

Abstract
Renal cell carcinoma (RCC) is polyresistant to chemo- and radiotherapy and biologicals, including TNF-related apoptosis-inducing ligand (TRAIL). Sorafenib, a multikinase inhibitor approved for the treatment of RCC, has been shown to sensitize cancer cells to TRAIL-induced apoptosis, in particular by down-regulation of the Bak-inhibitory Bcl-2 family protein Mcl-1. Here we demonstrate that sorafenib overcomes TRAIL resistance in RCC by a mechanism that does not rely on Mcl-1 down-regulation. Instead, sorafenib induces rapid dissipation of the mitochondrial membrane potential (ΔΨm) that is accompanied by the accumulation of reactive oxygen species (ROS). Loss of ΔΨm and ROS production induced by sorafenib are independent of caspase activities and do not depend on the presence of the proapoptotic Bcl-2 family proteins Bax or Bak, indicating that both events are functionally upstream of the mitochondrial apoptosis signaling cascade. More intriguingly, we find that it is sorafenib-induced ROS accumulation that enables TRAIL to activate caspase-8 in RCC. This leads to apoptosis that involves activation of an amplification loop via the mitochondrial apoptosis pathway. Thus, our mechanistic data indicate that sorafenib bypasses central resistance mechanisms through a direct induction of ΔΨm breakdown and ROS production. Activation of this pathway might represent a useful strategy to overcome the cell-inherent resistance to cancer therapeutics, including TRAIL, in multiresistant cancers such as RCC.
Keywords: apoptosis, B cell lymphoma 2 (Bcl-2) family, Bax, cancer, drug resistance, reactive oxygen species (ROS), Trail, Bak, sorafenib
Introduction
Apoptotic cell death induced by the death ligand tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)2 plays an important role in immune surveillance and is a major immune defense mechanism against tumor cells. Consequently, the use of TRAIL, which preferentially kills cancer cells while sparing non-cancerous tissue, is a promising concept in anticancer therapy. Recombinant TRAIL and agonistic antibodies against the TRAIL receptors are therefore tested in clinical phase I and II studies (1–3).
Binding of TRAIL to the death receptors DR4 (TRAIL-R1) and DR5 (TRAIL-R2) initiates receptor oligomerization and, via the adaptor protein Fas-associated protein with death domain (FADD), recruitment of the initiator caspase-8 to the death domain of the activated receptor (4, 5). Formation of this activation platform, the so-called death-inducing signaling complex, results in autocatalytic activation of caspase-8. In type I cells, active caspase-8 triggers, via direct proteolytic processing of caspase-3, a caspase cascade to induce apoptotic cell death. In type II cells, however, the E3-ligase X-linked inhibitor of apoptosis protein (XIAP) prevents accumulation of active caspase-3 by marking it for proteasomal degradation (6). Therefore, in type II cells, efficient caspase-3 activation upon death receptor signaling requires amplification via the mitochondrial apoptosis pathway. Activation of the mitochondrial pathway is achieved by mitochondrial outer membrane permeabilization (MOMP) (7). Upon MOMP, the XIAP inhibitor second mitochondria-derived activator of caspase/direct IAP-binding protein with low pI (Smac/DIABLO) is released from the mitochondrial intermembrane space into the cytosol (8), where it inhibits XIAP and thereby prevents degradation of active caspase-3 (9–11). Another important proapoptotic factor that is released into the cytosol upon MOMP is cytochrome c. Cytochrome c activates the adapter molecule APAF-1, resulting in the formation of the apoptosome, a multiprotein complex in which the initiator caspase-9 is activated (12) for processing of caspase-3 and amplification of the caspase cascade.
Upon TRAIL-R ligation, MOMP is induced by caspase-8-mediated cleavage and activation of BH3-interacting domain death agonist (Bid), a proapoptotic protein of the B cell lymphoma 2 (Bcl-2) family (13–15). The proteins of the Bcl-2 family are key regulators of MOMP and show homology in at least one of four Bcl-2 homology (BH1–4) domains. Antiapoptotic family members (e.g. Bcl-2, Bcl-xL, and Mcl-1) are characterized by the presence of all four BH domains. Proapoptotic members can be subdivided into the multidomain BH123 homologs (Bax, Bak, and Bok) and into the large BH3-only subfamily (e.g. Bid, Bim, Bad, Nbk/Bik, Puma, and Noxa) (16). The proapoptotic BH123 proteins Bcl-2 associated x protein (Bax) and Bcl-2 homologous antagonist/killer (Bak) drive MOMP and are neutralized by antiapoptotic family members. BH3-only proteins activate Bax and Bak to induce MOMP indirectly by inhibiting prosurvival Bcl-2 proteins and/or via direct interaction with Bax and Bak (17, 18).
Deregulation of these apoptosis signaling pathways accounts for resistance to anticancer therapies, including the biological agent TRAIL, which often serves as a prototypical targeted reagent to study apoptosis signaling in cancer cells. Strategies to overcome resistance to TRAIL-induced apoptosis comprise combinations with DNA-damaging therapies, including the use of chemotherapeutic drugs (19) and irradiation (20), or the inhibition of prosurvival signaling, e.g. the nuclear factor κB (NF-κB) pathway (21), inhibition of the proteasome (22, 23), or inhibition of histone deacetylases (24), all of which have been shown to sensitize tumor cells for TRAIL. In addition, BH3 mimetics, small molecules like ABT-737 or Obatoclax may potentiate TRAIL-mediated apoptosis through binding to the hydrophobic groove at the surface of antiapoptotic Bcl-2 proteins, thereby blocking their prosurvival function (25, 26). Furthermore, the multikinase inhibitor sorafenib sensitizes cancer cells toward TRAIL through alternative mechanisms, e.g. inhibition of STAT3 (27, 28), and in particular through down-regulation of the Bak inhibitor myeloid cell leukemia 1 (Mcl-1) (29, 30). Down-regulation of Mcl-1 enables TRAIL to kill cells via activation of Bak; thus, it can overcome TRAIL resistance of Bax-deficient cells (31).
Sorafenib is approved for the treatment of advanced renal cell carcinomas (RCCs) (32–35), a cancer entity that frequently shows resistance not only to conventional radio- and chemotherapy but also to experimental therapy with TRAIL (22). Here we show that sorafenib overcomes the TRAIL resistance of various RCC cell lines. Surprisingly, in RCC, sorafenib-induced down-regulation of Mcl-1 is not causative of the sensitization. Instead, sorafenib induces caspase- and Bax/Bak-independent depolarization of mitochondria accompanied by increased ROS accumulation. Accumulation of ROS then overcomes the failure of TRAIL to activate caspase-8 in RCC cells and thereby enables TRAIL to induce apoptosis.
Results
RCCs frequently display an impressive resistance to anticancer therapies, including application of the biological agent TRAIL. We therefore employed TRAIL as a well defined apoptosis inducer to evaluate strategies to overcome therapy resistance in RCC. To this end, we treated the three RCC cell lines RCC-KP, RCC-26, and RCC-GW, which had revealed high resistance toward TRAIL in dose-response experiments, with TRAIL (50 ng/ml for 24 h) or sorafenib (20 μm for 38 h) or preincubated cells for 14 h with sorafenib prior to TRAIL treatment. Induction of apoptosis was analyzed by flow cytometric detection of the relative cellular DNA content, and hypodiploid cells were assumed to be apoptotic. As expected, a TRAIL concentration of 50 ng/ml alone did not induce apoptotic DNA fragmentation in any of the three RCC cell lines (Fig. 1). 20 μm sorafenib also did not induce apoptosis in RCC-GW cells and only marginally induced apoptosis in RCC-KP and RCC-26 cells. Preincubation of cells with sorafenib, however, strongly sensitized all cell lines to TRAIL-induced apoptosis. In detail, 45% of RCC-KP, 44% of RCC-26, and 26% of RCC-GW cells showed a hypodiploid, i.e. apoptotic, phenotype upon combined treatment, whereas treatment with TRAIL or sorafenib as single agents resulted in less than 8% and 15% apoptotic cells, respectively.
Figure 1.

Sorafenib-mediated down-regulation of Mcl-1 is not involved in the sensitization to TRAIL-induced apoptosis. A, sorafenib (SF) overcomes the TRAIL resistance of RCC cell lines. The indicated RCC cells were incubated with 50 ng/ml TRAIL for 24 h or 20 μm sorafenib for 38 h or preincubated for 14 h with sorafenib before addition of TRAIL. Apoptotic (hypodiploid) cells were determined by FACS analysis of relative cellular DNA content. TRAIL did not induce apoptosis in any of the cell lines. Sorafenib did not induce apoptosis in RCC-GW cells and only marginally in RCC-KP and RCC-26 cells. Combined treatment with sorafenib and TRAIL potently induced apoptosis in all three cell lines. Top panel, representative flow cytometry histograms of PI-stained cells. The percentage of hypodiploid, i.e. apoptotic, cells is indicated between markers. Bottom panel, mean ± S.D. of triplicates. B, Western blotting analysis revealed down-regulation of Mcl-1 upon SF treatment in RCC-KW and RCC-26 cells. In contrast, XIAP expression levels were not reduced in any of the cell lines. C, RCC cells were either left untreated or transfected with non-targeting (c) or Mcl-1 siRNA, and knockdown of Mcl-1 was confirmed by Western blotting analysis. D, after Mcl-1 down-regulation, cells were treated with TRAIL, cultured for an additional 24 h, and apoptotic cells were assessed by FACS analysis. None of the cell lines were sensitized to TRAIL-induced apoptosis upon Mcl-1 down-regulation (mean ± S.D. of triplicates).
