

Abstract
The human cytomegalovirus (HCMV)-encoded viral cyclin-dependent kinase (v-CDK) UL97 phosphorylates the retinoblastoma (Rb) tumor suppressor. Here, we identify the other Rb family members p107 and p130 as novel targets of UL97. UL97 phosphorylates p107 and p130 thereby inhibiting their ability to repress the E2F-responsive E2F1 promoter. As with Rb, this phosphorylation, and the rescue of E2F-responsive transcription, is dependent on the L1 LXCXE motif in UL97 and its interacting clefts on p107 and p130. Interestingly, UL97 does not induce the disruption of all p107-E2F or p130-E2F complexes, as it does to Rb-E2F complexes. UL97 strongly interacts with p107 but not Rb or p130. Thus the inhibitory mechanisms of UL97 for Rb family protein-mediated repression of E2F-responsive transcription appear to differ for each of the Rb family proteins. The immediate early 1 (IE1) protein of HCMV also rescues p107- and p130-mediated repression of E2F-responsive gene expression, but it does not induce their phosphorylation and does not disrupt p107-E2F or p130-E2F complexes. The unique regulation of Rb family proteins by HCMV UL97 and IE1 attests to the importance of modulating Rb family protein function in HCMV-infected cells.
Keywords: cancer, cyclin-dependent kinase (CDK), herpesvirus, oncogene, phosphorylation, tumor suppressor gene, HCMV
Introduction
The retinoblastoma (Rb)5 family of proteins (also called the pocket protein family) consists of three members, Rb (1), p107 (2), and p130 (3). Although each protein has demonstrated function in attenuating cell cycle progression, only Rb (1, 4, 5) and p130 (6–10) act as tumor suppressors in vivo. The Rb family proteins display different expression patterns throughout the cell cycle (11, 12). p130 is expressed strongly in quiescent (G0) cells and at the beginning of the G1 phase of the cell cycle, but it declines as cells progress through the G1 phase. p107 is not detected in G0/G1 but accumulates in the S and G2 phases, whereas Rb is expressed at relatively constant levels throughout the cell cycle. All three Rb family proteins bind to and restrain the transcriptional potential of the E2F family of proteins that promote the transcription of multiple genes important for cell cycle progression (13, 14). Rb interacts with E2F1, E2F2, E2F3a, E2F3b, and E2F4, p107 with E2F1 and E2F4, and p130 with E2F4 and E2F5 (5, 13). In addition to the different expression profiles and E2F binding specificities, other differences exist between the Rb family proteins. Most notably, p107 and p130, but not Rb, associate with the MuvB core (LIN9, LIN37, LIN52, LIN54, and RBBP4) to form the DREAM complex (15, 16). The presence of three family members, and their overlapping yet unique functions, provides multiple levels of regulatory potential to cell cycle progression and tumor suppression.
The ability of Rb family proteins to interact with E2Fs is disrupted, and thus the transcriptional repressive function of Rb family proteins is inactivated by Rb family protein phosphorylation mediated by the cyclin-dependent kinases (CDKs) (17, 18). The Rb family proteins have amino acid motifs (abbreviated using the single letter code as RXL, where X depicts any amino acid) that interact with a hydrophobic patch (HP) on cyclin proteins, and this interaction contributes to CDK-dependent phosphorylation (19). Rb has 16 putative CDK phosphorylation sites (Ser/Thr-Pro motifs); p107 has 17, and p130 has 25. Mutant Rb, p107, or p130 proteins in which all or subsets of these sites are substituted with non-phosphorylatable residues are not inactivated by CDK-dependent phosphorylation (20). For Rb, phosphorylation at specific sites (Thr-373, Ser-608, Ser-612, Ser-788, Ser-795, Thr-821, and Thr-826) induces conformational changes that disrupt E2F binding through different mechanisms (21). For example, phosphorylation of Ser-608 prevents sequestering of the transactivation domains of the E2F proteins in the pocket domain of Rb by inducing an intramolecular conformational change in Rb (21–23). Ser-650 of p107 and Ser-672 of p130 are homologous to Rb Ser-608. Like Rb Ser-608, phosphorylation of Ser-650 weakens p107 pocket interactions with the E2F transactivation domain (24). Specific effects of p130 Ser-672 phosphorylation on E2F binding or transcriptional repression have not been reported.
