

Abstract
Disulfide bonds contribute to protein stability, activity, and folding in a variety of proteins, including many involved in bacterial virulence such as toxins, adhesins, flagella, and pili, among others. Therefore, inhibitors of disulfide bond formation enzymes could have profound effects on pathogen virulence. In the Escherichia coli disulfide bond formation pathway, the periplasmic protein DsbA introduces disulfide bonds into substrates, and then the cytoplasmic membrane protein DsbB reoxidizes DsbA's cysteines regenerating its activity. Thus, DsbB generates a protein disulfide bond de novo by transferring electrons to the quinone pool. We previously identified an effective pyridazinone-related inhibitor of DsbB enzymes from several Gram-negative bacteria. To map the protein residues that are important for the interaction with this inhibitor, we randomly mutagenized by error-prone PCR the E. coli dsbB gene and selected dsbB mutants that confer resistance to this drug using two approaches. We characterized in vivo and in vitro some of these mutants that map to two areas in the structure of DsbB, one located between the two first transmembrane segments where the quinone ring binds and the other located in the second periplasmic loop of DsbB, which interacts with DsbA. In addition, we show that a mutant version of a protein involved in lipopolysaccharide assembly, lptD4213, is synthetically lethal with the deletion of dsbB as well as with DsbB inhibitors. This finding suggests that drugs decreasing LptD assembly may be synthetically lethal with inhibitors of the Dsb pathway, potentiating the antibiotic effects.
Keywords: bacteria, bacterial genetics, disulfide, Escherichia coli (E. coli), inhibition mechanism, lptD, pyridazinone, redox, synthetic lethality
Introduction
Protein disulfide bonds are sulfur-sulfur chemical bonds that result from an oxidative process in which two electrons are removed from a protein, linking non-adjacent cysteines of the protein. Disulfide bonds contribute to protein stability, activity, and folding (1, 2). In bacteria, proteins containing structural disulfide bonds are rarely, if at all, found in cytoplasmic compartments; they are usually present in the cell envelope or the extracellular milieu (1). Many proteins involved in bacterial virulence (such as toxins, adhesins, flagella, fimbriae, pili, and types II and III secretion systems) require disulfide bonds (3). Pathways involved in catalyzing disulfide bond formation are therefore attractive targets for identifying small molecule inhibitors, because loss of such systems can undermine the activity of numerous bacterial virulence factors as do null mutations of the genes for these enzymes (4–11).
The enzymes that promote formation of protein disulfide bonds in Gram-negative bacteria are in the cell envelope. The periplasmic enzyme DsbA, a member of the thioredoxin family, oxidizes pairs of cysteines in substrate proteins through its Cys-Xaa-Xaa-Cys active site (12). The resulting reduced DsbA is reoxidized by the cytoplasmic membrane protein DsbB, regenerating DsbA's activity (13). DsbB itself is reoxidized by membrane-embedded quinones, from which electrons are transferred to the electron transport chain (14). However, in many of the Actinobacteria and Cyanobacteria the membrane protein VKOR (vitamin K epoxide reductase) instead of DsbB is required for the reoxidization of DsbA (15). Although VKOR has no overall amino acid sequence homology with DsbB, both proteins encode two extracytoplasmic soluble domains containing essential pairs of cysteines and are capable of reoxidizing DsbA fundamentally by the same mechanism (15, 16).
We have previously generated a methodology for identifying specific inhibitors of both bacterial DsbBs and a VKOR that is based on the functional homology between the two proteins (17). The assay for inhibition of disulfide bond formation utilizes a disulfide-sensitive β-galactosidase (β-Galdbs)4 assay. This approach allowed us to identify inhibitors of either enzyme by a single high-throughput screening procedure. By this approach, we have found a family of pyridazinone-related molecules that are effective inhibitors of DsbB proteins of several Gram-negative bacteria but do not inhibit a bacterial VKOR.
Because we have sought to develop DsbB inhibitors as antivirulents/antibiotics, we wanted to understand how resistance to these compounds might arise in vivo as well as how pyridazinones inhibit DsbB. To this end, we report here mutants of DsbB that confer resistance to that inhibition. We used two methods for direct selection of spontaneous mutants of DsbB resistant to one of the strongest pyridazinone inhibitors. These selections have failed to yield any mutants that have altered DsbB. However, when we randomly mutagenize by error-prone PCR a dsbB gene, which is carried on a high copy number plasmid, DsbB mutants resistant to our inhibitors can be isolated. Characterization of these DsbB mutant proteins shows that they all exhibit lower affinity toward ubiquinone and menadione, and two of them show higher turnover numbers. Our studies suggest that resistance of DsbB to pyridazinone inhibitors is difficult to obtain by spontaneous selections perhaps due to the effects of inhibitor-resistant mutations on the normal functioning of DsbB. The location within the DsbB protein of the amino acid changes that do confer resistance provides suggestions as to the mechanism of inhibition and regions of the protein that influence quinone binding.
Results
Selection of mutations that confer resistance to compound 12, a pyridazinone inhibitor of DsbB
We have developed a genetic selection for mutants resistant to inhibitors of DsbB that uses an Escherichia coli strain further sensitized to the inhibitor by the presence of an additional mutation (lptD4213). In Gram-negative bacteria, a set of lpt genes encodes proteins required for the transport and assembly of lipopolysaccharides (LPS) into the outer leaflet of the outer membrane. The Lpt proteins are essential for E. coli growth. LptD is an outer membrane β-barrel protein, which requires two disulfide bonds for its proper assembly and function, and it is involved in the last steps of LPS assembly (Fig. 1A) (18). Despite the essentiality of LptD, strains lacking a functional disulfide bond formation pathway remain viable under aerobic conditions presumably because background oxidation can lead to sufficient spontaneous disulfide bond formation in LptD and other essential proteins (19). We considered the possibility that a strain carrying the lptD4213 mutant allele (LptD4213) (20) might be hypersensitive to a loss of DsbA or DsbB. This mutant LptD, which lacks residues 330–352, displays major defects in protein maturation and assembly, and strains carrying such an allele show increased membrane permeability to detergents, bile salts, and antibiotics (21–24). In such a compromised strain, the loss of the disulfide bond machinery might be lethal.
Figure 1.

Synthetic lethality of dsbB and lptD4213. A, disulfide bond formation and Lps transport pathways converge because LptD requires two non-consecutive disulfide bonds that are essential for LptD assembly and functioning. Blue arrows show the process flow of LptD secretion to assembly. Black bold arrows show the flow of electrons in the disulfide bond formation pathway. SH, thiol groups representing cysteines; S–S, disulfide bonded cysteines; red, reduced; ox, oxidized; UQ, ubiquinone; Lps, lipopolysaccharides; OM, outer membrane; IM, inner membrane. B, growth on LB-Miller and M63 agar minimal media of lptDWTdsbBWT (HK295 strain), lptD4213dsbBWT (CL337 strain), and lptD4213ΔdsbB dsbBPBAD (CL380 strain). M63 glucose cultures with an A600 of 0.5 were serially diluted and spotted in the indicated agar media (10 μl). 0.2% arabinose was added to induce dsbB expression in CL380 strain. Plates were incubated for 2 days at 37 °C. C, structure of compound 12. D, growth on M63 minimal media broth in the presence of compound 12 of strains: HK295 (blue circles), CL337 (black circles), and CL380 (red circles). Values represent the average of at least three independent experiments with 95% confidence intervals in parentheses.
As a first step in testing for this potential synthetic lethality, we introduced a plasmid with a regulatable copy of dsbB cloned under the arabinose promoter (pCL67) into a dsbB+ strain encoding the lptD4213 allele (CL337 strain). We then attempted to transduce a deletion of the genomic copy of dsbB into the lptD4213 strain. Although ΔdsbB transductants were readily obtained when a second copy of dsbB was present, we were not able to isolate transductants in the strain encoding only one copy of dsbB unless cystine was added to the medium, which yielded a lower frequency than having two dsbB copies. Cystine is an oxidant that can mediate disulfide bond formation in the absence of the disulfide bond formation pathway (13). Furthermore, the strain carrying the plasmid with dsbB under an arabinose promoter (lptD4213ΔdsbB dsbBPBAD, CL380 strain) was able to grow on LB, although it did not grow on minimal media unless 0.2% arabinose was added (Fig. 1B). Thus, dsbB and lptD4213 are a synthetically lethal combination.
We then asked whether the lptD4213 mutant strain was sensitive to a particularly potent E. coli DsbB inhibitor, compound 12 (4,5-dichloro-2-[2-chlorophenyl]methylpiridazin-3-one, Fig. 1C). We have shown that compound 12 forms a covalent bond with the second cysteine of DsbB (Cys-44) (17). Ordinarily, quinones, the direct source of oxidation of DsbB, form a charge-transfer complex with Cys-44 of DsbB during the process of electron transfer between DsbA and DsbB (25). We have proposed that the inhibition of DsbB activity by pyridazinone compounds, including compound 12, results from the competition with quinone for the quinone-binding site leading to the covalent reaction with Cys-44, thus inactivating the protein (17). We observed that, unlike the strain with wild type lptD, the lptD4213 strain was highly sensitive to compound 12 as demonstrated by the inhibition of growth in a concentration-dependent manner (black circles, Fig. 1D). Because we have demonstrated that compound 12 targets DsbB (by interfering with DsbA reduction, exhibits a decrease in motility and an increase in the disulfide bond-sensitive β-galactosidase) (17), these data also indicated that the combination of the lptD4213 and dsbB inhibition results in a synthetic lethal interaction. In addition, the conditionally lethal strain lptD4213ΔdsbB dsbBPBAD was even more sensitive to compound 12 when no arabinose was present in liquid minimal media where disulfide bond formation is partly dependent on air oxidation (red circles, Fig. 1D).