It has been shown recently that sorafenib sensitizes tumor cells to TRAIL by down-regulation of XIAP and, in particular, by down-regulation of Mcl-1. Thus, we analyzed the influence of sorafenib on the expression levels of XIAP and Mcl-1 in RCC lines by immunoblotting. The expression levels of XIAP did not differ in control and sorafenib-treated RCC cell lines (Fig. 1B), indicating that XIAP down-regulation is not involved in the reversal of apoptosis resistance. However, the expression of Mcl-1 was slightly reduced in RCC-KP and RCC-26 cells (but not in RCC-GW cells), suggesting that sorafenib-induced Mcl-1 down-regulation might cause TRAIL sensitization. To investigate the relevance of reduced Mcl-1 expression for the sensitization of RCC cell lines, we analyzed TRAIL-induced apoptosis in the presence or absence of siRNA-mediated knockdown of Mcl-1. In contrast to other cell systems (31), knockdown of Mcl-1 (Fig. 1C), surprisingly, did not increase TRAIL-induced apoptosis in RCC compared with controls (Fig. 1D). These results indicate that sorafenib-mediated down-regulation of Mcl-1 is unlikely to cause sensitization of RCC cell lines to TRAIL.
To analyze the mechanism underlying the synergism of sorafenib and TRAIL in RCC, we investigated hallmarks of the mitochondrial apoptosis signaling cascade and death receptor signaling. Western blotting analysis showed that the death ligand TRAIL as a single agent failed to induce caspase-8 processing (Fig. 2A). Consistently, events located downstream of caspase-8 activation, such as Bid cleavage, caspase-3 processing, or the release of Smac and cytochrome c into the cytosol, were not detectable upon TRAIL treatment (Fig. 2A). Likewise, sorafenib alone did not induce caspase activation or release of proapoptotic factors into the cytosol. In contrast, a time course experiment revealed that preincubation of cells with sorafenib enabled TRAIL to activate caspase-8, as evidenced by the detection of the caspase-8 subunits p41/43 and p18 after 2–4 h of TRAIL treatment. In addition, after 4 h, a p30 subunit was detectable, indicating that caspase-8 was also processed via an alternative, previously described pathway (36). Activation of caspase-8 was accompanied by processing of Bid to its active form tBid/p15, release of Smac and cytochrome c, and processing of procaspase-3 to the active subunits (Fig. 2A). In accordance with the cleavage of Bid and the release of proapoptotic factors into the cytosol, immunofluorescence staining of cells with antibodies specific for active conformers of Bax and Bak, respectively, verified activation of both molecules in response to the combined incubation of cells with TRAIL and sorafenib (Fig. 2B). Thus, although TRAIL as a single agent was incapable of efficient caspase-8 activation, pretreatment of RCC cells with sorafenib enabled TRAIL to activate caspase-8, resulting in Bid cleavage and activation of the mitochondrial apoptosis pathway.
Figure 2.

Sorafenib enables TRAIL to activate caspase-8 and the mitochondrial apoptosis signaling pathway. A, after preincubation with sorafenib, cells were treated with TRAIL for the indicated periods of time, and caspase processing, Bid cleavage, and release of Smac and cytochrome c (Cyt c) were analyzed by immunoblotting. Treatment of cells with sorafenib or TRAIL as single agents for 16 h induced neither caspase processing nor Bid cleavage or release of SMAC or cytochrome c. In contrast, combined sorafenib/TRAIL treatment resulted in processing of caspase-8 and -3 to the respective active subunits that became detectable after 4 h. This was accompanied by Bid activation, indicated by increased tBid levels, and release of cytochrome c and Smac into the cytosol. B, cells were treated with SF, TRAIL, or a combination of both, and activation of Bax and Bak was analyzed by staining the cells with conformation-specific antibodies (Bax-NT and Bak-NT, respectively) and subsequent flow cytometry. As single agents, sorafenib and TRAIL failed to induce activation of Bax or Bak. However, preincubation of cells with sorafenib enables TRAIL to activate Bax as well as Bak. Left panel, representative flow cytometry histograms of immunostained cells. The percentage of cells positive for conformation-changed Bax or Bak is indicated between markers. Right panel, mean ± S.D. from triplicates. c, control.
To delineate the activation of the mitochondrial apoptosis pathway by combined treatment with sorafenib and TRAIL in more detail, we investigated loss of the mitochondrial membrane potential (ΔΨm). To this end, cells were incubated with the cationic dye JC-1, which exhibits specific mitochondrial localization and red fluorescence in vital cells and cytosolic localization and green fluorescence upon loss of ΔΨm. In control cells, fluorescence microscopy showed a red dotted staining pattern, indicating accumulation of JC-1 in the mitochondria. In contrast, upon sorafenib treatment, cells showed a dose-dependent increase in diffuse (cytosolic) green fluorescence (Fig. 3A). FACS analysis of JC-1 green fluorescence intensity revealed loss of ΔΨm in almost all cells upon treatment with sorafenib alone and, naturally, also upon combined treatment with sorafenib and TRAIL (Fig. 3B). Although sorafenib by itself was sufficient to induce loss of ΔΨm, it failed, as a single agent, to induce apoptosis in RCC cells (Fig. 2A).
Figure 3.

Sorafenib-induced mitochondrial depolarization is accompanied by accumulation of reactive oxygen species. A, RCC cell lines were treated with the indicated concentration of sorafenib and subsequently stained with JC-1 to analyze loss of ΔΨm. In control cells, JC-1 accumulates in mitochondria and forms red fluorescent J aggregates, reflecting high mitochondrial membrane potential, whereas green cytosolic fluorescence, indicating low mitochondrial membrane potential, increases with the concentration of sorafenib. B, RCC cells were treated with TRAIL and sorafenib as single agents or in combination. Measurement of green JC-1 fluorescence intensity by flow cytometry on a single-cell level revealed that sorafenib, but not TRAIL, as a single agent induces loss of ΔΨm in each of the RCC cell lines. C, cells were treated with 20 μm sorafenib alone or in combination with the free radical scavenger NAC. ROS accumulation was assessed by CellROX Green staining and fluorescence microscopy 6 h after treatment. Control cells showed a weak punctate green fluorescence pattern that changed to a bright nuclear fluorescence, demonstrating oxidation of CellROX Green by ROS and binding of the oxidized dye to nuclear DNA. Prevention of ROS accumulation by NAC inhibits nuclear fluorescence staining with CellROX green. In addition, cells with reduced ΔΨm were determined by JC-1 staining and flow cytometry. Treated cells were compared with controls. In contrast to ROS accumulation, sorafenib-induced loss of ΔΨm was not inhibited by NAC.
Mitochondrial dysfunction, such as loss of ΔΨm, is commonly associated with production of reactive oxygen species (ROS). To analyze whether sorafenib-induced depolarization of the mitochondria is accompanied by increased intracellular ROS levels, cells were incubated with the cell-permeable dye CellROX Green, which exhibits green fluorescence and binding to DNA only upon oxidation. Fluorescence microscopy of control cells showed a weak dotted green fluorescence pattern in all three cell lines, indicating basal ROS production at the mitochondria (Fig. 3C, left panel). Upon treatment with sorafenib, the staining pattern changed to bright nuclear fluorescence, demonstrating oxidation of CellROX Green and binding of oxidized CellROX Green to nuclear DNA. Co-incubation of sorafenib-treated cells with the antioxidative agent N-acetylcysteine (NAC) efficiently prevented bright nuclear fluorescence, indicating inhibition of ROS accumulation (Fig. 3C, left panel). However, although NAC efficiently prevented sorafenib-induced ROS accumulation, it failed to prevent sorafenib-induced depolarization of mitochondria, as shown by JC-1 staining and FACS analysis of sorafenib- and NAC-treated cells (Fig. 3C, right panel). Hence, sorafenib-induced mitochondrial depolarization is independent and occurs upstream of ROS accumulation.
To further study the role of caspases in the mechanism underlying sorafenib-mediated sensitization of RCC cells toward TRAIL-induced cell death, RCC cells were pretreated with sorafenib and subsequently exposed to TRAIL in the presence of the caspase-8 inhibitor Z-LETD-fmk, the caspase-3/7 inhibitor Z-DEVD-fmk, or the pan-caspase inhibitor Q-VD-OPh. After 6 h of incubation, cells were stained with annexin V-FITC/PI. Flow cytometric analysis revealed that all inhibitors potently reduced the proportion of apoptotic cells, with Q-VD-OPh showing the strongest effect (Fig. 4A), almost completely inhibiting sorafenib/TRAIL-induced apoptosis. Interestingly, FACS analysis of identically treated cells stained with JC-1 showed that Q-VD-OPh had no effect on sorafenib-induced mitochondrial depolarization (Fig. 4B). Also, like mitochondrial depolarization, sorafenib-induced ROS accumulation was not influenced by Q-VD-OPh and, thus, is independent of caspase activity, as shown by the bright CellROX Green staining induced by sorafenib in Q-VD-OPh-pretreated RCC cells (Fig. 4C). Sorafenib-induced depolarization of mitochondria and ROS accumulation thus are independent and functionally upstream of caspase activation.