Rb is phosphorylated not only by cellular CDKs but also by viral (v) CDKs. The β and γ families of the human herpesviruses encode v-CDKs that phosphorylate Rb. The v-CDK proteins are Epstein-Barr virus (EBV) BGLF4, human cytomegalovirus (HCMV) UL97, human herpesvirus type 6 (HHV-6) and type 7 (HHV-7) U69, and Kaposi's sarcoma-associated herpesvirus (KSHV) ORF36 (25). In addition to inactivation by phosphorylation, viruses also inactivate Rb family proteins through other mechanisms (26–29). For example, the adenovirus E1a protein and the simian virus 40 (SV40) T antigen bind Rb family proteins and disrupt their complexes with E2Fs. In addition, papillomavirus E7 and HCMV pp71 bind to Rb family proteins and induce their proteasomal degradation. In each of these instances, the hypophosphorylated form of Rb is targeted, and the reaction requires a specific motif on the viral protein with an amino acid sequence LXCXE. This LXCXE motif (30) associates in a well defined binding cleft (a shallow groove on the B box) found in all three Rb family proteins (31).
Unlike the cellular CDKs, the HCMV v-CDK UL97 utilizes an LXCXE motif, not a hydrophobic patch, to phosphorylate Rb (32). UL97 has three LXCXE motifs, the first one of which (called L1) is required for full UL97-mediated Rb phosphorylation and for the ability of UL97 to reverse Rb-imposed transcriptional repression of E2F-responsive promoter constructs in reporter assays (32). A recombinant HCMV in which wild-type UL97 is replaced with an L1 mutant UL97 (L1m) where the central cysteine of L1 is changed to a glycine (C151G) does not have a growth defect and does not show diminished induction of E2F-responsive genes (32, 33). This is likely because HCMV has multiple and possibly redundant mechanisms to inactivate Rb, p107, and p130 (26, 34–38). Indeed, the HCMV immediate early 1 (IE1) protein cooperates with UL97-L1m to relieve Rb-imposed E2F-responsive promoter repression, but it does not inactivate Rb by itself (32).
Our interest in UL97-mediated Rb protein inactivation stems from both the long-standing paradigm of using viral proteins to probe the mechanisms of cell cycle progression and oncogenesis, as well as our evolving understanding of an association between HCMV infection and human cancers. Although certainly not a classically defined tumor virus, HCMV is found in human tumors of the brain and breast (39–42), and the virus encodes multiple proteins that induce molecular events that represent the hallmarks of cancer (43–45). However, HCMV does not overtly transform cells in vitro. Thus it appears that, compared with tumor viruses such as human papillomavirus, HCMV modulates the cell cycle in a more nuanced way that does not lead to transformation but still supports viral infection. We suggest that understanding how HCMV modulates the cell cycle will illuminate potential ways the virus might contribute to cancer. But perhaps more importantly, an understanding of how HCMV manipulates the cell cycle without oncogenically transforming cells can direct us toward growth control pathways specific to cancer cells. Chemotherapeutic inhibition of such pathways might prove effective and better tolerated than more broadly acting drugs.
Here we extend our previous examination of how UL97 inactivates Rb to the Rb-related p107 and p130 proteins. We show that UL97 phosphorylates p107 and p130 in the context of an HCMV infection, when ectopically expressed, and during in vitro kinase reactions. The phosphorylation events require the L1 motif of UL97 and the clefts of p107 and p130, similar to Rb. However, UL97 does not disrupt all transcriptionally repressive complexes formed by p107 or p130, as it does for Rb (32). Furthermore, UL97 forms a stable complex with p107 but not p130 or Rb. Our data indicate that UL97 inactivates the different Rb proteins in different ways that are unique not only when compared with each other but also when compared with the mechanisms used by other viral and cellular proteins to inactivate Rb, p107, and p130.
