP1-TIRR complex dissociates after DNA damage, and this dissociation may be ataxia telangiectasia mutated-dependent. Similar to 53BP1, loss of TIRR restores PARPi resistance in BRCA1-deficient cells. Collectively, our data identified a novel 53BP1-TIRR complex in DNA damage response. TIRR may play both positive and negative roles in 53BP1 regulation. On the one hand, it stabilizes 53BP1 and thus positively regulates 53BP1. On the other hand, its association with 53BP1 prevents 53BP1 localization to sites of DNA damage, and thus TIRR is also an inhibitor of 53BP1.
Keywords: BRCA1, chemoresistance, DNA damage response, homologous recombination, protein-protein interaction, 53BP1, PARP inhibition, TIRR
Introduction
DNA double strand breaks (DSBs)4 are considered the most harmful type of DNA lesions, as one unrepaired DSB is sufficient to trigger permanent growth arrest and cell death. Two mechanistically distinct pathways have evolved to eliminate DSBs from the genome as follows: non-homologous DNA end-joining (NHEJ), which rejoins the broken ends of a severed DNA molecule (1); and homologous recombination (HR), which requires large stretches of identical or very similar DNA sequences elsewhere in the genome to serve as templates for DNA repair (2). Understanding DNA repair pathway choice will undoubtedly help us to design better therapy for cancer patients. Poly(ADP-ribose) polymerase inhibitors (PARPi) are being used in patients with BRCA1 or BRCA2 mutations because they show “synthetic lethality” with BRCA1 or BRCA2 deficiency (3, 4). Unfortunately, resistance to therapy is a major clinical problem. The results of two recent studies suggest that the loss of 53BP1, in the background of BRCA1 deficiency, can render these BRCA1-deficient cells resistant to PARPi (5, 6).
53BP1 participates in early DNA damage response. 53BP1 contains a highly phosphorylated N-terminal region, an ionizing radiation-induced foci (IRIF) region in the middle, and two BRCT domains at its C terminus. The IRIF region, which we previously mapped (7), not only contains the Tudor domain but also contains a ubiquitination-dependent recruitment (UDR) motif (8). Studies from Durocher and co-workers (8) suggest that accumulation of 53BP1 at DNA damage sites requires both of these domains and histone ubiquitination at H2AX Lys-13/Lys-15 sites, which is mediated by RNF8/RNF168 in the H2AX-dependent pathway. In addition, several research groups, including ours, further demonstrated that RIF1 and PTIP are key factors that act downstream of 53BP1 and counteract functions of BRCA1 in DNA repair (9–14). More recently, we showed that PTIP prevents HR repair by recruiting a nuclease Artemis to sites of DNA damage (15), whereas others and us revealed that RIF1 is involved in the prevention of end resection and HR repair by recruiting another DNA repair protein REV7 (also called MAD2L2) to sites of DNA breaks (16, 17). These studies uncovered events downstream of 53BP1 that are important for DNA repair. However, the upstream regulation of the 53BP1-dependent pathway remains unexplored.
In this study, we identified Nudt16L1, now named as TIRR (Tudor-interacting repair regulator), which specifically associates with the IRIF region of 53BP1. TIRR belongs to a family of nucleoside diphosphate-linked moiety X (Nudix) hydrolases, which are present in bacteria, viruses, eukaryotes, and archaea (18). Nudix proteins are characterized by a highly conserved 23-amino acid Nudix motif (Nudix box), GX5EX7REUXEEXGU, where U is an aliphatic or hydrophobic residue (18). Nudix proteins have protective, regulatory, and signaling functions in metabolism through their ability to remove a wide range of organic pyrophosphates from the cellular environment (18). However, different Nudix proteins may have distinct functions. Here, we further investigated the regulation and functional significance of TIRR/Nudt16L1 and its interaction with 53BP1 in DNA damage response.