Figure 4.
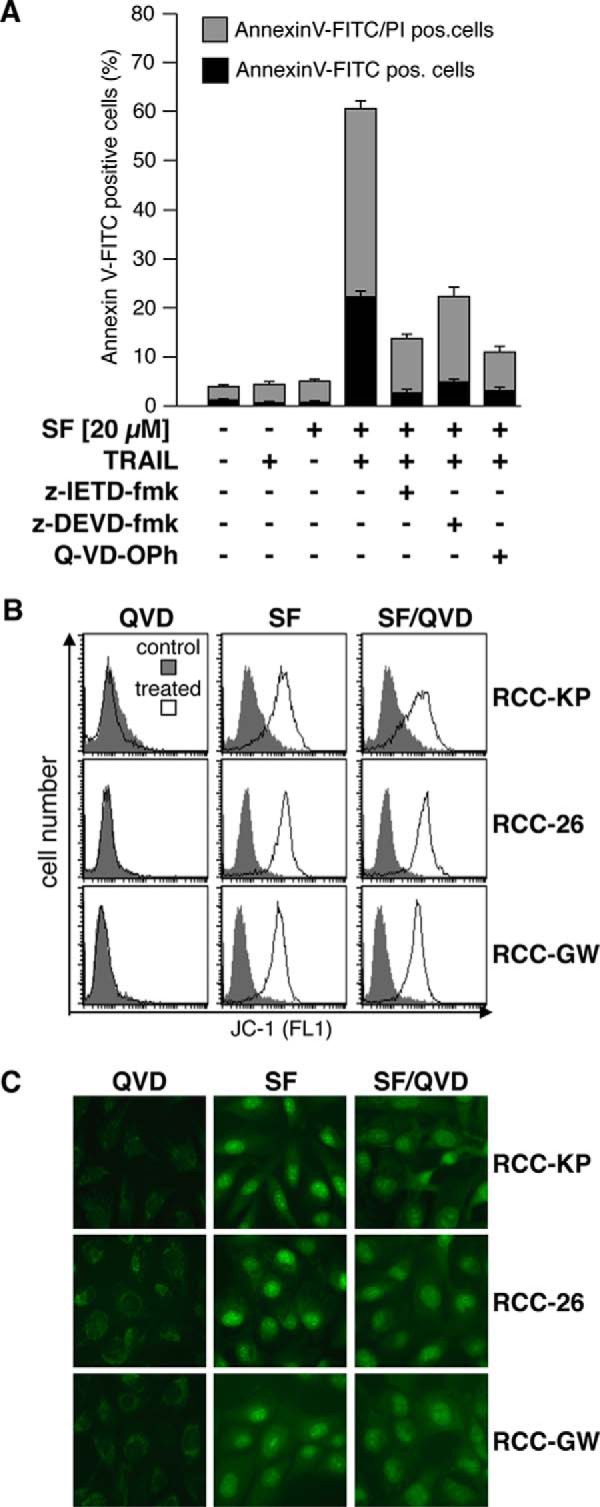
Sorafenib-induced depolarization and ROS production are independent of caspase activity. A, in addition to SF and TRAIL, the caspase-3/7 inhibitor Z-DEVD-fmk, the caspase-8 inhibitor Z-LETD-fmk, or the pan-caspase inhibitor Q-VD-OPh was added at a final concentration of 20 μm to the culture medium of RCC-26 cells After 6 h, cells were harvested, and apoptosis was analyzed by annexin V-FITC/PI staining and flow cytometry. Indicated are early (annexin V-FITC+/PI−) and late apoptotic (annexin V-FITC+/PI+) cells. Induction of apoptosis by combined sorafenib/TRAIL treatment is strongly reduced by Z-DEVD-fmk, Z-LETD-fmk, and Q-VD-OPh. B, cells were treated with sorafenib in the absence or presence of 20 μm Q-VD-OPh (QVD), and loss of ΔΨm was analyzed by JC-1 staining and FACS analysis. Treated cells were compared with controls. In contrast to apoptosis, the broad-range pan-caspase inhibitor Q-VD-OPh does not inhibit sorafenib-induced loss of ΔΨm. C, cells were treated as in B, and accumulation of ROS was analyzed by CellROX Green staining. The pan-caspase inhibitor Q-VD-OPh does not inhibit sorafenib-induced ROS accumulation.
TRAIL alone did not induce caspase-8 activation in RCC-26 cells but efficiently triggered processing of caspase-8 in cells preincubated with sorafenib (Fig. 2A). Therefore, we investigated whether sorafenib-induced accumulation of ROS is instrumental for caspase-8 activation upon TRAIL treatment. To this end, RCC cells were treated with sorafenib and TRAIL, and active caspase-8 was labeled with the fluorescent irreversible caspase-8 inhibitor FAM-LETD-fmk. Subsequent flow cytometric analysis revealed that the presence of NAC strongly reduced the number of cells positive for active caspase-8 labeling upon sorafenib/TRAIL treatment. For example, only 4% of RCC-26 control cells were positive for active caspase-8, whereas about 38% of sorafenib/TRAIL-treated cells were fluorescent for active caspase-8 (Fig. 5A). Inhibition of ROS accumulation by co-incubation with NAC strongly reduced the fraction of RCC-26 cells positive for active caspase-8 from 38% to 15% (Fig. 5A). NAC-mediated inhibition of sorafenib/TRAIL-induced caspase-8 processing was confirmed by immunoblotting. The processing of caspase-8 to its subunits in RCC-26 cells exposed to sorafenib and TRAIL was impaired by the ROS scavenger NAC (Fig. 5B), indicating that ROS production by sorafenib is crucial for TRAIL to activate caspase-8.
Figure 5.

Sorafenib-induced ROS accumulation enables TRAIL to activate caspase-8 and is responsible for sensitization to TRAIL-induced cell death. A, RCC cells were treated with sorafenib, TRAIL, and sorafenib/TRAIL in the absence or presence of NAC to prevent ROS accumulation. 8 h post-treatment, cells with active caspase-8 were detected by flow cytometry using the FAM-FLICA caspase-8 detection kit. The ROS scavenger NAC blocks sorafenib/TRAIL-induced caspase-8 activation. B, RCC-26 cells were treated as in A, and caspase-8 (C8) cleavage was analyzed by immunoblotting. Following combined treatment of sorafenib/TRAIL, pro-caspase-8 is processed to its active subunits. Proteolytic processing of caspase-8 is strongly reduced in the presence of the ROS scavenger NAC. C, induction of apoptosis was quantified by annexin V-FITC/PI staining and flow cytometry. Cells positive for annexin V-FITC (phosphatidylserine exposure) and negative for PI staining represent early apoptotic cells, whereas late apoptotic cells are positive for both annexin V-FITC and PI. Flow cytometric analysis revealed that induction of apoptosis by combined incubation with sorafenib and TRAIL is strongly reduced upon NAC-mediated inhibition of ROS accumulation. Panels show mean ± S.D. of triplicates.
We next asked whether the sensitizing effect of sorafenib toward TRAIL-induced apoptosis is mediated by excessive ROS production. Therefore, RCC cells were preincubated for 1 h with NAC or left untreated prior to addition of sorafenib for 14 h. Then TRAIL was added, and another 6 h later, induction of apoptosis was assessed by flow cytometric analysis of annexin V-FITC/PI-stained cells. In cells preincubated with NAC, the proportion of apoptotic cells (early apoptotic cells, annexin V-FITC+/PI−; late apoptotic cells, annexin V-FITC+/PI+) was reduced by ∼50% in each cell line, i.e. from 65% to 30% in RCC-KP cells, from 60% to 28% in RCC-26 cells, and from 36% to 19% in RCC-GW cells (Fig. 5C). These data demonstrate that induction of apoptosis by the combination of sorafenib and TRAIL is strongly reduced upon blocking of ROS accumulation. Because inhibition of ROS accumulation by NAC abrogates sorafenib-mediated sensitization of RCC cells toward TRAIL-induced apoptosis, we conclude that ROS accumulation is crucial for the TRAIL-sensitizing effect of sorafenib.
To test whether sorafenib-induced ROS production is a general mechanism that sensitizes cells to TRAIL-induced apoptosis, we incubated Mel-2a melanoma and DU145 prostate carcinoma cells with sorafenib and TRAIL. In both cell lines, sorafenib induced depolarization of the mitochondria and ROS formation (Fig. 6A). Moreover, sorafenib sensitized Mel-2a and DU145 cells to TRAIL-induced apoptosis, which was efficiently inhibited by the ROS scavenger NAC (Fig. 6B).
Figure 6.

Mitochondrial depolarization, ROS accumulation, and induction of apoptosis upon sorafenib/TRAIL treatment in cancer cell lines derived from different tissues. A, Mel-2a melanoma and DU145 prostate cancer cells were treated with sorafenib and stained with JC-1 or CellROX Green to analyze loss of ΔΨm and ROS accumulation, respectively. Similar to RCC cells, sorafenib induced mitochondrial depolarization and ROS accumulation in both cell lines. B, flow cytometric annexin V-FITC/PI staining revealed that induction of apoptosis by combined treatment with sorafenib and TRAIL was strongly reduced by NAC in Mel-2a and DU145 cells.