Results
v-CDK UL97 phosphorylates Rb family members p107 and p130 during HCMV infection
A previous report detected p130 but not p107 phosphorylation during HCMV infection (46). We observed the phosphorylation of both p107 and p130 during HCMV productive infection of primary human fibroblasts by monitoring upward shifts in electrophoretic mobility as well as detection by an antibody specific for Ser-672-phosphorylated p130 (Fig. 1A). The phosphorylated forms of p107 (Fig. 1B) are more definitively separated using phosphate affinity (Phos-tag) gel electrophoresis (47) as confirmed by phosphatase treatment (Fig. 1C), perhaps explaining why they were not observed in the previous study. The percentage of p107 found in the phosphorylated state increased after HCMV infection (Fig. 1B). Consistent with our previous work with Rb (32, 48), we show here that the HCMV UL97 protein is responsible for p107 and p130 phosphorylation during infection, as these Rb family members are not phosphorylated during infection with a UL97-null virus (Fig. 1, A and B). Although the viral IE1 protein was reported to possess kinase activity and to phosphorylate p107 and p130 in vitro (49), IE1 is present in UL97-null virus-infected cells, yet p107 and p130 remain hypophosphorylated (Fig. 1, A and B). We conclude that during HCMV infection, IE1 does not phosphorylate p107 or p130 but UL97 does.
Figure 1.

UL97 phosphorylates p107 and p130 during HCMV lytic infection. HFFs were serum-starved for 48 h then infected with HCMV or UL97-null virus (Δ97) at an m.o.i. of 1. Whole-cell lysates were prepared at the indicated times postinfection, and equal amounts of protein from each sample were loaded onto a normal gel (A) or phosphate affinity (phos-tag) gel (B) and subjected to Western blotting analysis with the indicated antibodies. The percentage of phosphorylated p107 (compared with total) quantified by ImageJ is presented below the panel. M, mock infection. C, lysates from HCMV-infected HFFs were treated (+) or not (−) with λ protein phosphatase (l-PPase) and analyzed as in B. D, Saos-2 cells were transfected with expression plasmids for HA-tagged p107 (HA-p107) together with either an empty vector (EV), one expressing V5-tagged UL97 (97), cyclin A2 (A2)/CDK2 (K2), cyclin E1 (E1)/CDK2, or cyclin D2 (D2)/CDK4 (K4). Lysates harvested 48 h after transfection were analyzed by Western blotting with the indicated antibodies. E, transfections were performed as in D with HA-tagged p130 (HA-p130), p130ΔCDK4, or p130-PM19A. F and G, lysates of Saos-2 cells transfected with plasmids expressing HA-tagged p107 (HA-p107) or p130 (HA-p130) were immunoprecipitated with anti-p107 or p130 antibodies. Either wild-type (WT) or kinase-dead (KD) UL97 was incubated with immune-enriched p107 or p130 substrates in the presence of [γ-32P]ATP. Kinase reactions were resolved by SDS-PAGE and transferred to a nitrocellulose membrane. They were visualized by both autoradiography and Western blotting with an anti-HA antibody for detection of HA-UL97 and HA-p107 and an anti-p130 antibody for detection of HA-p130. M.W., molecular weight. H, kinase reaction was conducted as in F except normal ATP and bacterially purified GST-tagged p130 were utilized. Samples were analyzed by Western blotting with the indicated antibodies. Experiments were performed in biological triplicate except those in D and E that were performed in biological duplicates.