The findings above suggested that selecting for growth of a strain that contains the lptD4213 allele and is exposed to the DsbB inhibitor, compound 12, could yield inhibitor-resistant mutants. We therefore plated the lptD4213 strain on M63 minimal media with 10 μm compound 12, a concentration of drug ∼10-fold higher than the minimal inhibitory concentration (MIC). Although mutants were obtained at a very low frequency (∼10−8) when exposed to the compound, none of them mapped to the dsbB gene but rather to the gene bamB. Twenty two of 51 colonies analyzed by PCR yielded a larger than expected product for the bamB region, and the sequence of all these indicated an insertion of an IS1 element in the gene. Whole-genome sequencing was performed in three of the colonies in which the bamB product was similar in size to wild type. Two of these encoded mutations within bamB (BamBE240* and BamBΔ252–255). Both mutations, bamB::IS1 and BamBΔ252–255, are known to be loss of function mutants of BamB that confer similar phenotypes (22). Therefore, these mutations most likely inactivated the outer membrane lipoprotein BamB, a scenario known to bypass the assembly defect of LptD4213 (22, 26–28).
Selection of mutations that confer resistance to compound 12 by PCR mutagenesis of the dsbB gene
Because our initial selection for dsbB mutants resistant to compound 12 did not yield any mutations in that gene, we decided to use the same LptD4213 strain to select for mutants resistant to compound 12 using a randomly mutagenized dsbB library. To do this, we mutagenized dsbB via error-prone PCR and cloned the resultant PCR products into a plasmid in which dsbB expression is under the control of an IPTG-inducible promoter. This pool of plasmids was then transformed into the conditionally lethal strain lptD4213ΔdsbB dsbBPBAD selecting for the presence of the plasmid using the antibiotic marker. The transformation yielded ∼4,500 independent colonies carrying both a plasmid with an arabinose-inducible wild type dsbB and a plasmid with an IPTG-inducible mutated dsbB. The colonies were scraped up, pooled together, and plated on selection plates of M63 glucose with 10 μm compound 12, which is ∼10-fold higher than the MIC observed for the strain carrying the two plasmids expressing wild type dsbB. Because glucose represses transcription of wild type dsbB from the PBAD promoter, these conditions select for resistant DsbBs expressed from the mutant library. We obtained 20 colonies and sequenced only the mutagenized dsbB gene from the IPTG-inducible plasmid (see “Materials and methods”). We found that 9 of 20 colonies (45%) had mutations in DsbB (Fig. 2A) and 6 of these 9 colonies encoded a change of Leu-25 to Pro in combination with a second mutation in residues Gln-134 or Glu-141, both located in the periplasmic loop that interacts with DsbA (Phe-64–Gly-65) just after Cys-130, which attacks the disulfide of Cys-41–Cys-44 of DsbB (29). However, 11 of 20 colonies (55%) did not have mutations in the mutagenized dsbB gene, and 5 of these 11 encoded mutations in the trc promoter region, possibly leading to increased DsbB expression.
Figure 2.

Compound 12-resistant mutations using two different selections. A, nucleotide changes found in dsbB after selection of LptD4213 growth on aerobic minimal media in the presence of 10 μm compound 12 (top). Nucleotide changes found in dsbB after selection on anaerobic minimal media in the presence of 2 μm compound 12 (bottom). The mutations studied in this work are indicated with an orange circle. B, location of the mutations (orange) in the structure of the DsbA-DsbB complex. DsbA is shown in green, DsbB in cyan, Cys-44 in red, and ubiquinone-1 (UQ1) in purple. PyMOL was used to visualize the structure (2ZUP) of the crystallized complex when Cys-30 of DsbA is forming a disulfide bond with Cys-104 of DsbB and Cys-41 and Cys-44 of DsbB are disulfide bonded. C, α-DsbB immunoblot analysis of strains carrying dsbB at λ att site under the control of trc204 promoter (CL591 to CL596 strains). Cells were grown for 4 h in M63 minimal medium with 0.2% glucose and 1 mm IPTG to induce expression of DsbB. Cells were TCA-precipitated, and protein pellets were resuspended in 100 mm Tris, 1% SDS buffer. β-Mercaptoethanol was used to reduce the proteins. 10 μg of total protein samples were loaded in 12% acrylamide gel. α-RpoA was used as a loading control. The relative amount was calculated using arbitrary levels given by Image Lab 5.2 software.
Anaerobic selection of mutations that confer resistance to compound 12
The Dsb pathway is not essential for aerobic growth of E. coli. However, under anaerobic conditions, dsbA and dsbB mutants do not grow.5 Compound 12 inhibits anaerobic growth of an E. coli wild type strain at 1 μm (17). We therefore sought to isolate mutants resistant to this inhibitor using a selection for anaerobic growth in the presence of 2 μm compound 12. We again observed that spontaneous resistant mutations arose at a very low frequency (∼10−7), and none of them mapped to the dsbB gene.5 However, whole-genome sequencing of four of these resistant mutants indicated that all of them encoded mutations in the gene encoding thioredoxin reductase (TrxBP16L, TrxBD287Y, TrxBS143F, and TrxBΔ231–236, V237I). TrxB is a critical component in the disulfide bond isomerization pathway, and mutations in this pathway have been shown to partially restore disulfide bond formation (30).
We again made use of the same library of plasmids containing the PCR-mutagenized dsbB and transformed them into an E. coli ΔdsbB strain, selecting aerobically for the presence of the plasmid using the antibiotic marker. The transformation yielded ∼3,000 independent colonies. This mutant pool was plated anaerobically on solid media containing M63 glucose with 40 mm fumarate, 2 μm compound 12 and solidified with 1% agarose. This concentration of compound 12 is twice the MIC normally seen under these conditions. From this selection, we isolated 82 resistant colonies and sequenced the dsbB gene of each (Fig. 2A). Most (92%) encoded mutant dsbB alleles. The most frequently isolated mutation was DsbBL25P similar to our lptD4213 selection, which could indicate a mutational hot spot that caused enrichment for this mutant in our library or a more effective resistance.
Characterization of five DsbB mutants
We observed that the mutations encoding resistance to compound 12 localized to two regions in the structure of DsbB, the quinone-binding site in the region of the first two transmembrane helices of DsbB and a segment of the periplasmic loop of the protein that interacts with DsbA during DsbA-DsbB complex formation (Fig. 2B).
We selected five of the mutants to study further (Fig. 3A) as follows: L25P (which was found in two different selections), A29V, K39E, P100S, and F106L, which included alterations of the two distinct regions, i.e. near the cysteines that bind to quinone and near the cysteines that bind to DsbA located in the periplasmic loop. We assessed the DsbB levels in the mutants to verify that the resistance to the drug was not due to an increased amount of DsbB. Four of the five mutants showed no difference in the amount of DsbB expression when 1 mm IPTG is added (Fig. 2C). The K39E mutant exhibited a 2-fold increase in DsbB levels for reasons that are not clear.
Figure 3.

Visualization of mutations in DsbB structure and binding prediction of pyridazinone inhibitors (compound 12). A, comparison of mutated residues in the structure of DsbB and in relation to ubiquinone. B, overlap of ubiquinone or menadione and compound 12 in the structure of DsbB. Residues studied in this work are highlighted in orange, DsbA in green, DsbB in cyan, Cys-44 in red, and ubiquinone-1 in purple. PyMOL was used to visualize the structure (2ZUP) of the crystallized complex.
To gain insights into the resistance displayed by DsbB mutants, we purified the proteins and analyzed their enzyme kinetics using an ubiquinone reduction assay (31). We observed that although the affinity toward ubiquinone (Km) of the wild type enzyme is 0.9 μm, the affinity of all five mutants exhibited 2–10-fold higher Km values (Table 1, 4th column), suggesting that the mutations directly impacted the binding of the ubiquinone substrate. Only DsbBK39E and DsbBF106L mutants showed 2-fold higher turnover rates (kcat) than the wild type enzyme (Table 1, 2nd column). We also tested DsbB mutants in a menadione (vitamin K3) reduction assay because menaquinones are used primarily during anaerobic growth (14). Similarly to ubiquinone, all mutants displayed higher Km values for menadione, with the greatest increases observed in DsbBL25P, DsbBA29V, and DsbBK39E (about 2–5-fold increases; Table 1, 5th column). In terms of turnover rate using menadione (Table 1, 3rd column), mutants DsbBL25P, DsbBP100S, and DsbBF106L displayed about one-third of the wild type rate, whereas DsbBK39E shows a 2-fold lower rate than wild type enzyme. Overall, the catalytic efficiency (kcat/Km) of the mutants was lower than wild type (Table 1, 6th and 7th columns).
Table 1.
In vitro and in vivo properties of DsbB mutants
Values represent the average of at least two independent experiments ± S.E. or the 95% confidence intervals in parentheses.
| Mutant |
kcata |
Kmb |
kcat/Km |
IC50 of compound 12 |
||||
|---|---|---|---|---|---|---|---|---|
| Ubiquinone | Menadione | Ubiquinone | Menadione | Ubiquinone | Menadione | In vitroc | In vivod | |
| μm | μm | |||||||
| DsbBWT | 2.8 ± 0.07 | 1.9 ± 0.05 | 0.94 ± 0.1 | 35.8 ± 2.8 | 2.99 | 5.3 × 10−2 | 0.033 (0.029–0.039) | 1.14 (1.09–1.18) |
| DsbBL25P | 2 ± 0.03 | 0.54 ± 0.05 | 2.3 ± 0.1 | 174 ± 25 | 0.88 | 0.31 × 10−2 | 0.173 (0.157–0.192) | 4.06 (3.84–5.52) |
| DsbBA29V | 3 ± 0.2 | 1.4 ± 0.05 | 10.8 ± 1.5 | 90.5 ± 6.5 | 0.28 | 1.5 × 10−2 | 1.697 (1.54–1.79) | 1.47 (1.39–1.56) |
| DsbBK39E | 5.5 ± 0.2 | 0.8 ± 0.1 | 3 ± 0.4 | 201 ± 38 | 1.81 | 0.39 × 10−2 | 0.071 (0.065–0.079) | 3.01 (2.75–3.30) |
| DsbBP100S | 2.1 ± 0.06 | 0.66 ± 0.03 | 3.6 ± 0.4 | 47 ± 5.1 | 0.58 | 1.4 × 10−2 | 0.033 (0.030–0.037) | 2.01 (1.93–2.1) |
| DsbBF106L | 6.2 ± 0.09 | 0.69 ± 0.08 | 3.6 ± 0.2 | 61.8 ± 15 | 1.73 | 1.1 × 10−2 | 0.055 (0.049–0.063) | 1.2 (1.05–1.37) |
a kcat expressed as nanomoles of ubiquinone-1 or menadione per nmol of DsbB per s.
b Km values represent ubiquinone-1 or menadione concentrations.
c In vitro inhibition was measured using 10 nm DsbB, 10 μm ubiquinone-1, and 20 μm reduced DsbA.
d In vivo inhibition was measured by growth inhibition of strain lptD4213ΔdsbB dsbBPtrc204 (CL409–410, CL416–417, and LI18–19 strains) in the presence of drugs.