During canonical activation of the intrinsic apoptosis pathway, the BH123 proteins Bax and Bak mediate the dissipation of the mitochondrial membrane potential. To elucidate whether Bax and Bak are also involved in the depolarization of mitochondria induced by sorafenib, we used HCT116-WT colon cancer cells and the isogenic knockout cell line HCT116-Bax−/−/Bak−/− (37). Fluorescence microscopy of JC-1-stained cells showed that sorafenib induced depolarization in both cell lines in a concentration-dependent manner (Fig. 7A), which became detectable already 1–3 min after the addition of sorafenib (fig. 7B). These results demonstrate that sorafenib-induced depolarization of mitochondria is a very early event and, more intriguingly, not mediated by Bax or Bak. Besides Bax and Bak, the opening of the mitochondrial permeability transition pore (MPTP) induces MOMP and causes loss of ΔΨm. However, cyclosporin A (CsA), which acts as a potent inhibitor of MPTP opening, did not prevent sorafenib-induced mitochondrial depolarization in HCT116-WT and HCT116-Bax−/−/Bak−/− cells (Fig. 7C), demonstrating that sorafenib-induced depolarization is independent of both Bax/Bak and the MPTP.
Figure 7.
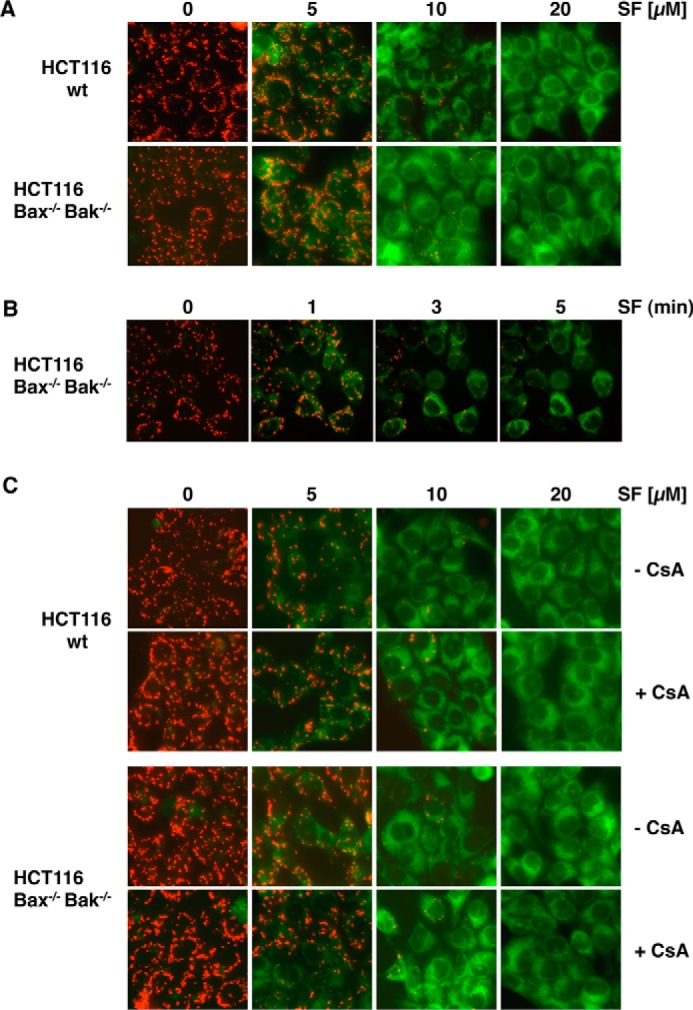
Sorafenib-induced mitochondrial depolarization upon sorafenib/TRAIL treatment is independent of Bax and Bak and the MPTP. A, HCT116-WT cells and isogenic HCT116-Bax−/−/Bak−/− cells were treated with sorafenib and stained with JC-1 to analyze loss of mitochondrial membrane potential. Fluorescence microscopy (top panels) showed decreased red and increased green fluorescence upon treatment with sorafenib in both cell lines, indicating that loss of ΔΨm is independent of Bax and Bak. B, HCT116-Bax−/−/Bak−/− cells were treated with sorafenib for the indicated time periods and stained with JC-1. Loss of ΔΨm is detectable 1–3 min after treatment in almost all cells. C, in addition to sorafenib, HCT116-WT and HCT116-Bax−/−/Bak−/− cells were preincubated with CsA to block MPTP opening. Upon treatment, cells were stained with JC-1 and analyzed by fluorescence microscopy. CsA failed to prevent sorafenib-induced mitochondrial depolarization in HCT116-WT and HCT116-Bax−/−/Bak−/− cells, demonstrating that sorafenib-induced depolarization is independent of both MPTP and Bax/Bak.
The rapid Bax/Bak- and MPTP-independent mitochondrial depolarization induced by sorafenib resembles the mechanism induced by classical uncouplers of the mitochondrial respiratory chain, like carbonyl cyanide m-chlorophenylhydrazone (CCCP). We therefore next asked whether CCCP sensitizes RCC-26 cells to TRAIL-induced apoptosis in a ROS-dependent mechanism. As supposed, flow cytometric analysis of JC-1-stained cells showed that CCCP treatment induced dissipation of ΔΨm (Fig. 8A), which was accompanied by ROS accumulation, as shown by CellROX staining (Fig. 8B). Furthermore, incubation with CCCP also increased TRAIL-induced apoptosis, evidenced by annexin V-FITC/PI staining (Fig. 8C). Both effects induced by CCCP, increased ROS accumulation and increased apoptosis induction by TRAIL, were considerably reduced by preincubation with NAC. However, in the presence of NAC, a small portion of cells was still positive for annexin V-FITC/PI, indicating that CCCP in combination with TRAIL might also, albeit to a lesser extent, induce necrosis-like cell death. Comparable with sorafenib, CCCP-induced mitochondrial depolarization was detectable 1–3 min upon treatment and independent of Bax and Bak (Fig. 7D).
Figure 8.

CCCP-induced mitochondrial depolarization and ROS accumulation sensitize RCC-26 cells to TRAIL. A, RCC-26 cells were treated with CCCP and stained with JC-1 to analyze loss of mitochondrial membrane potential. Fluorescence microscopy (top panels) and FACS analysis (bottom panels) showed loss of red fluorescence and increase in green fluorescence upon CCCP treatment in all cells, indicating loss of ΔΨm. B, RCC-26 cells were treated with CCCP alone or in combination with the free radical scavenger NAC. As shown by CellROX Green staining, CCCP-induced ROS accumulation was inhibited by NAC. C, in addition to TRAIL and CCCP, RCC-26 cells were pretreated with NAC. Induction of apoptosis was quantified by flow cytometric detection of cells positive for annexin V-FITC (phosphatidylserine exposure) and negative for PI staining (intact plasma membrane), i.e. early apoptotic, and detection of annexin V-FITC/PI positive (late apoptotic) cells. FACS measurement revealed that CCCP sensitizes cells to TRAIL-induced apoptosis. Sensitization to TRAIL was inhibited by NAC. Left panel, representative flow cytometry histograms. Right panel, mean ± S.D. of triplicates. D, HCT116-Bax−/−/Bak−/− cells were treated with CCCP for the indicated time periods and stained with JC-1. Loss of ΔΨm is detectable 1–3 min after treatment in almost all cells.
As mitochondrial depolarization is independent of Bax or Bak, we next asked whether, in RCC cells also, activation of caspase-8 and induction of apoptosis by combined treatment with TRAIL and sorafenib is independent of Bax and Bak. To this end, expression of Bax and Bak in RCC-26 cells was down-regulated by transfection with appropriate siRNAs, and cells were subsequently incubated with sorafenib and TRAIL. Despite efficient knockdown of Bax and Bak expression in RCC-26 cells (Fig. 9A), caspase-8 activation upon combined sorafenib/TRAIL treatment was not reduced (Fig. 9B), indicating that caspase-8 activation is Bax- and Bak-independent. However, despite unaltered caspase-8 activation, apoptosis induction by sorafenib/TRAIL was decreased but not completely inhibited by knockdown of Bax and Bak (Fig. 9C). In cells transfected with control siRNA, 60% of the cells were positive for annexin V-FITC staining upon sorafenib/TRAIL treatment. This was reduced to 40% upon transfection of the cells with Bax and Bak siRNA. Transfection of cells with Bak siRNA did not completely abolish Bak expression. Thus, activation of residual Bak, mediated by caspase-8-cleaved and -activated Bid, might account for the remaining induction of apoptosis. To completely block the mitochondrial amplification loop and to inhibit activation of residual Bak by tBid, we additionally down-regulated Bid expression to Bax and Bak (Fig. 10A). Similar to Bax/Bak knockdown, simultaneous knockdown of Bid, Bax, and Bak reduced, but did not completely block, sorafenib/TRAIL-induced apoptosis (Fig. 10B). This indicates that Bax/Bak-regulated events of mitochondrial apoptosis signaling only partially contribute to apoptosis mediated by the combination of sorafenib/TRAIL. Overall, induction of apoptosis upon sorafenib/TRAIL treatment appears to be rather mediated by a type I pathway, which might further boost downstream activation of the mitochondrial pathway.
Figure 9.