UL97 also phosphorylates p107 (Fig. 1D) and p130 (Fig. 1E) when transiently expressed by transfection of uninfected Saos-2 cells. Transfection of human cyclin/CDK pairs also resulted in p107 and p130 phosphorylation in these experiments. Although previous studies conducted in T98G cells concluded only cyclin D/CDK4 directed p130 Ser-672 phosphorylation (50), our experiments in Saos-2 cells detected phosphorylation of this residue after cyclin E, cyclin A, or UL97 transfection as well as after cyclin D transfection (Fig. 1E). UL97 could not phosphorylate residue 672 of a p130 mutant protein (p130ΔCDK4) in which three previously defined CDK4-dependent target sites (Thr-401, Ser-672, and Ser-1035) are substituted with alanines (Fig. 1E). However, UL97 still induced a slower migrating form of p130ΔCDK4 (Fig. 1E), suggesting that UL97, like the cellular CDKs, phosphorylates p130 at multiple sites, including Ser-672. UL97 and the cyclin/CDK pairs failed to hyperphosphorylate a mutant p130 protein (PM19A) (50) in which 18 of the 25 potential CDK phosphorylation sites (Ser/Thr-Pro motifs) were replaced with alanine residues (Fig. 1E) suggesting that, like Rb (32), most UL97 target sites on p130 are CDK consensus sites.
In vitro, UL97 autophosphorylates and phosphorylates p107 (Fig. 1F) and p130 (Fig. 1G) as determined by autoradiography. Western blotting with a p130 phospho-specific antibody (Fig. 1H) also detects UL97-mediated in vitro phosphorylation. A UL97 point mutant lacking kinase activity (51) fails to phosphorylate p107 (Fig. 1F) or p130 in vitro (Fig. 1, G and H). Combined, we conclude that UL97 is necessary and sufficient for p107 and p130 phosphorylation during HCMV infection.
Other v-CDKs also phosphorylate p107 and p130
All human herpesviruses encode a kinase orthologous to UL97. The orthologous kinases from the betaherpesviruses (HHV-6 and HHV-7) and gammaherpesviruses (EBV and KSHV) also display v-CDK activity and phosphorylate Rb, whereas the orthologs in the alphaherpesviruses (herpes simplex virus type 1 (HSV-1), HSV-2, and varicella zoster virus) do not (25). Similarly, we found that the alphaherpesvirus proteins were unable to phosphorylate p107 (Fig. 2A) or p130 (Fig. 2B), but the beta- and gammaherpesvirus proteins phosphorylated p107 (Fig. 2C) and p130 (Fig. 2D) with different efficiencies. Kinase-dead (KD) mutants of these proteins failed to phosphorylate p107 or p130, as expected. We conclude that the v-CDKs encoded by beta- and gammaherpesviruses phosphorylate p107 and p130. This ability highlights the functional relatedness of the v-CDKs as well as the importance of modifying Rb family protein function by phosphorylation for successful infection by these pathogenic human viruses. Understanding how UL97 (the representative v-CDK) targets and inactivates the Rb family proteins not only opens up opportunities for inhibiting this reaction as an antiviral intervention but also illustrates how the Rb family proteins might be inactivated during oncogenic transformation in virus-infected or -uninfected cancer cells.
Figure 2.

v-CDKs of EBV, HHV6, HHV7, and KSHV phosphorylate p107 and p130. Saos-2 cells were transfected with expression plasmids encoding HA-tagged p107 (A and C) or HA-tagged p130 (B and D) together with either an empty vector (EV) or an HA-tagged human herpesvirus protein kinase (HA-PK). Forty eight hours later, cells were harvested and subjected to Western blotting with the indicated antibodies. C, upper blot for HA-p107 corresponds to the blot after short exposure, and the bottom blot corresponds to the blot after long exposure. WT, wild type. KD, kinase-dead. The phosphorylation-dependent band shift of p107 was detected by phosphate affinity (phos-tag) gel electrophoresis. Experiments were performed in biological duplicate.