We also measured the inhibition of DsbB by compound 12 using an in vitro assay with purified components in the ubiquinone reduction assay (Table 1, 8th column). DsbBA29V displayed a 50-fold increase in the IC50, whereas DsbBL25P and DsbBK39E showed a 5- and 2-fold increase, respectively, under saturating concentrations of ubiquinone and DsbA. Under these conditions, neither DsbBP100S nor DsbBF106L showed an increase in the IC50 (see under “Discussion”).
Mutations isolated anaerobically conferred resistance aerobically to LptD4213 strain
We then asked whether the DsbB mutants obtained in the anaerobic selection also conferred resistance when tested in our aerobic model using the LptD4213 strain. We transformed the IPTG-inducible dsbB mutant plasmids obtained anaerobically into the lptD4213ΔdsbB strain carrying a plasmid with an arabinose-inducible wild type dsbB. We then determined whether these mutants were able to support growth of lptD4213ΔdsbB strain by curing the plasmid encoding the arabinose-inducible wild type dsbB (see “Materials and methods”). All DsbB mutants were able to support growth of lptD4213ΔdsbB strain indicating that the mutants selected anaerobically are also functional aerobically. These strains were then tested for growth in the presence or absence of compound 12 in minimal medium. The results are shown in Table 1 (9th column). DsbBL25P and DsbBK39E exhibited a 3–4-fold increase in the IC50, whereas the DsbBP100S mutant showed a modest increase. Hence, these mutants isolated anaerobically can also confer resistance to compound 12 aerobically. In contrast, DsbBA29V and DsbBF106L mutants did not show a significant increase of the IC50 under these conditions (see “Discussion”).
Compound 12-resistant mutants are also resistant to other pyridazinone analogs
We have previously performed structure/activity analysis with pyridazinones and found compound 12 to be one of the most effective inhibitors (17). Here, we explored other variations in the structure of the molecule in the hope of finding more effective compounds as well as gaining insight into the mechanism of action of this class of drugs. The results of this analysis are shown in Table 2 and described under “Materials and methods.” We found compound 36 more effective than compound 12, and compounds 37 and 38 were as effective as compound 12 in our β-galactosidase assay. Our results showed that changing both halogens (electron acceptors) in the pyridazinone ring to methyl groups (electron donors) makes the drug ineffective; similarly, changing the halogen at position 5 to methyl (Table 2, see Fig. 3B for atom numbers) while changing the halogen at position 4 to methyl only decreases the inhibitor efficacy (Table 2), thus suggesting that the halogen at position 5 is the leaving group in the covalent interaction with Cys-44 of DsbB. To confirm this possibility, we analyzed the inhibition of DsbB by mass spectrometry. When compounds 12 or 38 are incubated with the DsbB-DsbAC33A complex, a mass decrease of 36.7 and 36.8 Da, respectively, is observed indicating a loss of a chloride (plus a proton) (Table 3). Although with compounds 36 or 37, the measured molecular mass of the complex compound is decreased by 80.9 and 81.4 Da, respectively, indicating a loss of a bromide (plus a proton) (Table 3). These data provide evidence that the halogen at position 5 of the pyridazinone ring is the leaving group when covalently binding to Cys-44 of DsbB.
TABLE 2.
Relative inhibition of DsbB by other pyridazinone drugs
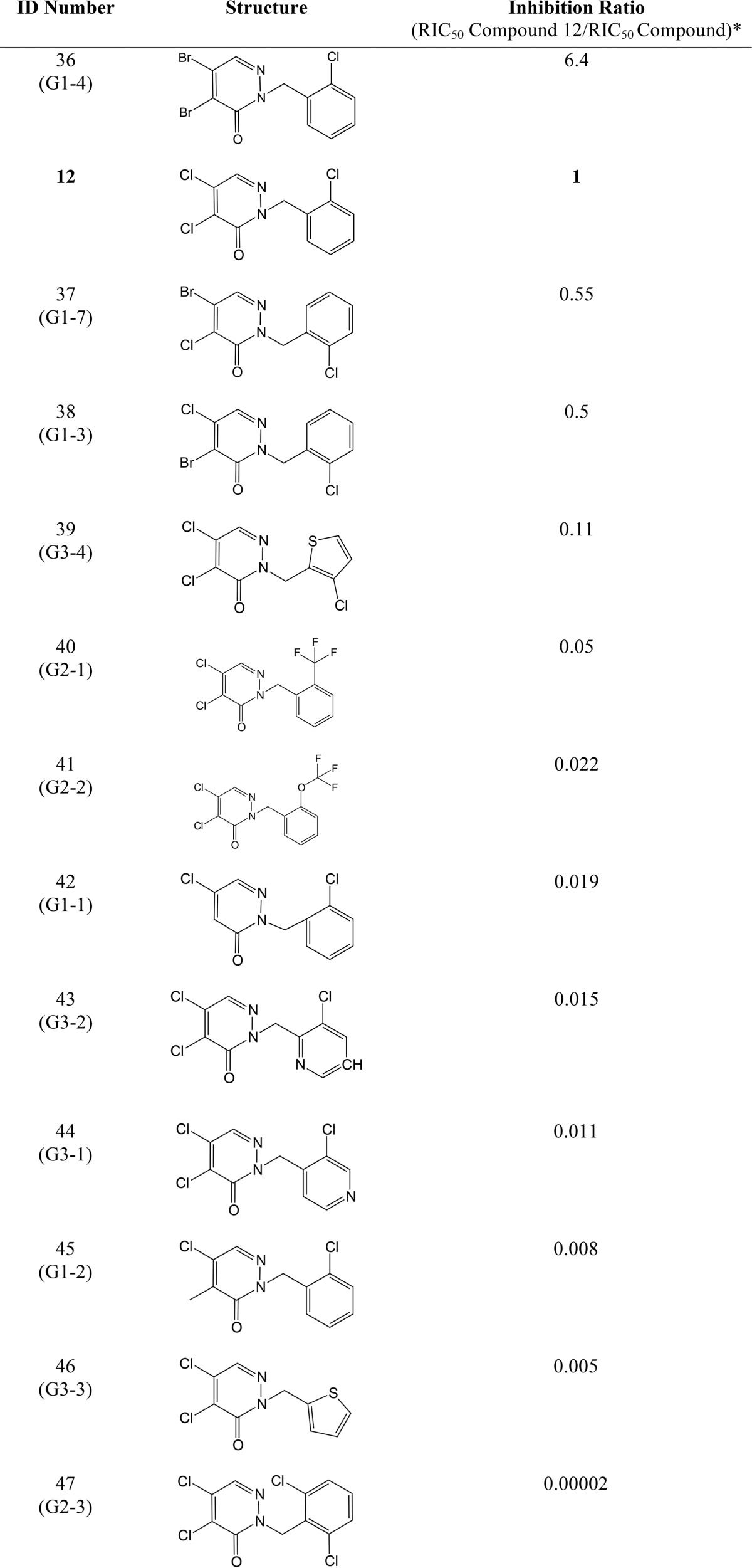
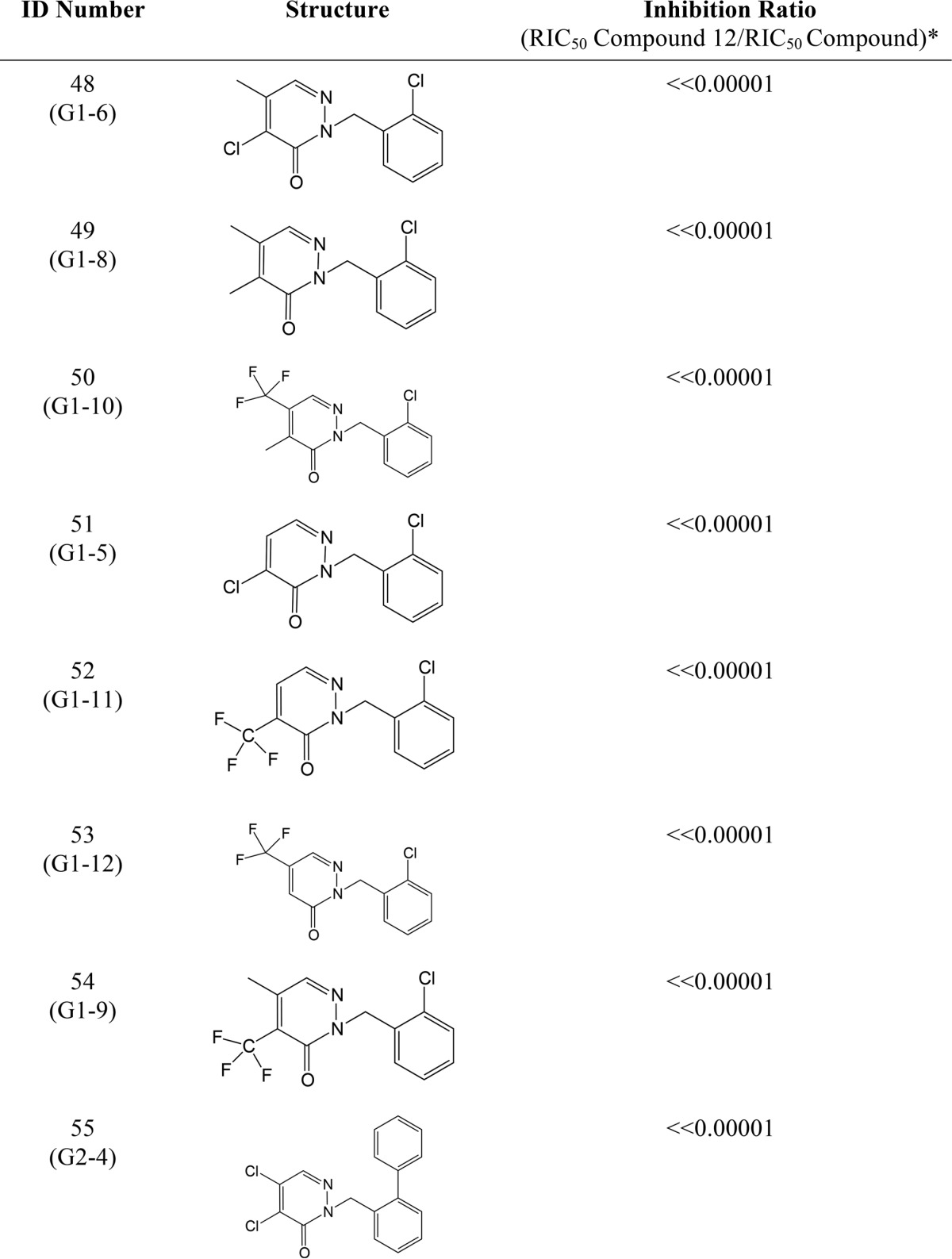
* The RIC50 values were obtained using β-galactosidase activity, which is a measure of the inhibition of DsbB in E. coli expressing β-Galdbs. The more DsbB inhibition of a drug the more β-galactosidase activity will be observed in cells, so one can calculate the concentration that gives 50% of inhibition (RIC50) of the total activity observed in a ΔdsbB strain and use that concentration to get the fold-increase by dividing the RIC50 of compound 12 (0.16 μm, 95% confidence interval 0.13–0.20 μm) between the RIC50 of the tested drug. Thus, a drug more potent than compound 12 will have a higher ratio and vice versa. The results were obtained using data of at least three independent experiments.