Results
TIRR is a novel 53BP1-interacting protein
We and others have shown that a central region containing the Tudor domain and ubiquitination-dependent recruitment (UDR) motif of 53BP1 is required for the accumulation of 53BP1 at sites of DNA breaks (7, 8), which we named the IRIF region (Fig. 1A). Indeed, distinct nuclear foci of this IRIF region of 53BP1 could be readily detected in cells after irradiation (Fig. 1B). To understand how 53BP1 localization is regulated before and after DNA damage, we carried out tandem affinity purification with use of lysates derived from 293T cells stably expressing the SFB (S protein, FLAG, and streptavidin-binding peptide tag)-tagged IRIF region of 53BP1. Mass spectrometry analysis revealed that TIRR is a novel 53BP1-associated protein (Fig. 1C). TIRR is a 211-amino acid protein containing Nudix motif with unknown function in DNA damage response. To verify that TIRR indeed associates with 53BP1, we repeated tandem affinity purification using 293T cell lines stably expressing SFB-tagged TIRR and identified 53BP1 as a TIRR-associated protein (Fig. 1D). To further confirm this interaction, we generated anti-TIRR antibodies and showed that endogenous TIRR strongly associates with 53BP1 (Fig. 1E). Furthermore, using a bacterially expressed and purified GST-fused IRIF region of 53BP1 protein, we showed that the 53BP1 IRIF region binds to TIRR in vitro (Fig. 1F), indicating that the IRIF region of 53BP1 is sufficient for its binding to TIRR. Taken together, these data suggest that TIRR is a bona fide 53BP1-binding protein.
Figure 1.

TIRR is 53BP1-associated protein. A, schematic presentation of 53BP1 and its IRIF region that is used in this study. B, 53BP1 IRIF region form foci following DNA damage. 293T cells stably expressing SFB-53BP1 IRIF region grown on coverslips were treated with 10 Gy of IR; cells were fixed, and immunostaining was carried out using indicated antibodies. C and D, lists of 53BP1 IRIF region or TIRR-associated proteins identified in soluble fraction by mass spectrometry analysis. E, endogenous 53BP1 interacts with TIRR. HeLa cell lysates were prepared and immunoprecipitated (IP) with TIRR antibody. Immunoprecipitates were blotted using antibodies as indicated. F, 53BP1 IRIF region specifically binds to TIRR. Beads coated with bacterially expressed GST-fused 53BP1 IRIF region were incubated with cell lysates containing exogenously expressed SFB-tagged TIRR. Immunoblotting experiments were carried out using the indicated antibodies. WB, Western blotting.
IRIF region of 53BP1 is required for the 53BP1-TIRR interaction
To further confirm that the IRIF region of 53BP1 is responsible for its interaction with TIRR, we generated an IRIF region deletion mutant of 53BP1. As shown in Fig. 2A, deletion of the IRIF region of 53BP1 abolishes the 53BP1-TIRR interaction. In addition, the 53BP1 D1521R mutation disrupts the Tudor domain, whereas the 53BP1 L1619A mutant abolishes the UDR activity, both of which impair the ability of 53BP1 to form IR-induced foci (8). We observed that 53BP1 D1521R mutant dramatically reduced 53BP1-TIRR interaction, whereas 53BP1 L1619A mutant did not notably affect its binding to TIRR (Fig. 2B). We next sought to identify the region(s) of TIRR that is responsible for its interaction with 53BP1 IRIF region. Using SFB-tagged wild-type TIRR and a series of truncation and internal deletion mutants of TIRR (Fig. 2C), we showed that deletion of the Nudix motif abolished the TIRR-53BP1 interaction, indicating that the Nudix motif of TIRR is important for the binding of TIRR to 53BP1 (Fig. 2D).
Figure 2.