Caspase-8 activation and apoptosis upon combined treatment with sorafenib and TRAIL is independent of Bax and Bak. A, RCC-26 cells were transfected with non-targeting or Bax- and Bak-targeting siRNA. After 24 h, protein extracts were prepared and analyzed by Western blotting to confirm down-regulation of the targeted proteins. c, control. B, after down-regulation of Bax and Bak, cells were treated with TRAIL and sorafenib. Cells with active caspase-8 were detected by flow cytometry using the FAM-FLICA caspase-8 detection kit. Measurement revealed that knockdown of Bax and Bak did not inhibit caspase-8 activation by combined treatment of cells with sorafenib and TRAIL. C, cells were treated as in B, and apoptosis was quantified by flow cytometric detection of annexin V-FITC/PI-positive cells. Down-regulation of Bax and Bak decreased but did not prevent sorafenib/TRAIL-induced apoptosis.
Figure 10.
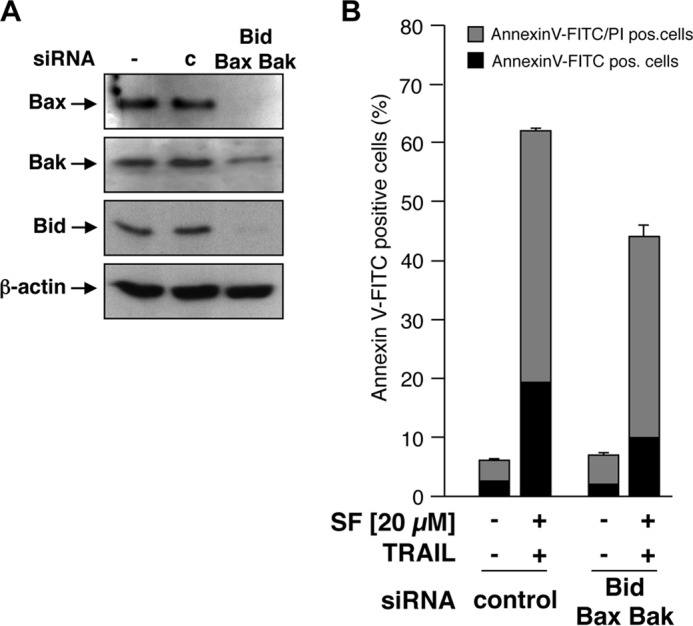
Combined knockdown of Bax, Bak, and Bid does not prevent sorafenib/TRAIL-induced apoptosis. A, RCC-26 cells were transfected with non-targeting or Bax-, Bak-, and Bid-targeting siRNA. After 24 h, protein extracts were prepared and analyzed by Western blotting to confirm the down-regulation of the targeted proteins. c, control. B, after combined down-regulation of Bax, Bak, and Bid, cells were treated with TRAIL and sorafenib, and the proportion of annexin V-FITC/PI-positive cells was detected by flow cytometry. Measurement revealed that the combined knockdown of Bax, Bak, and Bid attenuated, but did not prevent, sorafenib/TRAIL-induced apoptosis.
Discussion
Resistance to classical chemotherapy or targeted treatment modalities, including TRAIL, is the major obstacle in oncology. RCC is one of the most chemoresistant tumor types, and only the introduction of multitargeted kinase inhibitors has provided some, rather limited, therapeutic progress. Here we provide evidence for an interesting new mechanism of how sorafenib may be utilized to overcome resistance to apoptosis induction. As a model agent, we employed the death ligand TRAIL, which induces apoptosis through a Bax/Bak-controlled (type II) mitochondrial pathway in most solid tumors.
The multikinase inhibitor sorafenib is approved for the treatment of advanced hepatocellular and renal cell carcinoma and shows promising antitumor activity as a single agent. However, primary and acquired drug resistance decreases the rate and duration of tumor response and impedes survival benefits (38, 39). Nevertheless, the multiple targets of sorafenib make it an attractive choice for combination therapy with TRAIL, and, consequently, sorafenib was shown to sensitize various cancer cells to TRAIL-induced apoptosis (30, 40, 41). Here we present a new mechanism by which sorafenib overcomes the TRAIL resistance of RCC cells. Increased ROS levels, generated upon sorafenib-induced mitochondrial depolarization, enable TRAIL to activate caspase-8 and to induce cell death.
The anticancer effect of sorafenib is assumed to rely on its ability to inhibit cancer cell proliferation and to block angiogenesis through inhibition of Raf, PDGF receptor, and VEGF receptor (38). In addition, sorafenib induces apoptosis and sensitizes tumor cells to chemo- and radiotherapy by modulating STAT3 (42), Akt (43), NF-κB (29, 44), and apoptosis signaling pathways. Particularly sorafenib-mediated down-regulation of Mcl-1 seems to be crucial to sensitize cancer cells to TRAIL-induced cell death (29, 45). Down-regulation of Mcl-1, a specific Bak antagonist, lowers the apoptotic threshold and enables TRAIL to induce apoptosis via a Bak-dependent mitochondrial pathway. Hence, even TRAIL resistance caused by Bax deficiency is overcome by sorafenib-induced Mcl-1 down-regulation (31). However, RCC cells treated with sorafenib do not show consistently decreased levels of Mcl-1, indicating that, in these cells, down-regulation of Mcl-1 is unlikely to be involved in overcoming TRAIL resistance. Furthermore, the specific siRNA-mediated knockdown of Mcl-1 did not affect TRAIL resistance in RCC cells.
In this study, we further demonstrate that sorafenib sensitizes RCC cells to apoptosis induced by TRAIL through a mechanism that depends on sorafenib-induced ROS accumulation. This conclusion is supported by the prevention of sorafenib-induced ROS accumulation by NAC, which results in the inhibition of sorafenib/TRAIL-induced apoptosis. Importantly, NAC does not inhibit mitochondrial depolarization, indicating that depolarization per se is insufficient to enhance TRAIL-induced apoptosis and that, rather, generation of ROS downstream of mitochondrial depolarization provokes sensitization toward TRAIL. It has been shown that, in FADD-deficient acute lymphoblastic leukemia cells, ROS induction is a critical regulator of necroptotic cell death induced by a Smac mimetic in combination with TNFα. Consistent with the definition of necroptosis, this cell death is caspase-independent (46, 47). In contrast, ROS-mediated cell death induced by sorafenib/TRAIL is blocked by the caspase inhibitor Q-VD-OPh, indicating induction of apoptotic cell death.
ROS production has also been reported previously during sorafenib-mediated cytotoxicity in hepatocarcinoma and EBV-transformed B cells (48, 49). The increased ROS production might be a consequence of activation of a NAD(P)H oxidase (48) or sorafenib-induced inactivation of mitochondrial complex I (NADH dehydrogenase) (50). In neuroblastoma cells, down-regulation of complex I, induced by sorafenib, results in caspase- and Bcl-2 independent loss of ΔΨm that is accompanied by increased ROS levels and induction of cell death (50). In contrast to neuroblastoma cells, however, in which the decrease of ΔΨm became evident after 12 h of treatment, in RCC cells, depolarization was already detectable 1–3 min after addition of sorafenib. This almost instant depolarization makes a mechanism involving down-regulation of the NADH dehydrogenase most unlikely.
Interestingly, it has been shown that sorafenib, in contrast to the kinase inhibitors imatinib, dasatinib, and sunitinib, can directly impair mitochondrial function. Sorafenib can act as an inhibitor of the oxidative phosphorylation complexes and/or as an uncoupler of oxidative phosphorylation (51), and both mechanisms might explain the rapid sorafenib-induced mitochondrial depolarization. Although the exact mechanism of ROS induction is elusive, uncoupling of oxidative phosphorylation by sorafenib is compatible with our finding that sorafenib-mediated depolarization is independent of Bax, Bak, and opening of the mitochondrial permeability transition pore. In line with this hypothesis, uncoupling of oxidative phosphorylation is accompanied by generation of ROS and results in sensitization toward TRAIL, as shown for the classical uncoupler CCCP. CCCP sensitizes human colon carcinoma cells to TRAIL-induced apoptosis by enhancing caspase-8 activation via generation of ROS (52). ROS, in turn, modulate apoptosis signaling by assisting in the activation of initiator caspases (53). Likewise our results indicate that ROS act as upstream signaling molecules that facilitate caspase-8 activation after receptor ligation, ultimately resulting in induction of apoptosis in RCC. ROS accumulation, in contrast to cell death, is not inhibited by caspase inhibitors. ROS accumulation is therefore caspase-independent and represents an upstream event in sorafenib/TRAIL-induced apoptosis. Furthermore, sorafenib/TRAIL-triggered caspase-8 activation is inhibited by NAC, indicating that ROS accumulation plays a causal role in facilitating caspase-8 activation by TRAIL.