TABLE 3.
Summary of deconvoluted masses obtained from ESI-MS analysis of non-reduced proteins treated with compounds
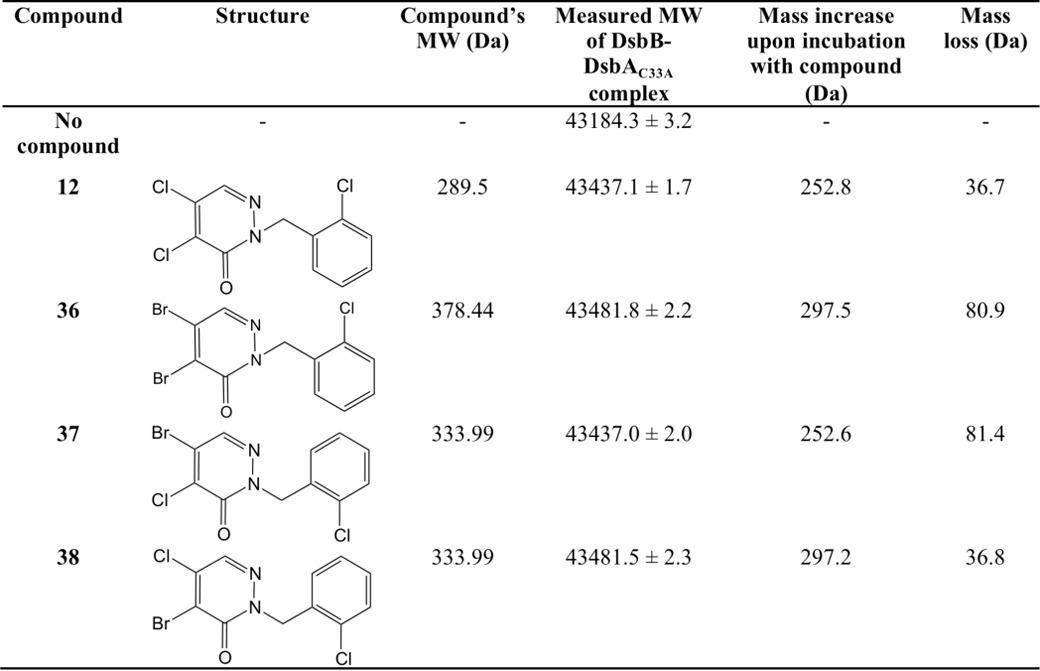
We determined whether the residues important for resistance to compound 12 also led to resistance in other strong inhibitors of the pyridazinone family. We tested the five DsbB mutants in vivo using the LtpD4213 strain and found that, like compound 12, all are resistant to the other pyridazinone inhibitors. DsbBL25P and DsbBK39E showed 2–5-fold increases in the IC50 for all of the pyridazinone inhibitors (Table 4). In addition, DsbBA29V, DsbBP100S, and DsbBF106L showed an almost 2-fold increase in the IC50 for at least two of the inhibitors tested (Table 4). Thus, we observed at least some level of cross-resistance to pyridazinones for all DsbB mutants.
TABLE 4.
DsbB mutants give in vivo resistance against pyridazinone-related molecules
Values represent the average of three independent experiments with the 95% confidence intervals in parenthesis. Boldface numbers have more than a 2-fold increase compared with wild type. Underlined numbers have 1.8–1.9-fold increase of IC50. Values that do not have a 95% confidence interval is due to the lack of higher concentrations tested given the poor solubility of some compounds in minimal media.
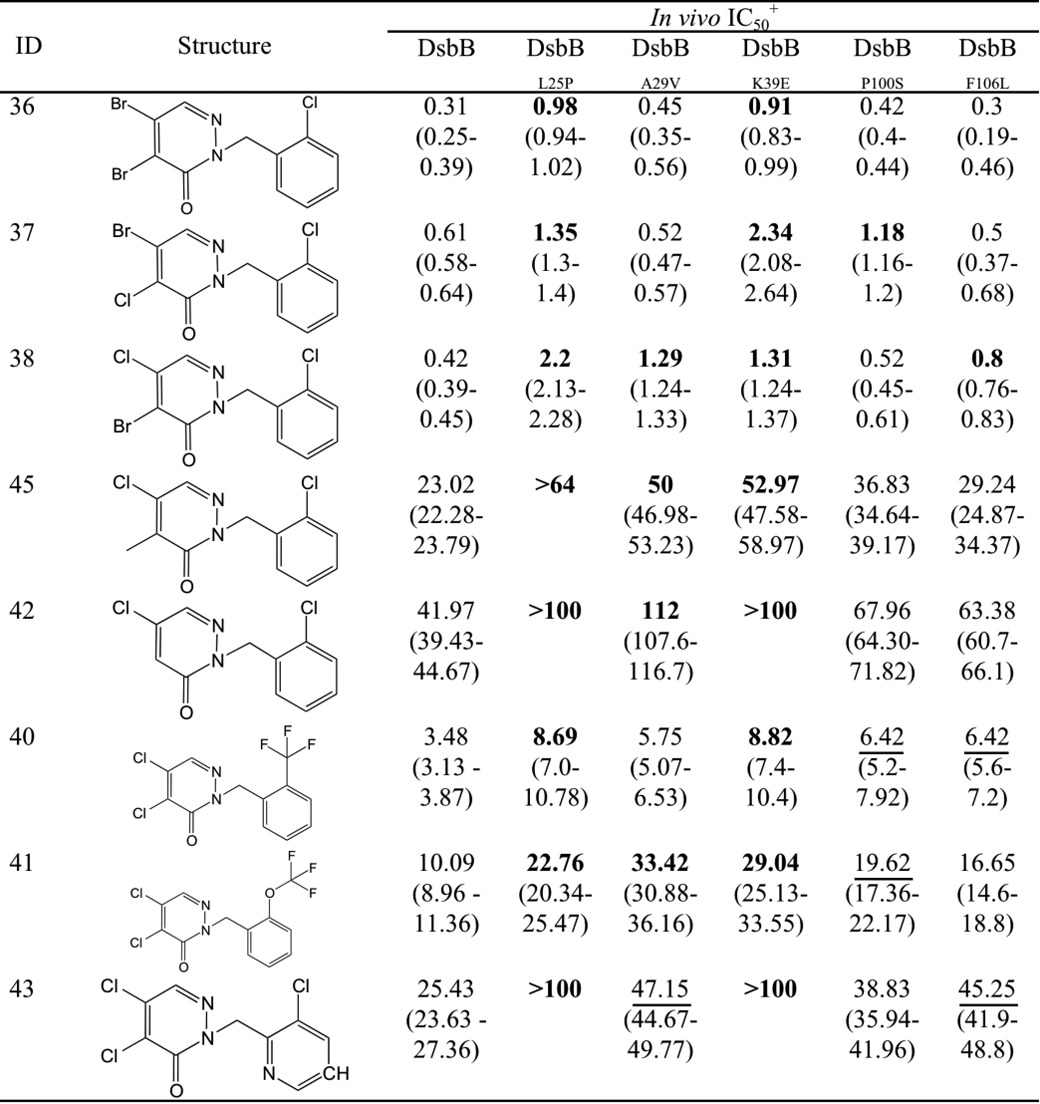
+ In vivo inhibition was measured by growth inhibition of strain lptD4213 ΔdsbB dsbBPtrc204 (CL409–10, CL416–7 and LI18–19 strains) in the presence of drugs.