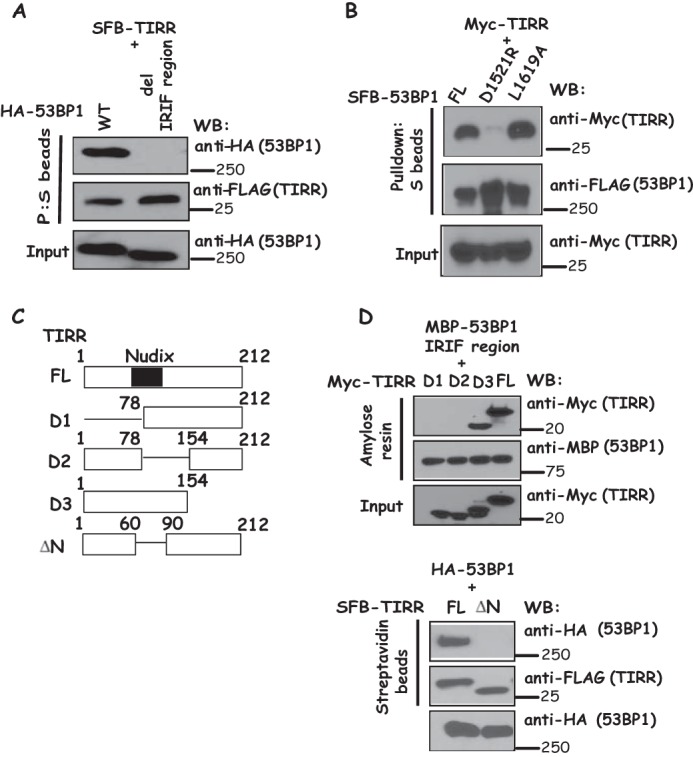
Mapping the binding regions on 53BP1 and TIRR. A, 53BP1 IRIF region is required for 53BP1-TIRR interaction. 293T cells were transfected with plasmids encoding SFB-tagged TIRR together with plasmids encoding wild-type or IRIF region deletion mutant of HA-tagged 53BP1. Immunoprecipitation reactions were conducted using S-protein beads and then subjected to Western blotting (WB) using the indicated antibodies. B, disruption of the Tudor domain of 53BP1 decreased the binding of 53BP1 to TIRR. 293T cells were transfected with plasmids encoding SFB-tagged TIRR together with plasmids encoding wild-type, D1521R, and L1619A mutant of HA-tagged 53BP1, respectively. Immunoprecipitation reactions were conducted using S-protein beads and then subjected to Western blotting using indicated antibodies. C, schematic presentation of wild-type and deletion mutants of TIRR used in this study. Nudix domain is indicated as a blue rectangle (residues 60–90). D, Nudix motif of TIRR is required for its binding to 53BP1. Beads coated with bacterially expressed MBP-fused 53BP1 IRIF region were incubated with cell lysates containing exogenously expressed Myc-tagged wild-type or deletion mutants of TIRR. Moreover, 293T cells were transfected with plasmids encoding HA-tagged 53BP1 together with plasmids encoding wild-type deletion mutants of SFB-tagged TIRR. Immunoprecipitation reactions were conducted using S-protein beads. Immunoblotting experiments were carried out using indicated antibodies.
53BP1-TIRR forms a stable complex and dissociates after DNA damage
We first studied the consequence of TIRR overexpression on DNA damage-induced focus formation of 53BP1 by transfecting HeLa cells with SFB-tagged versions of TIRR or control Nudt15. As shown in Fig. 3A, the IR-induced focus formation by 53BP1 was abrogated by TIRR but not Nudt15 overexpression, suggesting that TIRR may regulate the recruitment of 53BP1 to sites of DNA breaks following IR.
Figure 3.