When caspase-8 is activated, however, additional sorafenib-induced mechanisms are likely to amplify TRAIL-induced caspase-8 activation and apoptosis in RCC cells. Such an amplification of caspase-8 activation via the mitochondrial pathway upon depolarization and ROS accumulation has been shown for TRAIL-resistant human colon carcinoma cell lines. Depolarization and ROS accumulation induced by CCCP sensitizes these cells to TRAIL-induced caspase-8 activation and apoptosis, but complete processing of caspase-8 in response to TRAIL requires a functional mitochondrial amplification loop (52). In RCC cells, the initial step in overcoming TRAIL resistance seems to be increased TRAIL-induced caspase-8 activation facilitated by sorafenib-mediated ROS accumulation, which enables the death receptors to kill cells via a type I, largely Bax/Bak-independent apoptosis pathway. However, participation of the mitochondrial cell death pathway in RCC cell lines upon sorafenib/TRAIL treatment is obvious because of the reduced induction of apoptosis upon Bax and Bak knockdown. Thus, it is likely that enhancement of the mitochondrial amplification loop, e.g. by down-regulation of Mcl-1, further boosts sorafenib/TRAIL-induced apoptosis.
Both the death ligand TRAIL and sorafenib have been proposed as promising antitumor agents, although their therapeutic use is restricted because of primary and acquired resistance of tumors. However, we show here that the combined use of TRAIL and sorafenib can overcome resistance of various RCC cell lines. Based on the presented data, we suggest a new pathway that depends on sorafenib-induced ROS accumulation, which, in turn, mediates increased caspase-8 activation (Fig. 11). This mechanism explains the sensitization of RCC cells to TRAIL-induced cell death by sorafenib. Importantly, we show that sorafenib-induced depolarization and ROS accumulation are not confined to RCC and might be more generally involved in sorafenib/TRAIL-induced apoptosis. Thus, the combined use of TRAIL and sorafenib might be a useful strategy to broaden the therapeutic efficacy of both agents. Interestingly, although our finding of sorafenib-induced ROS production in mitochondria is so far restricted to in vitro settings, elevated serum levels of advanced oxidation protein products have been recently associated with improved overall survival of sorafenib-treated patients (48).
Figure 11.
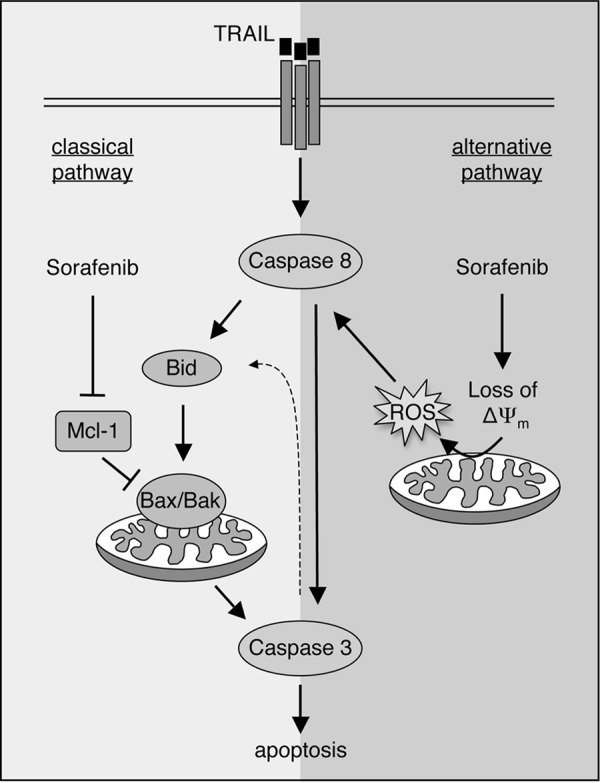
Sensitization of cancer cells toward TRAIL-induced apoptosis by sorafenib. Sensitization of cancer cells to TRAIL-induced apoptosis is mediated by a classical mechanism, including down-regulation of Mcl-1, which activates the Bax/Bak-triggered mitochondrial caspase cascade. Alternatively, as shown in this study, sorafenib can overcome TRAIL resistance by Bax/Bak-independent dissipation of the mitochondrial membrane potential, resulting in ROS accumulation and increased activation of caspase-8.
Experimental procedures
Cell culture
RCC lines were cultured as described previously (54). In brief, cells were grown in RPMI 1640 medium supplemented with 10% FCS, 100 units/ml penicillin, and 0.1 mg/ml streptomycin (Life Technologies). HCT116 wild-type cells and the isogenic double knockout subline HCT116-Bax−/−/Bak−/− were kindly provided by Dr. R. J. Youle, NIH (Bethesda, MD) (37). Cells were grown in DMEM supplemented with 10% FCS, 100,000 units/liter penicillin, and 0.1 g/liter streptomycin at 37 °C with 5% CO2 in a humidified atmosphere. Media and culture reagents were from Invitrogen.
Antibodies and reagents
Anti-Smac mAb (2954), anti-caspase-8 (1C12) mAb (9746), anti-Bid (2002), and anti-XIAP mAb (3B6) (2045) were purchased from Cell Signaling Technology, Inc. Anti-Bak mAb (clone TC102) was from Calbiochem (Merck). Anti-Bax mAb (clone YTH-2D2) was purchased from Trevigen (Gaithersburg, MD), and anti-Mcl-1 (H-260) was from Santa Cruz Biotechnology. Anti-caspase-3 antibody (AF605) was from R&D Systems (Wiesbaden-Nordenstadt, Germany), and anti-actin mAb (AC-74) was from Sigma-Aldrich (Taufkirchen, Germany). Secondary anti-rabbit, anti-goat, and anti-mouse HRP-conjugated antibodies were from Promega (Mannheim, Germany) or Southern Biotechnology Associates (Birmingham, AL). RNase A was from Carl Roth GmbH (Karlsruhe, Germany). Recombinant human TRAIL was from R&D Systems. Sorafenib (BAY 43-9006) was from Enzo Life Sciences GmbH (Lörrach, Germany). NAC was from Sigma-Aldrich. The pan-caspase inhibitor Q-VD-OPh, the caspase-3 inhibitor Z-DEVD-fmk, and the caspase-8 inhibitor Z-LETD-fmk were purchased from Merck.
Small interfering RNA
SMARTPool On-Target plus Mcl-1, Bid, Bax, Bak, and control siRNAs were purchased from Dharmacon. Transfection of cells was carried out by use of DharmaFECT transfection reagent 1 according to the instructions of the manufacturer. Down-regulation of the respective proteins was confirmed by immunoblotting 24 h after transfection.
Immunoblotting
After trypsination, cells were washed twice with ice-cold PBS and lysed in 10 mm Tris-HCl (pH 7.5), 137 mm NaCl, 1% Triton X-100, 2 mm EDTA, 1 μm pepstatin, 1 μm leupeptin, and 100 μm PMSF. Protein concentration was determined using the Thermo Scientific Pierce BCA protein assay kit (Life Technologies). Equal amounts of protein were separated by SDS-PAGE, electroblotted onto a nitrocellulose membrane, and visualized as described previously (55). For analysis of cytochrome c and SMAC release, cytosolic extracts were prepared according to a method described previously (55). Briefly, after induction of apoptosis, cells were harvested, washed in PBS, equilibrated in hypotonic buffer (20 mm HEPES (pH 7.4), 10 mm KCl, 2 mm MgCl2, and 1 mm EDTA) supplemented with 100 μm PMSF and 0.75 mg/ml digitonin (Sigma-Aldrich) and incubated on ice for 3 min. Debris was pelleted by centrifugation at 10,000 × g at 4 °C for 5 min, and the supernatant was subjected to Western blotting analysis.
Mitochondrial outer membrane permeabilization
Cells were harvested by trypsination and collected by centrifugation at 300 × g at 4 °C for 5 min. Mitochondrial outer membrane permeabilization was assessed by staining the cells with JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanin iodide, Life Technologies). JC-1 is a cationic dye that exhibits membrane potential-dependent accumulation and formation of red fluorescent J aggregates in mitochondria with a high membrane potential. At low membrane potential (upon MOMP), JC-1 shows a cytosolic green fluorescence. Mitochondrial permeability transition was analyzed by flow cytometry, and cells with increased green fluorescence were assumed to be cells with reduced ΔΨm.
Detection of apoptotic cell death by flow cytometry
DNA fragmentation was analyzed as described previously (56). Briefly, cells were harvested and collected by centrifugation at 300 × g for 5 min, washed with PBS at 4 °C, and fixed in PBS/2% (v/v) formaldehyde on ice for 30 min. After fixation, cells were incubated with ethanol/PBS (2:1, v/v) for 15 min on ice, pelleted, and resuspended in PBS containing 40 μg/ml RNase A. After incubation for 30 min at 37 °C, cells were pelleted and finally resuspended in PBS containing 50 μg/ml propidium iodide. Relative cellular DNA content was quantified using a FACScan (BD Biosciences), and the fraction of hypodiploid cells was calculated using CELLQuest software. Data are given in percent hypodiploid (subG1), which reflects the number of apoptotic cells. Alternatively, cell death was determined by staining cells with annexin V-FITC and propidium iodide (PI) (57). Briefly, cells were washed twice with cold PBS and resuspended in 10 mm HEPES (pH 7.4), 140 mm NaCl, and 2.5 mm CaCl2 at 1 × 106 cells/ml. To 100 μl of the solution (1 × 105 cells), 5 μl of annexin V-FITC (BD Biosciences) and 10 μl PI (20 mg/ml, Sigma-Aldrich) were added. Analysis was performed using a FACScan and CELLQuest software (BD Biosciences).