Discussion
In the oxidation pathway that introduces disulfide bonds into proteins in the bacterial periplasm, DsbA cysteines need to be reoxidized to start a new catalytic cycle. The cytoplasmic membrane protein DsbB performs this task. DsbB is a cellular machine that generates a protein disulfide bond de novo at the expense of electrons to be transferred to ubiquinone (14, 32). During the transfer and interaction of DsbA with DsbB, the latter undergoes conformational changes (29). In this work, we have selected DsbB mutants that confer resistance to a pyridazinone inhibitor and are located in two prominent areas in the structure of DsbB, one located between the two first transmembrane segments where the quinone ring fits. These mutants, DsbBL25P, DsbBA29V, and DsbBK39E, show a higher Km value for quinones as one might expect given that they are in the region of the quinone binding (Fig. 2B). Surprisingly, the other area is located in the second periplasmic loop of DsbB known to interact with DsbA. It has been shown that this segment from Pro-100 to Phe-106 is accommodated deep in the hydrophobic groove of DsbA's structure (29, 33). The fact that we find mutants in this region and that DsbBP100S and DsbBF106L mutants exhibit an increase in the Kmvalue for quinone despite not being located within the quinone-binding site suggests that this region also shapes the DsbB-quinone interaction. This model is in agreement with the fact that this segment of DsbB has to be mobile because it contains the Cys-104 residue that forms a disulfide with Cys-30 of DsbA and participates in the exchange of disulfides with Cys-41–Cys-44 of DsbB to finally oxidize DsbA (34).
Levels of DsbB were assessed in the mutants to demonstrate that the resistance to compound 12 is not due to changes in DsbB amount (Fig. 2C). Although levels remained unchanged in four of the five mutants, due to unknown reasons DsbBK39E showed a 2-fold increase in DsbB. Although we cannot rule out the possibility that the increase in DsbB levels may contribute to the resistance of DsbBK39E, the purified mutant displayed significantly different kinetics than the wild type enzyme, suggesting that the resistance conferred by the mutation is at least partially due to its effects on enzyme activity. This study highlights the decrease in quinone affinity rendering the mutants less susceptible to inhibition. The mutations may selectively inhibit access of the compound to Cys-44 while allowing limited passage of quinone. However, two mutants DsbBK39E and DsbBF106L also show an increase in kcat implying that the reaction pathway is possibly altered by a change in the rate-limiting step, which resolves the DsbA-DsbB complex, releasing reduced quinone and oxidized Cys-41–Cys-44 of DsbB. It is possible that the mutations affected this step as well by restructuring the region in the DsbA-DsbB complex in a way that the compound cannot reach Cys-44.
We showed that DsbBL25P, DsbBA29V, and DsbBK39E mutants confer in vitro resistance to compound 12 observed as a 5-, 50-, and 2-fold increase in the IC50, respectively (Table 1, 8th column). One explanation for the finding that DsbBP100S and DsbBF106L did not show an increase of the IC50 value is that these mutants may have also affected the Km value for DsbA. Even though DsbA is in excess in the conditions used in vitro (20 μm), this may not represent the physiological conditions, and/or the fact that DsbB is away from the membrane in the in vitro experiments may affect the hydrophobic environment of the membrane required to observe resistance. Future work is needed to determine whether this is the case.
Besides being found as a resistant mutant in two different selections and having a 2- and 5-fold increase in Km for ubiquinone and menadione, respectively, a decrease in kcat, and a decrease in catalytic efficiency (kcat/Km), the DsbBL25P mutant conferred in vivo resistance to all the inhibitory pyridazinones tested, and this resistance resulted in a 2–5-fold increase in the in vivo IC50 (Tables 1 and 4). Similarly, the DsbBK39E mutant exhibited a 2-fold increase in kcat when using ubiquinone, and a 3- and 5-fold increase in Km for ubiquinone and menadione, and it provided as well resistance to all inhibitory pyridazinones tested in vivo, resulting in 2–4-fold increases in the IC50 (Tables 1 and 4). Therefore, Leu-25 and Lys-39 may be associated with the binding of the pyridazinone ring and the phenyl ring, which is common to all the inhibitors tested. Overlapping the pyridazinone ring with the quinone ring in the DsbB structure (Fig. 3B), we observed that the phenyl ring is possibly oriented to the hydrophobic groove of the membrane where the methyl-butenyl group of ubiquinone-1 orients, although the pyridazinone ring may sit near Cys-44 on the periplasmic facing surface. Therefore, it is possible that Lys-39 may be responsible for binding to the pyridazinone ring and Leu-25 to the phenyl ring.
It is important to note that the DsbB mutants were selected to be at least 2-fold resistant to compound 12 under anaerobic conditions in which the abundant quinone is menaquinone (35, 36). Therefore, one explanation for the finding that some mutants conferring resistance to compound 12 anaerobically do not do so aerobically (DsbBA29V and DsbBF106L) may depend on the amount and redox state of the quinone species available in the strain. It has been shown that changes in oxygen levels alter the composition and the redox states of the quinone pool (ubiquinone-8, menaquinone-8, and demethylmenaquinone-8) (36). Another possibility is that the amount of DsbA changes among growth conditions. Nevertheless, we did not observe any change in the DsbA levels of the mutants compared with wild type grown aerobically or anaerobically (data not shown). One additional observation is that these two mutants conferred in vivo resistance to the lptD4213 strain to compounds that are less potent inhibitors than compound 12, with the exception of compound 38 (Table 4).
We asked whether there exist variants of DsbB enzymes that might be resistant to pyridazinones by doing a bioinformatic search among the different E. coli-sequenced genomes available. From the 52 DsbB protein sequences analyzed (that share 90% or greater identity), the five residues presented in this work were conserved and similar to wild type DsbB (data not shown). We also looked at the conservation of these five residues among other DsbB proteins from Gram-negative bacteria, specifically the ones that we know from our previous work are inhibited by pyridazinone-related molecules (17). The identity between DsbB proteins from Salmonella enterica sv. typhimurium, Klebsiella pneumoniae, Vibrio cholerae, and Haemophilus influenzae ranges from 85 to 41% when compared with E. coli DsbB. Among these organisms, four of the five residues were conserved overall when aligned to wild type E. coli DsbB, except for Lys-39. However, DsbB proteins from Pseudomonas aeruginosa, Acinetobacter baumannii, and Francisella tularensis, which share ∼20% of identity with E. coli DsbB, demonstrated little or no conservation in the five residues studied. Moreover, P. aeruginosa DsbB1 encodes a Val-29 variant; similarly, P. aeruginosa DsbB2 and A. baumannii DsbB encode a Glu-39-resistant variant studied in this work. Nevertheless, we have shown that these proteins are still sensitive to compound 12 and related pyridazinones (17). Thus, it is possible that each enzyme may have slight differences in the structure and therefore differences in binding to pyridazinone drugs, which is in agreement with our previous observation that the extent of inhibition changes among different pyridazinone inhibitors (17).
All mutant DsbBs were able to functionally complement the lptD4213dsbB− strain for growth, indicating that the mutants are functional enzymes not only anaerobically but also aerobically. Similarly, the DsbB mutants were also able to complement two other dsbB− phenotypes. They restored motility of the dsbB mutant, and they also lacked β-galactosidase activity when the β-Galdbs is expressed in the strain (data not shown). However, all of the mutants obtained displayed lower catalytic efficiencies than the wild type enzyme.
Our finding that the combination of an lptD4213 mutation and a dsbB null mutation are synthetically lethal leads us to suggest that any mutation or drug that decreases LptD assembly may also be synthetic lethal with the Dsb pathway (dsbA or dsbB mutants). Consequently, this finding suggests that combinations of drugs that target these two pathways can potentiate their antibiotic effect. This also suggests that inhibitors of the Dsb pathway may help to study LptD assembly by searching for mutations that confer resistance to these small molecules in order to identify additional genetic factors involved in LptD assembly (22).
The mutants studied here have a modest level of resistance (2–5-fold increase in IC50) to pyridazinone molecules in E. coli growth. It may be that greater changes in resistance are costly to the enzyme and thus to bacterial growth. Two different spontaneous genetic selections for resistance to the pyridazinone inhibitor, anaerobic selection for growth and growth of the lptD4213 mutant, indicate that the frequency with which resistance arises is quite low. Obtaining such mutations was only made possible by PCR mutagenesis of a plasmid-encoded dsbB, which artificially increased the mutation rate. Although the environment in infections may generate different conditions for selection, these initial results raise the possibility that resistance problems during infections may possibly be avoided. Our findings may provide insights to the development of more effective pyridazinone drugs that do not bind covalently and are also important for understanding the nature of resistance, which may also hold some clinical relevance. This suggests that further development of pyridazinones as potential antivirulents/antibiotics may be warranted.
Materials and methods
Bacterial strains and growth conditions
The strains and plasmids used in this study are listed in Tables 5 and 6, respectively. Standard molecular biology techniques and P1 transduction were used for the construction of strains and expression vectors (37, 38). All strains were grown in LB Miller agar or in M63 0.2% glucose liquid and agar media at 37 °C. Minimal M63 with 0.2% glucose and 40 mm fumarate solidified with 1% agarose plates were prepared for anaerobic growth experiments by placing in a Coy anaerobic chamber (85% N2, 10% H2, 5% CO2) to equilibrate for several days before use. The antibiotic concentrations used were as follows: ampicillin 100 μg/ml (for plasmid copy), 25 μg/ml (for chromosomal copy), or 10 μg/ml (for LptD4213 strain), kanamycin 40 μg/ml, tetracycline 10 μg/ml, and chloramphenicol 10 μg/ml.
Table 5.