53BP1-TIRR complex is required for the expression of both 53BP1 and TIRR, and this complex dissociates following DNA damage. A, overexpression of TIRR reduced 53BP1 foci formation following IR. Cells were transfected with constructs encoding tagged TIRR or Nudt15 and treated with 10 Gy of IR. Immunostaining experiments were performed using anti-53BP1 and anti-FLAG antibodies. B, depletion of TIRR decreases 53BP1 protein level. MCF10A cells were infected with control or TIRR-specific gRNAs. Cells were harvested and immunoblotted with the indicated antibodies. C, Nudix motif of TIRR is critical for 53BP1 expression. TIRR knock-out cells were reconstituted with wild-type TIRR or Nudix motif deletion mutant of TIRR. Cell lysates were immunoblotted with indicated antibodies. D, knock-out 53BP1 decreases TIRR protein level. MCF10A cells and MCF10A-derived 53BP1 knock-out cells were treated with or without IR (10 Gy). Cells were harvested and immunoblotted with indicated antibodies. E, IRIF region of 53BP1 is required for TIRR stability. 53BP1 knock-out cells were reconstituted with wild-type 53BP1 or an IRIF deletion mutant of 53BP1. Cell lysates were immunoblotted with indicated antibodies. F–H, 53BP1 and TIRR dissociate after DNA damage. F and G, beads coated with bacterially expressed MBP-TIRR fusion protein were incubated, respectively, with cell lysates containing exogenously expressed SFB-tagged 53BP1 IRIF region mock-treated or treated with ATMi (KU60019) with or without IR (20 Gy) (F) or HeLa cell lysates treated with or without IR (20 Gy) (G). Immunoblotting experiments were carried out using the indicated antibodies. Alternatively, cell lysates with or without IR (20 Gy) were immunoprecipitated with anti-TIRR antibodies and immunoblotted with indicated antibodies (H). Dissociation of 53BP1 with TIRR does not require TIRR modification following DNA damage (I). Beads coated with bacterially expressed MBP-53BP1 IRIF region fusion protein were incubated with cell lysates containing exogenously expressed SFB-tagged TIRR mock-treated or treated with ATMi (KU60019) with or without IR (20 Gy). Immunoblotting experiments were carried out using indicated antibodies. WB, Western blotting.
Given that TIRR interacts with 53BP1, we wanted to know the functional significance of this interaction. Occasionally subunits of a stable protein complex would rely on each other for their expression. Therefore, we knocked out or depleted each protein and examined the expression of other components. We observed that depletion of TIRR by gRNAs led to a decrease of 53BP1 protein levels (Fig. 3B). Moreover, the expression of gRNA-resistant wild-type TIRR restored 53BP1 protein levels in TIRR knock-out cells, whereas the expression of TIRR with a deletion of Nudix motif did not restore 53BP1 expression (Fig. 3C), indicating that the 53BP1-TIRR interaction is required for TIRR expression. Likewise, knock-out of 53BP1 led to a decrease in TIRR protein expression (Fig. 3D). This reduction of TIRR protein level was observed in 53BP1 knock-out cells with or without IR (Fig. 3D). Similarly, as shown in Fig. 3E, the expression of gRNA-resistant wild-type 53BP1 restored TIRR protein level in 53BP1 knock-out cells, whereas the expression of IRIF deletion mutant of 53BP1 failed to do so, indicating that the 53BP1-TIRR interaction is critical for TIRR expression.
We further investigated how 53BP1-TIRR interaction may be regulated after DNA damage. In this regard, we used bacterially expressed and purified TIRR protein and showed that its interaction with the 53BP1 IRIF region was drastically decreased after IR, and this IR-induced dissociation of TIRR with 53BP1 could be prevented if cells were treated with ATM inhibitor (Fig. 3F). Moreover, as shown in Fig. 3, G and H, we observed a dissociation between endogenous 53BP1 and TIRR following IR. Conversely, using the bacterially expressed IRIF region of 53BP1, we showed that its interaction with TIRR was not notably changed with or without IR and/or ATM inhibitor treatment (Fig. 3I). Together, these data indicate that damage-induced and ATM-dependent phosphorylation of 53BP1 likely controls the dissociation of 53BP1-TIRR following DNA damage.