Detection of Bax/Bak conformational change
Activation of Bax and Bak, i.e. their conformational change and exposure of an N-terminal epitope, was detected by the use of primary antibodies specific for the Bax or Bak N-terminal domains (Bax-NT from Upstate Biotechnology and Bak-NT from Merck) and fluorescently labeled secondary antibodies. After fluorescent staining, cells were subjected to flow cytometry (58). In brief, 1 × 105 cells were harvested, washed in PBS, and fixed for 30 min with 0.5% paraformaldehyde in PBS. After washing, the cells were resuspended in PBS supplemented with 1% FCS and 0.1% saponin and incubated with the Bax-NT- or Bak-NT-specific antibody. Thereafter, cells were washed in PBS and incubated with a secondary FITC-labeled goat-anti-rabbit or goat-anti-mouse IgG (H + L) antibodies (Jackson ImmunoResearch Laboratories, Newmarket, UK) at a final concentration of 1 μg/ml for 30 min at 4 °C in the dark. After washing and resuspension, cells were immediately analyzed by flow cytometry.
Caspase-8 activation
Cells with active caspase-8 were detected using the FAM-FLICATM caspase-8 assay kit (Immunochemistry Technology) according to the instructions of the manufacturer. This kit employs a carboxyfluorescein-labeled fluoromethyl ketone peptide inhibitor of caspase-8 (FAM-LETD-fmk) that is cell-permeable and a non-cytotoxic fluorochrome.
Detection of ROS
ROS in cells were analyzed by use of the fluorogenic CellROX® Green reagent (Invitrogen, Life Technologies) according to the instructions of the manufacturer. In brief, 1 × 105 cells were plated in a 12-well plate and incubated with the indicated drugs. Upon treatment, CellROX® reagent was added to a final concentration of 5 μm, and cells were incubated for 60 min at 37 °C. Microscopy images were taken using an Axiovert 200 fluorescence microscope (Carl Zeiss, Inc.) equipped with a Hamamatsu ORCA-ER digital camera using Openlab software (Improvision).
Author contributions
B. G. performed experiments, analyzed experiments, and wrote the manuscript. Anja Richter and Antje Richter performed and analyzed experiments. R. P. analyzed data and reviewed the manuscript. F. E. performed experiments, analyzed experiments, and wrote the manuscript. K. S. O. analyzed data and wrote the manuscript. P. T. D. supervised the work, analyzed experiments, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by the Stiftung Urologische Forschung Berlin, Deutsche Krebshilfe, and Deutsches Konsortium für Translationale Krebsforschung (DKTK) - Deutsches Krebsforschungszentrum. The authors declare that they have no conflicts of interest with the contents of this article.
- TRAIL
- tumor necrosis factor-related apoptosis-inducing ligand
- FADD
- Fas-associated protein with death domain
- XIAP
- X-linked inhibitor of apoptosis protein
- MOMP
- mitochondrial outer membrane permeabilization
- BH
- Bcl-2 homology
- RCC
- renal cell carcinoma
- ΔΨm
- mitochondrial membrane potential
- ROS
- reactive oxygen species
- NAC
- N-acetylcysteine
- Z-LETD-fmk
- benzyloxycarbonyl-LETD-fluoromethyl ketone
- Z-DEVD-fmk
- benzyloxycarbonyl-DEVD-fluoromethyl ketone
- PI
- propidium iodide
- MPTP
- mitochondrial permeability transition pore
- CsA
- cyclosporin A
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- JC-1
- 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanin iodide
- NT
- N-terminal
- SF
- sorafenib
- FAM-LETD-fmk
- carboxyfluorescein LETD fluoromethyl ketone
- Q-VD-OPh
- quinolyl-valyl-O-methylaspartyl-[-2,6-difluorophenoxy]-methyl ketone.
References
- 1. Falschlehner C., Ganten T. M., Koschny R., Schaefer U., and Walczak H. (2009) TRAIL and other TRAIL receptor agonists as novel cancer therapeutics. Adv. Exp. Med. Biol. 647, 195–206 [DOI] [PubMed] [Google Scholar]
- 2. Gonzalvez F., and Ashkenazi A. (2010) New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 29, 4752–4765 [DOI] [PubMed] [Google Scholar]
- 3. Yang A., Wilson N. S., and Ashkenazi A. (2010) Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical translation. Curr. Opin. Cell Biol. 22, 837–844 [DOI] [PubMed] [Google Scholar]
- 4. Chaudhary P. M., Eby M., Jasmin A., Bookwalter A., Murray J., and Hood L. (1997) Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity 7, 821–830 [DOI] [PubMed] [Google Scholar]
- 5. Kischkel F. C., Lawrence D. A., Chuntharapai A., Schow P., Kim K. J., and Ashkenazi A. (2000) Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 12, 611–620 [DOI] [PubMed] [Google Scholar]
- 6. Suzuki Y., Nakabayashi Y., and Takahashi R. (2001) Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. U.S.A. 98, 8662–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Green D. R., and Kroemer G. (2004) The pathophysiology of mitochondrial cell death. Science 305, 626–629 [DOI] [PubMed] [Google Scholar]
- 8. Du C., Fang M., Li Y., Li L., and Wang X. (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42 [DOI] [PubMed] [Google Scholar]
- 9. Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K. J., Debatin K. M., Krammer P. H., and Peter M. E. (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17, 1675–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jost P. J., Grabow S., Gray D., McKenzie M. D., Nachbur U., Huang D. C., Bouillet P., Thomas H. E., Borner C., Silke J., Strasser A., and Kaufmann T. (2009) XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460, 1035–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gillissen B., Richter A., Richter A., Overkamp T., Essmann F., Hemmati P. G., Preissner R., Belka C., and Daniel P. T. (2013) Targeted therapy of the XIAP/proteasome pathway overcomes TRAIL-resistance in carcinoma by switching apoptosis signaling to a Bax/Bak-independent “type I” mode. Cell Death Dis. 4, e643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zou H., Li Y., Liu X., and Wang X. (1999) An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 274, 11549–11556 [DOI] [PubMed] [Google Scholar]
- 13. Yamada H., Tada-Oikawa S., Uchida A., and Kawanishi S. (1999) TRAIL causes cleavage of bid by caspase-8 and loss of mitochondrial membrane potential resulting in apoptosis in BJAB cells. Biochem. Biophys. Res. Commun. 265, 130–133 [DOI] [PubMed] [Google Scholar]
- 14. Luo X., Budihardjo I., Zou H., Slaughter C., and Wang X. (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94, 481–490 [DOI] [PubMed] [Google Scholar]
- 15. Slee E. A., Keogh S. A., and Martin S. J. (2000) Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ. 7, 556–565 [DOI] [PubMed] [Google Scholar]
- 16. Daniel P. T., Schulze-Osthoff K., Belka C., and Güner D. (2003) Guardians of cell death: the Bcl-2 family proteins. Essays Biochem. 39, 73–88 [DOI] [PubMed] [Google Scholar]
- 17. van Delft M. F., and Huang D. C. (2006) How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 16, 203–213 [DOI] [PubMed] [Google Scholar]
- 18. Bender T., and Martinou J. C. (2013) Where killers meet: permeabilization of the outer mitochondrial membrane during apoptosis. Cold Spring Harb. Perspect. Biol. 5, a011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmelz K., Wieder T., Tamm I., Müller A., Essmann F., Geilen C. C., Schulze-Osthoff K., Dörken B., and Daniel P. T. (2004) Tumor necrosis factor α sensitizes malignant cells to chemotherapeutic drugs via the mitochondrial apoptosis pathway independently of caspase-8 and NF-κB. Oncogene 23, 6743–6759 [DOI] [PubMed] [Google Scholar]
- 20. Marini P., Schmid A., Jendrossek V., Faltin H., Daniel P. T., Budach W., and Belka C. (2005) Irradiation specifically sensitises solid tumour cell lines to TRAIL mediated apoptosis. BMC Cancer 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keane M. M., Rubinstein Y., Cuello M., Ettenberg S. A., Banerjee P., Nau M. M., and Lipkowitz S. (2000) Inhibition of NF-κB activity enhances TRAIL mediated apoptosis in breast cancer cell lines. Breast Cancer Res. Treat. 64, 211–219 [DOI] [PubMed] [Google Scholar]
- 22. Brooks A. D., Jacobsen K. M., Li W., Shanker A., and Sayers T. J. (2010) Bortezomib sensitizes human renal cell carcinomas to TRAIL apoptosis through increased activation of caspase-8 in the death-inducing signaling complex. Mol. Cancer Res. 8, 729–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen K. F., Yeh P. Y., Hsu C., Hsu C. H., Lu Y. S., Hsieh H. P., Chen P. J., and Cheng A. L. (2009) Bortezomib overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance in hepatocellular carcinoma cells in part through the inhibition of the phosphatidylinositol 3-kinase/Akt pathway. J. Biol. Chem. 284, 11121–11133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inoue S., Mai A., Dyer M. J., and Cohen G. M. (2006) Inhibition of histone deacetylase class I but not class II is critical for the sensitization of leukemic cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 66, 6785–6792 [DOI] [PubMed] [Google Scholar]