Strain list used in this study
| Strain | Genotype | Reference |
|---|---|---|
| E. coli strains | ||
| HK295 | MC1000 Δara714 leu+ | 34 |
| HK310 | HK295 ΔdsbB (Kmr) | 34 |
| HK320 | HK295 ΔdsbB | 34 |
| NR698 | MC4100 LptD4213 | 22 |
| GC208 | MC4100 carB::Tn10 (Tcr) | 40 |
| JR6 (FSH94) | BL21 C43 (DE3) ΔdsbB (Kmr) | 42 |
| JR7 (FSH95) | BL21 C43 (DE3) ΔdsbB (Kmr) pWM76 (DsbBC8A C49V 6His, (Ampr) | 42 |
| FSH69 | Lemo21(DE3, Cmr) pFL39 (6HisDsbA, Kmr) | 17 |
| CL337 | HK295 LptD4213 | This study |
| CL380 | HK295 LptD4213 ΔdsbB (Kmr) pCL67 (dsbBPBAD, Cmr) | This study |
| CL410 | HK295 LptD4213 ΔdsbB (Kmr) pCL23 (dsbBPtrc204, Ampr) | This study |
| CL417 | HK295 LptD4213 ΔdsbB (Kmr) pBOM230 (DsbBL25P under Ptrc204, Ampr) | This study |
| LI18 | HK295 LptD4213 ΔdsbB (Kmr) pBOM252 (DsbBA29V under Ptrc204, Ampr) | This study |
| LI19 | HK295 LptD4213 ΔdsbB (Kmr) pBOM253 (DsbBK39E under Ptrc204, Ampr) | This study |
| CL416 | HK295 LptD4213 ΔdsbB (Kmr) pBOM228 (DsbBP100S under Ptrc204, Ampr) | This study |
| CL409 | HK295 LptD4213 ΔdsbB (Kmr) pBOM231 (DsbBF106L under Ptrc204, Ampr) | This study |
| CL591 | HK295ΔdsbB λatt::DsbBWT (Ptrc204, Ampr) | This study |
| CL592 | HK295ΔdsbB λatt::DsbBL25P (Ptrc204, Ampr) | This study |
| CL594 | HK295ΔdsbB λatt::DsbBA29V (Ptrc204, Ampr) | This study |
| CL595 | HK295ΔdsbB λatt::DsbBK39E (Ptrc204, Ampr) | This study |
| CL593 | HK295ΔdsbB λatt::DsbBP100S(Ptrc204, Ampr) | This study |
| CL596 | HK295ΔdsbB λatt::DsbBF106Y(Ptrc204, Ampr) | This study |
| Plasmids | ||
| pTrc99A | Expression vector, pBR322 origin, Ampr | |
| pDSW204 | Promoter down mutation in −35 of pTrc99A (Ptrc204), (Ampr) | 39 |
| pBAD45 | Arabinose-inducible vector (PBAD), pSC101 origin, Cmr | 45 |
| pET28a | Expression vector, T7lac promoter, N-terminal and C-terminal His tag, thrombin cleavage site, pBR322 origin, Kmr | EMD |
| pWM76 | pQE70-DsbBC8A C49V-6His (Ampr) | 42 |
| pFL39 | pET28a-6His-DsbA cloned at NdeI-XhoI | 17 |
| pCL23 | pDSW204-dsbB cloned at NcoI-SacI, DsbBWT (MV-DsbB2–176) | This study |
| pCL67 | pBAD45-dsbB cloned at EcoRI-HindIII, DsbBWT | This study |
| pBOM228 | pDSW204-dsbBT77C cloned at NcoI-SacI, DsbBL25P | This study |
| pBOM252 | pDSW204-dsbBC89T cloned at NcoI-SacI, DsbBA29V | This study |
| pBOM253 | pDSW204-dsbBA118G cloned at NcoI-SacI, DsbBK39E | This study |
| pBOM230 | pDSW204-dsbBC301T cloned at NcoI-SacI, DsbBP100S | This study |
| pBOM231 | pDSW204-dsbBT319C cloned at NcoI-SacI, DsbBF106L | This study |
| pLI1 | pWM76-dsbBT77C, DsbBL25P | This study |
| pLI2 | pWM76-dsbBC89T, DsbBA29V | This study |
| pLI3 | pWM76-dsbBA118G, DsbBK39E | This study |
| pLI4 | pWM76-dsbBC301T, DsbBP100S | This study |
| pLI6 | pWM76-dsbBT319C, DsbBF106L | This study |
Table 6.
Primers used in this study
| ID | Sequence | Restriction site |
|---|---|---|
| Cl6 | ATGCCATAGCATTTTTATCC | |
| Cl8 | GATTTAATCTGTATCAGG | |
| Cl13 | CTCCATGGTGTTGCGATTTTTGAACCAATG (Adds V after M) | NcoI |
| Cl14 | CGGAGCTCTTAGCGACCGAACAGATCACGTT | SacI |
| Cl24 | GGCGCACTCCCGTTCTGGATAATGT | |
| Cl25 | GGTCAGGTGGGACCACCGCGCTACT | |
| Cl55 | CTGCGTCGAGTTTACGCTTGCCCCTGTA | |
| Cl56 | GGGATCCAGCAACAATGGCAGATGAA | |
| Cl84 | TGAGTTCTACCTGCCATATTACTGG | |
| Cl85 | TTATCCCAACCGTTCAGCTTCCGCT | |
| Cl105 | GTCGTGAATTCATGTTGCGATTTTTGAACCAATG | EcoRI |
| Cl106 | CGTAAGCTTTTAGCGACCGAACAGATCACGTT | HindIII |
| Cl117 | AGGTGAAGGGTGGGCTGCCATTGTTGC | |
| Cl118 | GGTTAAATAACGTGGATTTTCCTACGTTAGGGCGCCCGA | |
| Cl119 | GTTGGGGTTTTACGGCTTTGCCGTTTAATA | |
| Cl120 | AGCGAGCCATATTTGATGAGATCGATAGCG | |
| Cl129 | TACTGGCTGCGACAGACGGCGA | |
| Cl130 | CAGCAAAACTTTGAATATCCACTTATGCTGA | |
| Cl131 | CTTTCCTGCAATAACAGAGGAT | |
| Cl132 | GATGGCGTTACTGTACGTAAAGTGATT | |
| L25P-f | GCTCTGGCACCGGAACTGACG | |
| L25P-r | AGTAAACGCCATCAACAGC | |
| A29V-f | GAACTGACGGtGCTGTGGTTC | |
| A29V-r | CAGTGCCAGAGCAGTAAAC | |
| K39E-f | GATGTTACTGGAACCTTGCGTG | |
| K39E-r | ACATGCTGGAACCACAGC | |
| P100S-f | CTATCCTTCGTCGTTTGCCAC | |
| P100S-r | AGCTGAAGCATGGTGTGC | |
| F106L-f | CACCTGTGATCTTATGGTTCG | |
| F106-r | GCAAACGGCGAAGGATAG | |
| Cl225 | GCAGGAGTCTATGAACACGTTTCAGTGAAACCATTTAAGAAAGTGTTCTGAGTGTAGGCTGGAGCTGCTTC | |
| Cl226 | CAAACAAGAACACGGTTGCAAAAACCGTGCCCTTAAATATTGAATCTCTATATGGGAATTAGCCATGGTCC | |
| Cl230 | CAAACAAGAACACGGTTGCAAAAACCGTGCCCTTAAATATTGAATCTCTATGTGTAGGCTGGAGCTGCTTC | |
| Cl231 | GTGCACATTTTCTGAACATACATGCAGCGCG | |
| Cl240 | CCCGAACAAGGAGTTGTGCCCGTGT |
dsbB mutagenesis and construction of mutant library
A mutagenic PCR of the dsbB gene using primers Cl13 and Cl14 was generated using the first seven mutagenic conditions of Diversify mutagenesis kit (Clontech) that on average generates 2–5.8 mutations/kb. The amplification conditions used were 94 °C (30 s) as denaturing temperature, 55 °C (30 s) as annealing, and 68 °C (30 s) as extension repeated for 25 cycles. The products were reamplified using Taq platinum (Thermo Fisher Scientific) to produce more of the PCR product. PCR products of all reactions were then mixed, column-purified, NcoI-SacI-digested, and ligated to a digested pDSW204 plasmid (39). 1 μl of the ligation reaction was transformed into highly competent XL1-Blue cells (Agilent Technologies). A sample of the colonies obtained after selection on ampicillin plates was collected for plasmid preparation used to confirm efficiency of ligation by PCR and digestion. Given that 9 of 10 colonies did have the expected insert, the rest of the ligation reaction (49 μl) was transformed into DH10β highly competent cells (New England Biolabs). The transformation yielded ∼3,000 colonies, which were scraped up and grown overnight in M63 glucose for plasmid preparation. Plasmid preparations were frozen at −20 °C until use.
Construction of a conditionally lethal strain lptD4213ΔdsbB
lptD4213 (amino acid deletion from 330–352) mutant was constructed in E. coli MC1000 strain by transducing the mutation from the MC4100 strain, NR698 (22). First, the lptD gene was linked to a tetracycline resistance cassette (at carB gene, 20–25% linkage) by making a P1 lysate from the GC208 strain (40). This lysate was then used to infect the NR698 strain (lptD4213 mutation) selecting for transductants in tetracycline plates. The lptD4213 transductants linked to tetracycline cassette were verified by the size of the PCR product of part of lptD gene (1.5 kb), and the mutants have a 68-bp smaller PCR product due to the deletion of 23 amino acids using primers Cl84 and Cl85. It was also noticed that all small colonies had lptD4213 mutation, and the regular size colonies had wild type lptD. Thus, this was used in later selections to distinguish between them. A P1 lysate from one verified transductant in the previous step was prepared to infect HK295 (MC1000) strain. After verifying the presence of lptD4213 mutation in HK295, the tetracycline cassette linker was removed from the strain by P1 transduction of wild type strain and selecting on minimal M63 media, because the carB mutation makes the cells arginine and uracil auxotrophs on minimal media (41). The colonies that grew in minimal glucose media were again verified by PCR and sequenced to have the lptD4213 mutation; one colony was selected for further experiments (CL337 strain).
To construct the conditionally lethal strain lptD4213ΔdsbB dsbBPBAD, first a plasmid expressing dsbB under the regulation of arabinose promoter (pCL67) was transformed into the lptD4213 strain (CL337). The deletion of dsbB gene from HK310 strain was then P1-transduced to the lptD4213 strain selecting on LB kanamycin plates supplemented without or with 0.2% arabinose. Kanamycin-resistant colonies were obtained in both cases, and the transduction of the dsbB deletion was verified by PCR using primers Cl55–56; all checked colonies did have the correct product size (1-kb dsbBWT versus 1.6-kb dsbB::Km). This result indicated that the basal levels of expression from arabinose promoter were enough to complement growth in rich medium. One colony with confirmed deletion was isolated and used for further work (CL380 strain). When the dsbB deletion was transduced to lptD4213 with no other copy of the dsbB gene, no colonies with the correct deletion of dsbB were obtained unless the transductants were plated on LB with 1 mm cystine; however, the transduction frequency was lower than the frequency observed in the strain with two copies of dsbB. The growth of CL380 strain was tested in minimal media plates. M63 glucose with 0.2% arabinose allowed growth of the CL380 strain, whereas the strain was not able to grow on M63 minimal media plates lacking arabinose. However, this strain is able to grow in liquid M63 minimal media with no arabinose under shaking conditions where oxygen may contribute to background oxidation.