TIRR participates in DNA repair pathway
Because TIRR forms a stable complex with 53BP1, we speculated that TIRR may regulate 53BP1 expression and therefore play an indirect role in facilitating NHEJ and preventing BRCA1-dependent DNA end resection and HR repair. Indeed, loss of TIRR led to reduced 53BP1 expression (Fig. 3D) and also rendered BRCA1-deficient cells resistant to PARP inhibition (Fig. 4A). Furthermore, damage-induced RPA2 foci (marker for end resection) or RAD51 foci (marker for HR repair) were reduced in BRCA1-depleted cells, but they were re-established in BRCA1/TIRR co-depleted cells (Fig. 4, B and C), indicating that similar to 53BP1, co-depletion of TIRR together with BRCA1 promote end resection and RAD51-dependent HR repair. Moreover, although expression of gRNA-resistant wild-type TIRR restored 53BP1 expression (Fig. 3E) and therefore rescued PARPi sensitivity in TIRR knock-out cells, reconstitution with TIRR with a deletion of the Nudix motif (53BP1 binding region) fails to rescue 53BP1 expression (Fig. 3E) or PARPi sensitivity following PARPi treatment (Fig. 4D), suggesting that the 53BP1-TIRR interaction is critical not only for 53BP1 expression but also for antagonizing BRCA1-mediated DNA repair. In agreement with our hypothesis that the phenotypes of TIRR depletion noted above are due to its effect on 53BP1 expression, we observed that ectopically expression of 53BP1 restored PARP sensitivity in TIRR/BRCA1 knock-out cells (Fig. 4E), suggesting that TIRR contributes to DNA repair at least in part via its regulation of 53BP1 expression.
Figure 4.

TIRR participates in 53BP1-dependent DNA damage response. Loss of TIRR renders BRCA1-deficient cells resistant to PARP inhibition (A). HeLa cells were transfected with indicated gRNAs and treated with 1 μm PARPi (Olaparib). Colony formation was quantified relative to colonies formed in untreated cells from the same setting. Data are represented as the mean ± S.E. (n = 3). *, p < 0.05. TIRR prevents end resection and RAD51-dependent HR repair in the absence of BRCA1 (B and C). HeLa cells were transfected with indicated gRNAs. At 3 h post-irradiation (10 Gy), cells were processed for RAD51 (B) or RPA (C) immunofluorescence staining. RAD51 (B) or RPA (C) foci were quantified (at least 400 cells were counted for each experiment). Data are represented as the mean ± S.E. (n = 3). *, p < 0.05. 53BP1-TIRR interaction is important for cellular response to PARPi (D). TIRR knock-out MCF10A cells were reconstituted with gRNA-resistant wild-type or Nudix motif deletion mutant of SFB-tagged TIRR. These cells were then infected with indicated control or BRCA1 gRNAs and treated with 1 μm PARPi. Colony formation was quantified relative to colonies formed in untreated cells from the same setting. Data are represented as the mean ± S.E. (n = 3). *, p < 0.05. TIRR functions in DNA repair via its ability to regulate 53BP1 expression (E). Control cells or TIRR knock-out cells with or without 53BP1 overexpression were treated with control or BRCA1 siRNA. Cells were treated with olaparib at various concentrations for 6 days, and viable cell count was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. *, p < 0.05. TIRR is required for cell survival following ionizing radiation (F). HeLa cells transfected with control siRNA (siCTR) or TIRR specific siRNAs (siTIRR#1 and siTIRR#2) were mock-treated or irradiated. Cell survival following irradiation was measured by clonogenic assay according to the “Experimental procedures.” Western blotting analysis was performed to verify the depletion of TIRR following siRNA transfection (lower panel). *, p < 0.05. G, revised model of TIRR-53BP1 interaction and its dissociation in regulating DNA repair and cellular response to PARPi following DNA damage.
53BP1 is clearly involved in DNA damage response and is required for cell survival following IR (20). Because TIRR depletion reduced 53BP1 expression (Fig. 3D), we anticipate that TIRR loss should also lead to IR sensitivity, which is supported by our experimental data shown in Fig. 4F. These data suggest that the 53BP-TIRR complex is required for cell survival in response to DNA damage.
Discussion
Two main pathways, NHEJ and HR, exist in eukaryotic cells for the repair of DSBs. A major question in the field is, mechanistically, how cells modulate the pathway choice for DSB repair. A number of studies demonstrated that 53BP1 determines DNA repair pathway choice and counteracts HR repair in the absence of BRCA1 (5, 6). Recently, we and others showed that, mechanistically, 53BP1 acts as an adaptor protein and controls two downstream sub-pathways that are mediated, respectively, by PTIP-Artemis and RIF1-REV7 to promote end-joining and suppress HR repair (9–14). However, questions remain regarding the upstream regulation of the 53BP1 pathway.