- 25. Huang S., and Sinicrope F. A. (2008) BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res. 68, 2944–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mott J. L., Bronk S. F., Mesa R. A., Kaufmann S. H., and Gores G. J. (2008) BH3-only protein mimetic obatoclax sensitizes cholangiocarcinoma cells to Apo2L/TRAIL-induced apoptosis. Mol. Cancer Ther. 7, 2339–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang S., and Sinicrope F. A. (2010) Sorafenib inhibits STAT3 activation to enhance TRAIL-mediated apoptosis in human pancreatic cancer cells. Mol. Cancer Ther. 9, 742–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen K. F., Tai W. T., Liu T. H., Huang H. P., Lin Y. C., Shiau C. W., Li P. K., Chen P. J., and Cheng A. L. (2010) Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin. Cancer Res. 16, 5189–5199 [DOI] [PubMed] [Google Scholar]
- 29. Ricci M. S., Kim S. H., Ogi K., Plastaras J. P., Ling J., Wang W., Jin Z., Liu Y. Y., Dicker D. T., Chiao P. J., Flaherty K. T., Smith C. D., and El-Deiry W. S. (2007) Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 12, 66–80 [DOI] [PubMed] [Google Scholar]
- 30. Abdulghani J., Allen J. E., Dicker D. T., Liu Y. Y., Goldenberg D., Smith C. D., Humphreys R., and El-Deiry W. S. (2013) Sorafenib sensitizes solid tumors to Apo2L/TRAIL and Apo2L/TRAIL receptor agonist antibodies by the Jak2-Stat3-Mcl1 axis. PLoS ONE 8, e75414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gillissen B., Wendt J., Richter A., Richter A., Müer A., Overkamp T., Gebhardt N., Preissner R., Belka C., Dörken B., and Daniel P. T. (2010) Endogenous Bak inhibitors Mcl-1 and Bcl-xL: differential impact on TRAIL resistance in Bax-deficient carcinoma. J. Cell Biol. 188, 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilhelm S., Carter C., Lynch M., Lowinger T., Dumas J., Smith R. A., Schwartz B., Simantov R., and Kelley S. (2006) Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5, 835–844 [DOI] [PubMed] [Google Scholar]
- 33. Iyer R., Fetterly G., Lugade A., and Thanavala Y. (2010) Sorafenib: a clinical and pharmacologic review. Expert Opin. Pharmacother. 11, 1943–1955 [DOI] [PubMed] [Google Scholar]
- 34. Kane R. C., Farrell A. T., Saber H., Tang S., Williams G., Jee J. M., Liang C., Booth B., Chidambaram N., Morse D., Sridhara R., Garvey P., Justice R., and Pazdur R. (2006) Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 12, 7271–7278 [DOI] [PubMed] [Google Scholar]
- 35. Bracarda S., Caserta C., Sordini L., Rossi M., Hamzay A., and Crinò L. (2007) Protein kinase inhibitors in the treatment of renal cell carcinoma: sorafenib. Ann. Oncol. 18, vi22–25 [DOI] [PubMed] [Google Scholar]
- 36. Hoffmann J. C., Pappa A., Krammer P. H., and Lavrik I. N. (2009) A new C-terminal cleavage product of procaspase-8, p30, defines an alternative pathway of procaspase-8 activation. Mol. Cell Biol. 29, 4431–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang C., and Youle R. J. (2012) Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1's inhibitory effect on Bak. Oncogene 31, 3177–3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ibrahim N., Yu Y., Walsh W. R., and Yang J. L. (2012) Molecular targeted therapies for cancer: sorafenib mono-therapy and its combination with other therapies (review). Oncol. Rep. 27, 1303–1311 [DOI] [PubMed] [Google Scholar]
- 39. Berasain C. (2013) Hepatocellular carcinoma and sorafenib: too many resistance mechanisms? Gut 62, 1674–1675 [DOI] [PubMed] [Google Scholar]
- 40. Chen K. F., Chen H. L., Shiau C. W., Liu C. Y., Chu P. Y., Tai W. T., Ichikawa K., Chen P. J., and Cheng A. L. (2013) Sorafenib and its derivative SC-49 sensitize hepatocellular carcinoma cells to CS-1008, a humanized anti-TNFRSF10B (DR5) antibody. Br. J. Pharmacol. 168, 658–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nojiri K., Sugimoto K., Shiraki K., Tameda M., Inagaki Y., Ogura S., Kasai C., Kusagawa S., Yoneda M., Yamamoto N., Takei Y., Nobori T., and Ito M. (2013) Sorafenib and TRAIL have synergistic effect on hepatocellular carcinoma. Int. J. Oncol. 42, 101–108 [DOI] [PubMed] [Google Scholar]
- 42. Yang F., Brown C., Buettner R., Hedvat M., Starr R., Scuto A., Schroeder A., Jensen M., and Jove R. (2010) Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol. Cancer Ther 9, 953–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oh S. J., Erb H. H., Hobisch A., Santer F. R., and Culig Z. (2012) Sorafenib decreases proliferation and induces apoptosis of prostate cancer cells by inhibition of the androgen receptor and Akt signaling pathways. Endocr. Relat. Cancer 19, 305–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuo Y. C., Lin W. C., Chiang I. T., Chang Y. F., Chen C. W., Su S. H., Chen C. L., and Hwang J. J. (2012) Sorafenib sensitizes human colorectal carcinoma to radiation via suppression of NF-κB expression in vitro and in vivo. Biomed. Pharmacother. 66, 12–20 [DOI] [PubMed] [Google Scholar]
- 45. Meng X. W., Lee S. H., Dai H., Loegering D., Yu C., Flatten K., Schneider P., Dai N. T., Kumar S. K., Smith B. D., Karp J. E., Adjei A. A., and Kaufmann S. H. (2007) Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization. J. Biol. Chem. 282, 29831–29846 [DOI] [PubMed] [Google Scholar]
- 46. Laukens B., Jennewein C., Schenk B., Vanlangenakker N., Schier A., Cristofanon S., Zobel K., Deshayes K., Vucic D., Jeremias I., Bertrand M. J., Vandenabeele P., and Fulda S. (2011) Smac mimetic bypasses apoptosis resistance in FADD- or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia 13, 971–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schenk B., and Fulda S. (2015) Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 34, 5796–5806 [DOI] [PubMed] [Google Scholar]
- 48. Coriat R., Nicco C., Chéreau C., Mir O., Alexandre J., Ropert S., Weill B., Chaussade S., Goldwasser F., and Batteux F. (2012) Sorafenib-induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo. Mol. Cancer Ther. 11, 2284–2293 [DOI] [PubMed] [Google Scholar]
- 49. Park G. B., Choi Y., Kim Y. S., Lee H. K., Kim D., and Hur D. Y. (2014) ROS-mediated JNK/p38-MAPK activation regulates Bax translocation in sorafenib-induced apoptosis of EBV-transformed B cells. Int. J. Oncol. 44, 977–985 [DOI] [PubMed] [Google Scholar]
- 50. Bull V. H., Rajalingam K., and Thiede B. (2012) Sorafenib-induced mitochondrial complex I inactivation and cell death in human neuroblastoma cells. J. Proteome Res. 11, 1609–1620 [DOI] [PubMed] [Google Scholar]
- 51. Will Y., Dykens J. A., Nadanaciva S., Hirakawa B., Jamieson J., Marroquin L. D., Hynes J., Patyna S., and Jessen B. A. (2008) Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol Sci. 106, 153–161 [DOI] [PubMed] [Google Scholar]
- 52. Izeradjene K., Douglas L., Tillman D. M., Delaney A. B., and Houghton J. A. (2005) Reactive oxygen species regulate caspase activation in tumor necrosis factor-related apoptosis-inducing ligand-resistant human colon carcinoma cell lines. Cancer Res. 65, 7436–7445 [DOI] [PubMed] [Google Scholar]
- 53. Perez-Cruz I., Cárcamo J. M., and Golde D. W. (2007) Caspase-8 dependent TRAIL-induced apoptosis in cancer cell lines is inhibited by vitamin C and catalase. Apoptosis 12, 225–234 [DOI] [PubMed] [Google Scholar]
- 54. Sturm I., Stephan C., Gillissen B., Siebert R., Janz M., Radetzki S., Jung K., Loening S., Dörken B., and Daniel P. T. (2006) Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 13, 619–627 [DOI] [PubMed] [Google Scholar]
- 55. von Haefen C., Wieder T., Essmann F., Schulze-Osthoff K., Dörken B., and Daniel P. T. (2003) Paclitaxel-induced apoptosis in BJAB cells proceeds via a death receptor-independent, caspases-3/-8-driven mitochondrial amplification loop. Oncogene 22, 2236–2247 [DOI] [PubMed] [Google Scholar]
- 56. Gillissen B., Essmann F., Hemmati P. G., Richter A., Richter A., Oztop I., Chinnadurai G., Dörken B., and Daniel P. T. (2007) Mcl-1 determines the Bax dependency of Nbk/Bik-induced apoptosis. J. Cell Biol. 179, 701–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gillissen B., Essmann F., Graupner V., Stärck L., Radetzki S., Dörken B., Schulze-Osthoff K., and Daniel P. T. (2003) Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J. 22, 3580–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hemmati P. G., Müer A., Gillissen B., Overkamp T., Milojkovic A., Wendt J., Dörken B., and Daniel P. T. (2010) Systematic genetic dissection of p14ARF-mediated mitochondrial cell death signaling reveals a key role for p21CDKN1 and the BH3-only protein Puma/bbc3. J. Mol. Med. 88, 609–622 [DOI] [PubMed] [Google Scholar]