Selection of DsbB mutants using lptD4213 strain
For spontaneous resistant mutants, CL337 cells from overnight culture were washed twice with M63 minimal media, and ∼109 cells were plated in M63 glucose media plates with 10 μm compound 12 (10-fold higher the MIC). Plates were incubated for 2 days at 37 °C. 51 colonies were purified in M63 minimal media plates to characterize them. We amplified dsbB (primers Cl55–56) and dsbA (primers Cl129–130) genes by PCR from 25 colonies and sequencing of these gave a wild type sequence. We noticed that some of the selected mutants did confer resistance to bile salts, and because these mutations had been previously studied (21), we amplified and sequenced also bamB (primers Cl117–118), bamD (primers Cl119–120), and lptE (primers Cl131–132). 22 of 51 colonies analyzed by PCR did have a higher size product of the bamB gene, and the sequence of all these indicated an insertion of IS1 element in the gene. Whole-genome sequencing was performed in three of the colonies that did not have a higher size bamB product. Two of these did have mutations that inactivated bamB (one had a stop codon insertion in amino acid 240 and the other was a deletion of amino acids 252–255).
For selection of dsbB mutants, the plasmid mutant library was used to transform the conditionally lethal strain, CL380 (lptD4213ΔdsbB dsbBPBAD). This strain was more sensitive to ampicillin; therefore, a lower concentration of ampicillin was used (10 μg/ml) to select for transformants containing the plasmid library. The transformation gave around 4,500 independent colonies, which were scraped and saved in glycerol stocks. After growing the library on LB broth, cells were washed twice with M63 minimal media, and ∼108 cells were plated on M63 glucose minimal media containing 10 μm compound 12 to select for resistant mutants. After 2 days of growth at 37 °C, colonies appeared and were purified on LB plates with no antibiotic. A PCR product of the mutagenized dsbB gene was amplified using primers Cl24–25 (prime only to pDSW204) to sequence.
Anaerobic selection of DsbB mutants
Purified plasmids from the mutagenized library were transformed into ΔdsbB cells (HK320) and plated aerobically on LB with ampicillin. The transformation yielded around 3,000 colonies, which were scraped and saved as glycerol stocks to use for further selection. The mutant library obtained in ΔdsbB mutant was grown aerobically in M63 0.2% glucose to an A600 of 0.6. Cells were washed, and ∼107 cells were plated on M63, 0.2% glucose, 40 mm fumarate, 1% agarose plates with 2 μm compound 12. Plates were then incubated at 37 °C in a Coy anaerobic chamber (85% N2, 10% H2, 5% CO2) for 3 days. The resistant colonies were purified under the same conditions and then cultured aerobically to isolate plasmids. Plasmids were transformed back into ΔdsbB cells, and growth of the resultant transformants was tested anaerobically under selective conditions to confirm that the plasmid carried the resistance mutation. The dsbB gene was then sequenced with primers Cl24-Cl25 to identify the mutations.
Using lptD4213 strain to confirm resistance of the studied DsbB mutants
To confirm resistance of the five selected mutations, the plasmids pBOM228, -30, -31, -52, and -53 were used to transform the CL380 strain. The resultant strains were then plated on LB with 0.4% arabinose plates to select for cells cured of the plasmid with the wild type copy of dsbB (pCL67). Because the overexpression of dsbB causes cell toxicity, those cells able to grow under arabinose are most likely the cells that have lost the arabinose-inducible plasmid. Purified colonies were checked for loss of chloramphenicol resistance and were verified by PCR with primers Cl24 and Cl25 that prime only to pDSW204 but not to pBAD plasmid and with primers Cl6 and Cl8 that prime only to pBAD but not to pDSW204 plasmid. The dsbB mutations were confirmed by sequencing.
Growth assays of lptD4213 dsbB mutants in the presence of various pyridazinone drugs
Strains were grown overnight in minimal M63 0.2% glucose media with 5 μm IPTG (Enzo Life Sciences Inc.) to induce the expression of dsbB. Overnight cultures of bacteria were diluted to an A600 of 0.02 in M63 0.2% glucose minimal media, and 200 μl of diluted cultures were aliquoted in 96-well plates (Thermo Fisher Scientific). Serial dilutions of the drug or DMSO were added in a volume of 2 μl (1% DMSO final concentration). The plates were covered with breathable films (VWR Scientific) and then incubated for 19 h at 37 °C and 900 rpm in an orbital shaker (Multitron ATR). The A600 from at least three independent experiments was read to determine the growth, and this was used to calculate the IC50 values (concentration that gives 50% inhibition of growth without drug) with 95% confidence intervals using GraphPad Prism (La Jolla, CA) in the function of non-linear regression (log inhibitor versus response with variable slope, normalized response).
Purification of DsbB proteins and enzyme kinetics
The five mutations in DsbB were generated by site-directed mutagenesis of plasmid pWM76 using the primers listed in Table 2. Then DsbB proteins were purified as described before (42). Purified proteins were at least 90% as judged from SDS-PAGE (supplemental Fig. 1). Determination of kinetic properties and IC50 values was done as described before with slight changes (17). Briefly, various amounts of inhibitors were mixed with 10 nm DsbB in phosphate buffer (pH 6.5) containing 0.1% n-dodecyl-β-d-maltopyranoside (Affymetrix Inc.), 100 mm NaCl and ubiquinone-5 (Sigma, 1–50 μm for kinetic constants and 10 μm for inhibition assays) or menadione (Sigma, 0.5–128 μm). Reactions were started at room temperature by the addition of small amounts of highly concentrated DsbA solution to give a final concentration of 20 μm. Initial velocities of DsbB-catalyzed quinone reduction were measured at 275 nm for ubiquinone and 260 nm for menadione.
Structure-activity relationship approach of related pyridazinones
Given that a substructure analysis with pyridazinones helped us previously to identify more effective inhibitors such as compound 12 (17), we decided to explore more variations in the core of the drug to validate our understanding of the drug inhibition and to find more effective inhibitors. The molecules were designed first by substituting the chlorine atoms at positions 4 and 5 of the pyridazinone ring by other halogen atoms such as bromine and by other groups that unlike halogens could act as nucleophile (electron donor) rather than electrophile (electron acceptor), i.e. methyl groups. Second, we substituted the benzyl group at position 2 by different rings such as thiophene and pyridine and finally changed/added substituents in the benzyl ring at the ortho position. Molecules shown in Table 2 were synthesized by Sundia MediTech Co., Ltd. (China, purity over 95% analyzed by NMR and LC-MS). The chemical synthesis protocols are presented at the end of supplemental Information. Compound 12 was purchased from Enamine (Ukraine, purity over 95% analyzed by LC-MS).
To test inhibition, all compounds were tested in β-galactosidase assays as described previously (17). Briefly, the relative inhibitory concentration 50 (RIC50) values obtained in the β-galactosidase activity assay are a measure of the inhibition of DsbB in E. coli expressing β-Galdbs and were used to compare the potency of the drugs. The more DsbB inhibition of a drug the more β-galactosidase activity will be observed, so the concentration that gives 50% inhibition (RIC50) of the total activity observed in a ΔdsbB strain can be used to get the increase in potency by dividing the RIC50 of compound 12 (0.16 μm, 95% confidence interval 0.13–0.20 μm) between the RIC50 of the tested compound. Thus, drugs more effective than compound 12 will have higher ratios. The results shown in Table 3 were obtained using data of at least three independent experiments.
DsbB immunoblots
Each plasmid containing dsbB mutants was integrated into the chromosome of the strain HK320 by λInCh method generating strains CL591–596 (43). To determine DsbB expression levels, strains CL591 to CL596 were grown aerobically in M63 minimal media with 1 mm IPTG until log phase. The lack of IPTG makes DsbB levels undetectable when dsbB is under trc204 promoter (data not shown). Proteins were TCA-precipitated, run on reducing SDS-PAGE, and immunoblotted against anti-DsbB (44). DTT was used for reducing disulfide bonds.
Author contributions
C. L. performed lptD and substructure experiments. B. M. M. performed anaerobic selection. L. M. and C. L. performed β-gal and growth assays. L. I. and F. H. purified proteins and performed in vitro and mass spectrometry assays. N. Q. T. purified a protein. D. B. performed bioinformatics analysis. C. L., B. M. M., and F. H. analyzed and interpreted the data. C. L., B. M. M., D. B., and J. B. discussed the data. C. L. and J. B. wrote the paper.
Supplementary Material
Acknowledgments
We thank Su Chiang and Jinbo Lee for helpful advice on medicinal chemistry. We also thank Dan Kahne for helpful discussions and kindly providing LptD strains.
This work was supported in part by National Institutes of Health Grant GMO41883 from NIGMS (to J. B. and D. B.), by the Blavatnik Biomedical Accelerator at Harvard University (to J. B.), and by an industry research agreement with F. Hoffmann-La Roche Ltd. and F. Hoffmann-La Roche Inc. (to J. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. S1, Schemes S1–S5, and additional information.
B. M. Meehan. C. Landeta, D. Boyd, and J. Beckwith, manuscript in preparation.
- β-Galdbs
- disulfide-bond sensitive β-galactosidase LacZ fused to the membrane protein MalF, which localizes LacZ in the periplasm making it sensitive to disulfide bond formation
- MIC
- minimal inhibitory concentration
- PBAD
- arabinose promoter
- Ptrc204
- promoter down mutation in −35 of pTrc99A promoter
- IPTG
- isopropyl β-d-1-thiogalactopyranoside.