In this study, we identified TIRR as 53BP1-binding protein that specifically associates with 53BP1 via its IRIF region, which contains the Tudor domain and UDR motif. We further confirmed that TIRR associates with 53BP1 via the IRIF region and showed that this interaction requires the intact Tudor domain of 53BP1. TIRR belongs to the Nudix motif-containing protein family of phosphohydrolase. We showed that the Nudix motif of TIRR is indispensable for the TIRR-53BP1 interaction. In addition, overexpression of TIRR blocks 53BP1 recruitment to DSBs following IR. Moreover, we showed that TIRR expression level decreased in 53BP1 KO cells and vice versa. These data suggest that 53BP1 and TIRR may form stable complexes and that loss of each would destabilize the other protein in this complex. Indeed, similar to loss of 53BP1, loss of TIRR renders the BRCA1-deficient cells resistant to PARPi, suggesting that TIRR is required for 53BP1 expression and therefore specifically involved in the 53BP1-dependent repair pathway. Interestingly, TIRR dissociates with 53BP1 following IR, and this dissociation could be prevented with treatment of ATM inhibitor, suggesting that ATM-dependent phosphorylation of 53BP1 may be critical for the regulation of 53BP1-TIRR complex after DNA damage. Thus, TIRR may play both positive and negative roles in 53BP1 regulation. On the one hand, it stabilizes 53BP1 and thus positively regulates 53BP1 and its dependent repair functions. On the other hand, its association with 53BP1 prevents 53BP1 localization to sites of DNA damage. Therefore, TIRR is also an inhibitor of 53BP1 and participates in the regulation of 53BP1 localization before and after DNA damage.
Our current working hypothesis is that 53BP1 normally resides in the nuclei as protein complexes with TIRR, which stabilizes 53BP1. After DNA damage, 53BP1 dissociates with TIRR, and translocates to DNA damage sites. At sites of DNA breaks, 53BP1 not only associates with methylated and ubiquitinated histones, as reported previously (8, 21, 22), but also binds to other chromatin proteins, which may retain 53BP1 at damage sites and function with 53BP1 to carry out DNA repair and cellular response to PARP inhibitors (Fig. 4G). Besides regulating 53BP1 expression, TIRR may also be recruited to sites of DNA damage via other mechanisms and participate in DNA damage response beyond its association with 53BP1 (Fig. 4G). We will further explore this possibility in future studies.
Materials and methods
Cell culture and plasmids
HeLa, HEK293T, and MCF10A cells were purchased from American Type Culture Collection (ATCC) and cultured under conditions specified by the manufacturer. TIRR cDNA was subcloned into pDONR201 as entry vector and subsequently transferred to gateway-compatible destination vectors for the expression of triple-epitope tag SFB, HA epitope, or Myc epitope-tagged fusion proteins. All deletion mutants were generated by site-directed mutagenesis and verified by sequencing.
Antibodies
Anti-TIRR antibody was raised by immunization of rabbit with MBP-TIRR fusion protein. Antisera was affinity-purified using the AminoLink Plus immobilization and purification kit (Pierce). 53BP1 antibody was described previously (13, 19). The monoclonal anti-FLAG M2 and anti-β-actin antibodies were purchased from Sigma. The anti-Myc (9E10) antibody was obtained from Covance.
Immunofluorescence staining
Cells grown on coverslips were mock-treated or irradiated with a JL Shepherd Cs137 source (10 Gy) and allowed to recover for 4 h. Cells were fixed in 3% paraformaldehyde solution for 10 min and then permeabilized in 0.5% Triton X-100-containing solution for 5 min at room temperature. Cells were incubated with primary antibodies diluted in 5% goat serum at 37 °C for 30 min. Coverslips were washed and incubated either with FITC or rhodamine for 30 min at 37 °C. Cells were then stained with DAPI to visualize nuclear DNA. The coverslips were mounted onto glass slides with anti-fade solution and visualized using Nikon Eclipse E800 fluorescence microscope.