References
- 1. Kadokura H., Katzen F., and Beckwith J. (2003) Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72, 111–135 [DOI] [PubMed] [Google Scholar]
- 2. Kadokura H., and Beckwith J. (2010) Mechanisms of oxidative protein folding in the bacterial cell envelope. Antioxid. Redox Signal. 13, 1231–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heras B., Shouldice S. R., Totsika M., Scanlon M. J., Schembri M. A., and Martin J. L. (2009) DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7, 215–225 [DOI] [PubMed] [Google Scholar]
- 4. Lasica A. M., Wyszynska A., Szymanek K., Majewski P., and Jagusztyn-Krynicka E. K. (2010) Campylobacter protein oxidation influences epithelial cell invasion or intracellular survival as well as intestinal tract colonization in chickens. J. Appl. Genet. 51, 383–393 [DOI] [PubMed] [Google Scholar]
- 5. Gonzalez M. D., Lichtensteiger C. A., and Vimr E. R. (2001) Adaptation of signature-tagged mutagenesis to Escherichia coli K1 and the infant-rat model of invasive disease. FEMS Microbiol. Lett. 198, 125–128 [DOI] [PubMed] [Google Scholar]
- 6. Miki T., Okada N., and Danbara H. (2004) Two periplasmic bisulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J. Biol. Chem. 279, 34631–34642 [DOI] [PubMed] [Google Scholar]
- 7. Rosadini C. V., Wong S. M., and Akerley B. J. (2008) The periplasmic disulfide oxidoreductase DsbA contributes to Haemophilus influenzae pathogenesis. Infect. Immun. 76, 1498–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim S. H., Park S. Y., Heo Y. J., and Cho Y. H. (2008) Drosophila melanogaster-based screening for multihost virulence factors of Pseudomonas aeruginosa PA14 and identification of a virulence-attenuating factor, HudA. Infect. Immun. 76, 4152–4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Totsika M., Heras B., Wurpel D. J., and Schembri M. A. (2009) Characterization of two homologous disulfide bond systems involved in virulence factor biogenesis in uropathogenic Escherichia coli CFT073. J. Bacteriol. 191, 3901–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Himpsl S. D., Lockatell C. V., Hebel J. R., Johnson D. E., and Mobley H. L. (2008) Identification of virulence determinants in uropathogenic Proteus mirabilis using signature-tagged mutagenesis. J. Med. Microbiol. 57, 1068–1078 [DOI] [PubMed] [Google Scholar]
- 11. Straskova A., Pavkova I., Link M., Forslund A. L., Kuoppa K., Noppa L., Kroca M., Fucikova A., Klimentova J., Krocova Z., Forsberg A., and Stulik J. (2009) Proteome analysis of an attenuated Francisella tularensis dsbA mutant: identification of potential dsbA substrate proteins. J. Proteome Res. 8, 5336–5346 [DOI] [PubMed] [Google Scholar]
- 12. Bardwell J. C., McGovern K., and Beckwith J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67, 581–589 [DOI] [PubMed] [Google Scholar]
- 13. Bardwell J. C., Lee J. O., Jander G., Martin N., Belin D., and Beckwith J. (1993) A pathway for disulfide bond formation in vivo. Proc. Natl. Acad. Sci. U.S.A. 90, 1038–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bader M., Muse W., Ballou D. P., Gassner C., and Bardwell J. C. (1999) Oxidative protein folding is driven by the electron transport system. Cell 98, 217–227 [DOI] [PubMed] [Google Scholar]
- 15. Dutton R. J., Boyd D., Berkmen M., and Beckwith J. (2008) Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X., Dutton R. J., Beckwith J., and Boyd D. (2011) Membrane topology and mutational analysis of Mycobacterium tuberculosis VKOR, a protein involved in disulfide bond formation and a homologue of human vitamin K epoxide reductase. Antioxid. Redox Signal. 14, 1413–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Landeta C., Blazyk J. L., Hatahet F., Meehan B. M., Eser M., Myrick A., Bronstain L., Minami S., Arnold H., Ke N., Rubin E. J., Furie B. C., Furie B., Beckwith J., Dutton R., and Boyd D. (2015) Compounds targeting disulfide bond forming enzyme DsbB of Gram-negative bacteria. Nat. Chem. Biol. 11, 292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruiz N., Chng S.-S., Hiniker A., Kahne D., and Silhavy T. J. (2010) Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc. Natl. Acad. Sci. U.S.A. 107, 12245–12250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meehan B. M., Landeta C., Boyd D., and Beckwith J. (2017) The essential cell division protein FtsN contains a critical disulfide bond in a non-essential domain. Mol. Microbiol. 103, 413–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sampson B. A., Misra R., and Benson S. A. (1989) Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122, 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eggert U. S., Ruiz N., Falcone B. V., Branstrom A. A., Goldman R. C., Silhavy T. J., and Kahne D. (2001) Genetic basis for activity differences between vancomycin and glycolipid derivatives of vancomycin. Science 294, 361–364 [DOI] [PubMed] [Google Scholar]
- 22. Ruiz N., Falcone B., Kahne D., and Silhavy T. J. (2005) Chemical conditionality: A genetic strategy to probe organelle assembly. Cell 121, 307–317 [DOI] [PubMed] [Google Scholar]
- 23. Chng S.-S., Xue M., Garner R. A., Kadokura H., Boyd D., Beckwith J., and Kahne D. (2012) Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science 337, 1665–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee J., Xue M., Wzorek J. S., Wu T., Grabowicz M., Gronenberg L. S., Sutterlin H. A., Davis R. M., Ruiz N., Silhavy T. J., and Kahne D. E. (2016) Characterization of a stalled complex on the β-barrel assembly machine. Proc. Natl. Acad. Sci. U.S.A. 113, 8717–8722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Inaba K., Takahashi Y. H., Fujieda N., Kano K., Miyoshi H., and Ito K. (2004) DsbB Elicits a red-shift of bound ubiquinone during the catalysis of DsbA oxidation. J. Biol. Chem. 279, 6761–6768 [DOI] [PubMed] [Google Scholar]
- 26. Wu T., Malinverni J., Ruiz N., Kim S., Silhavy T. J., and Kahne D. (2005) Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121, 235–245 [DOI] [PubMed] [Google Scholar]
- 27. Vuong P., Bennion D., Mantei J., Frost D., and Misra R. (2008) Analysis of YfgL and YaeT interactions through bioinformatics, mutagenesis, and biochemistry. J. Bacteriol. 190, 1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tellez R. Jr., and Misra R. (2012) Substitutions in the BamA β-barrel domain overcome the conditional lethal phenotype of a ΔbamB ΔbamE strain of Escherichia coli. J. Bacteriol. 194, 317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inaba K., Murakami S., Nakagawa A., Iida H., Kinjo M., Ito K., and Suzuki M. (2009) Dynamic nature of disulphide bond formation catalysts revealed by crystal structures of DsbB. EMBO J. 28, 779–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rietsch A., Belin D., Martin N., and Beckwith J. (1996) An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 93, 13048–13053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bader M. W., Xie T., Yu C. A., and Bardwell J. C. (2000) Disulfide bonds are generated by quinone reduction. J. Biol. Chem. 275, 26082–26088 [DOI] [PubMed] [Google Scholar]
- 32. Kobayashi T., and Ito K. (1999) Respiratory chain strongly oxidizes the CXXC motif of DsbB in the Escherichia coli disulfide bond formation pathway. EMBO J. 18, 1192–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Inaba K., Murakami S., Suzuki M., Nakagawa A., Yamashita E., Okada K., and Ito K. (2006) Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 127, 789–801 [DOI] [PubMed] [Google Scholar]
- 34. Kadokura H., and Beckwith J. (2002) Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. EMBO J. 21, 2354–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Newton N. A., Cox G. B., and Gibson F. (1971) The function of menaquinone (vitamin K2) in Escherichia coli K-12. Biochem. Biophys. Acta 244, 155–166 [DOI] [PubMed] [Google Scholar]
- 36. Shestopalov A. I., Bogachev A. V., Murtazina R. A., Viryasov M. B., and Skulachev V. P. (1997) Aeration-dependent changes in composition of the quinone pool in Escherichia coli. Evidence of post-transcriptional regulation of the quinone biosynthesis. FEBS Lett. 404, 272–274 [DOI] [PubMed] [Google Scholar]
- 37. Miller J. H. (1992) A short course in bacterial genetics and a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 38. Sambrook J., and Russell D. W. (eds) (2001) Molecular Cloning: A Laboratory Manual, Vols. 1–3, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 39. Weiss D. S., Chen J. C., Ghigo J. M., Boyd D., and Beckwith J. (1999) Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 181, 508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chimalakonda G., Ruiz N., Chng S.-S., Garner R. A., Kahne D., and Silhavy T. J. (2011) Lipoprotein LptE is required for the assembly of LptD by the β-barrel assembly machine in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 108, 2492–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gigot D., Crabeel M., Feller A., Charlier D., Lissens W., Glansdorff N., and Piérard A. (1980) Patterns of polarity in the Escherichia coli car AB gene cluster. J. Bacteriol. 143, 914–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Regeimbal J., Gleiter S., Trumpower B. L., Yu C. A., Diwakar M., Ballou D. P., and Bardwell J. C. (2003) Disulfide bond formation involves a quinhydrone-type charge-transfer complex. Proc. Natl. Acad. Sci. U.S.A. 100, 13779–13784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boyd D., Weiss D. S., Chen J. C., and Beckwith J. (2000) Towards single-copy gene expression systems making gene cloning physiologically relevant: Lambda InCh, a simple Escherichia coli plasmid-chromosome shuttle system. J. Bacteriol. 182, 842–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kadokura H., Bader M., Tian H., Bardwell J. C., and Beckwith J. (2000) Roles of a conserved arginine residue of DsbB in linking protein disulfide-bond-formation pathway to the respiratory chain of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 97, 10884–10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guzman L. M., Belin D., Carson M. J., and Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose P(BAD) promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.