Co-immunoprecipitation and Western blotting
Cells were lysed with NTEN buffer (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40) containing 20 mm NaF, and 1 μg/ml pepstatin A and aprotinin on ice for 20 min. Clear cell lysates were incubated with either protein A-agarose beads coupled with anti-TIRR antibody or streptavidin-Sepharose beads (Amersham Biosciences) for 3 h at 4 °C. Beads were then washed and boiled in 2× Laemmli buffer and separated on SDS-PAGE. PVDF membranes were blocked in 5% milk in TBST buffer and then probed with antibodies as indicated.
Tandem affinity purification
293T cells were transfected with plasmids encoding SFB-53BP1 IRIF region or SFB-TIRR. Cell lines stably expressing tagged proteins were selected, and the expression of exogenous proteins was confirmed by immunoblotting and immunostaining. For tandem affinity purification, a total of 20 10-cm dishes of 293T cells stably expressing SFB-53BP1 IRIF region or SFB-TIRR were collected and lysed with NETN buffer (see above) on ice for 20 min. Crude lysates were incubated with 200 μl of streptavidin-Sepharose beads for 3 h at 4 °C. The beads were washed three times with NETN buffer and then eluted with 2 mg/ml biotin (Sigma) for 1 h twice at 4 °C. The eluates were incubated with 100 μl of S-protein agarose (Novagen) for 2 h at 4 °C and then washed three times with NETN buffer. The proteins bound to beads were eluted by boiling with 2× Laemmli buffer, resolved by SDS-PAGE, and visualized by Coomassie Blue staining. The identities of eluted proteins were revealed by mass spectrometry analysis) (performed by the Taplin Biological Mass Spectrometry Facility, Harvard University).
Clonogenic survival assays
IR sensitivity assay was carried out as described previously (19). Briefly, a total of 1 × 103 cells were seeded onto 60-mm dish in triplicate and treated with ionizing radiation with indicated doses the next day. Cells were then incubated for 14 days. Resulting colonies were fixed and stained with Coomassie Blue. Numbers of colonies were counted using a GelDoc with Quantity One software (Bio-Rad). Results were the averages of data obtained from three independent experiments.
Pulldown assays using bacterially expressed fusion proteins
GST or MBP fusion proteins were expressed in Escherichia coli and purified. GST or MBP fusion proteins were immobilized on either glutathione-Sepharose 4B beads or amylose resin and incubated with lysates prepared from cells transiently transfected with plasmids encoding the indicated proteins. The samples were subjected to SDS-PAGE and analyzed by Western blotting.
Author contributions
A. Z. and B. P. performed all the experiments with help from P. H. J. C. and Z. G. analyzed the data. J. C. and Z. G. drafted the manuscript. J. C. and Z. G. supervised the project. All authors reviewed the results and approved the final version of the manuscript.
Note added in proof
There was some textual overlap under the “Experimental procedures” between the version of this article that was published as a Paper in Press on February 17, 2017 and Gong, Z. and Chen, J. (2011) J. Biol. Chem. 286, 22308–22313. The text overlap has now been removed.
This work was supported in part by National Institutes of Health Grants CA089239, CA092312, and CA100109 (to J. C.), an Ovarian Cancer Research Fund Alliance Grant 373376 (to Z. G.), and National Institutes of Health Grant CA192052 from NCI (to Z. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- DSB
- double strand break
- PARPi
- poly(ADP-ribose)polymerase inhibitor
- PTIP
- Pax transactivation domain-interacting protein
- TIRR
- Tudor-interacting repair regulator
- IRIF
- ionizing radiation-induced foci formation
- NHEJ
- non-homologous DNA end-joining
- HR
- homologous recombination
- UDR
- ubiquitination-dependent recruitment
- Gy
- gray
- ATM
- ataxia telangiectasia mutated
- IR
- ionizing radiation
- MBP
- maltose-binding protein
- RPA
- replication protein A.
References
- 1. Lieber M. R. (2010) The mechanism of double strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Heyer W. D., Ehmsen K. T., and Liu J. (2010) Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 44, 113–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., and Helleday T. (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 [DOI] [PubMed] [Google Scholar]
- 4. Farmer H., McCabe N., Lord C. J., Tutt A. N., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M., Jackson S. P., Smith G. C., and Ashworth A. (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 [DOI] [PubMed] [Google Scholar]
- 5. Bouwman P., Aly A., Escandell J. M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q., Haffty B. G., Tommiska J., Blomqvist C., Drapkin R., Adams D. J., et al. (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bunting S. F., Callén E., Wong N., Chen H. T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L., Xu X., Deng C. X., Finkel T., Nussenzweig M., Stark J. M., and Nussenzweig A. (2010) 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ward I. M., Minn K., Jorda K. G., and Chen J. (2003) Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J. Biol. Chem. 278, 19579–19582 [DOI] [PubMed] [Google Scholar]
- 8. Fradet-Turcotte A., Canny M. D., Escribano-Díaz C., Orthwein A., Leung C. C., Huang H., Landry M. C., Kitevski-LeBlanc J., Noordermeer S. M., Sicheri F., and Durocher D. (2013) 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Callen E., Di Virgilio M., Kruhlak M. J., Nieto-Soler M., Wong N., Chen H. T., Faryabi R. B., Polato F., Santos M., Starnes L. M., Wesemann D. R., Lee J. E., Tubbs A., Sleckman B. P., Daniel J. A., et al. (2013) 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153, 1266–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chapman J. R., Barral P., Vannier J. B., Borel V., Steger M., Tomas-Loba A., Sartori A. A., Adams I. R., Batista F. D., and Boulton S. J. (2013) RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double strand break resection. Mol. Cell 49, 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Virgilio M., Callen E., Yamane A., Zhang W., Jankovic M., Gitlin A. D., Feldhahn N., Resch W., Oliveira T. Y., Chait B. T., Nussenzweig A., Casellas R., Robbiani D. F., and Nussenzweig M. C. (2013) Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Escribano-Díaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J. T., Tkáč J., Cook M. A., Rosebrock A. P., Munro M., Canny M. D., Xu D., and Durocher D. (2013) A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 [DOI] [PubMed] [Google Scholar]
- 13. Feng L., Fong K. W., Wang J., Wang W., and Chen J. (2013) RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 288, 11135–11143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zimmermann M., Lottersberger F., Buonomo S. B., Sfeir A., and de Lange T. (2013) 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339, 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang J., Aroumougame A., Lobrich M., Li Y., Chen D., Chen J., and Gong Z. (2014) PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 28, 2693–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boersma V., Moatti N., Segura-Bayona S., Peuscher M. H., van der Torre J., Wevers B. A., Orthwein A., Durocher D., and Jacobs J. J. (2015) MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 521, 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu G., Chapman J. R., Brandsma I., Yuan J., Mistrik M., Bouwman P., Bartkova J., Gogola E., Warmerdam D., Barazas M., Jaspers J. E., Watanabe K., Pieterse M., Kersbergen A., Sol W., et al. (2015) REV7 counteracts DNA double strand break resection and affects PARP inhibition. Nature 521, 541–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McLennan A. G. (2006) The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 63, 123–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gong Z., Cho Y. W., Kim J. E., Ge K., and Chen J. (2009) Accumulation of Pax2 transactivation domain interaction protein (PTIP) at sites of DNA breaks via RNF8-dependent pathway is required for cell survival after DNA damage. J. Biol. Chem. 284, 7284–7293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ward I. M., Minn K., van Deursen J., and Chen J. (2003) p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23, 2556–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Botuyan M. V., Lee J., Ward I. M., Kim J. E., Thompson J. R., Chen J., and Mer G. (2006) Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huyen Y., Zgheib O., Ditullio R. A. Jr, Gorgoulis V. G., Zacharatos P., Petty T. J., Sheston E. A., Mellert H. S., Stavridi E. S., and Halazonetis T. D. (2004) Methylated lysine 79 of histone H3 targets 53BP1 to DNA double strand breaks. Nature 432, 406–411 [DOI] [PubMed] [Google Scholar]