L1 motif of UL97 and the clefts of p107 and p130 are required for p107 and p130 phosphorylation
UL97 could potentially interact with Rb family proteins (physically or functionally) through two distinct interfaces: one or more of three LXCXE motifs in UL97 (L1, L2, or L3) interacting with the binding cleft of the Rb family proteins, or the HP of UL97 interacting with RXL motifs in the Rb family proteins. We previously showed that of all these potential interfaces, only the first LXCXE motif (L1) of UL97 and the cleft of Rb were required for UL97-mediated Rb hyperphosphorylation (32). The rest of the motifs were dispensable. We achieved essentially identical results with p107 and p130. UL97 L2 (C428G) and L3 (C693G) mutants that carry the same central cysteine to a glycine substitution as the L1 mutant phosphorylated p107 (Fig. 3A) and p130 (Fig. 3B) as well as wild type in transfection assays, but the L1 mutant did not. Mutation of all three LXCXE motifs (Tri) also impaired p107 (Fig. 3A) and p130 (Fig. 3B) phosphorylation. Similar results were obtained upon infection of fibroblasts with recombinant HCMVs harboring the UL97 mutant alleles (Fig. 3C). Each LXCXE motif mutant virus and the triple LXCXE mutant virus grow like wild type (32, 33). Both the L1 and Tri mutant viruses showed reduced p107 and p130 phosphorylation compared with wild type (Fig. 3C). The L1 mutation-specific decreases in p130 phosphorylation were less dramatic during infection than transfection but were reproducible and statistically significant (Fig. 3D). It is possible the limited p107 and p130 phosphorylation observed in the presence of UL97-L1 mutant results from some residual activity of the mutant kinase toward the substrates. For example, the L1 mutant still efficiently phosphorylates Rb on some cyclin/CDK target sites (32). Alternatively, this restricted phosphorylation may be the result of a cellular kinase.
Figure 3.

Phosphorylation of p107 and p130 by UL97 requires the L1 LXCXE motif of UL97 and the cleft of p107 and p130. A and B, Saos-2 cells were transfected with plasmids encoding HA-tagged p107 (A) or p130 (B) together with either an empty vector (EV), an expression plasmid for wild-type UL97 (WT), or an expression plasmid for UL97 alleles with the following mutations: L1, C151G; L2, C428G; L3, C693G; Tri, C151G/C428G/C693G; HP, W368A; quadruple (Qd), C151G/W368A/C428G/C693G; kinase-dead (KD), K355Q. After 48 h, cells were harvested and subjected to Western blotting with the indicated antibodies. C, serum-starved HFFs were infected with HCMV or the indicated LXCXE mutant viruses at an m.o.i. of 1. Whole-cell lysates were subjected to Western blotting analysis with the indicated antibodies. M, mock infection. D, level of p130 phosphorylated at Ser-672 normalized to total p130 was quantitated from wild-type or L1 mutant UL97 virus-infected cells at 48 h post-infection in experiments identical to those in C except at an m.o.i. of 2. Values are presented relative to the value in wild-type virus-infected cells (set at 1). Error bars denote the standard deviation. *, p < 0.05. E, Saos-2 cells were transfected with expression plasmids encoding V5-tagged wild-type UL97 together with either wild-type HA-tagged p107, a p107 cleft mutant (C846F), or a p107 RXL mutant. Forty eight hours later, lysates were harvested and subjected to Western blotting with the indicated antibodies. F, Saos-2 cells were transfected with expression plasmids encoding V5-tagged wild-type UL97 together with either wild-type HA-tagged p130 or a p130 cleft mutant (C894F). Forty eight hours later, lysates were harvested and subjected to Western blotting with the indicated antibodies. G, Saos-2 cells were transfected with expression plasmids encoding HA-tagged wild-type UL97 together with either wild-type HA-tagged p107, a p107 cleft mutant (N935F), or a p107 RXL mutant. Forty eight hours later, lysates were harvested and subjected to Western blotting with the indicated antibodies. H, Saos-2 cells were transfected with expression plasmids encoding HA-tagged wild-type UL97 together with either wild-type HA-tagged p130, a p130 cleft mutant (N1010F), or a p130 RXL mutant. Forty eight hours later, lysates were harvested and subjected to Western blotting with the indicated antibodies. I, Saos-2 cells were transfected with an expression plasmid for cyclin E1 together with HA-tagged wild-type or RXL mutant p107 or p130 expression plasmids. Lysates harvested 48 h after transfection were subjected to immunoprecipitation (IP) with the HA antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. J, Saos-2 cells were transfected with expression plasmids encoding cyclin E1 and CDK2 together with HA-tagged wild-type p107, a p107 RXL mutant, wild-type p130, or a p130 RXL mutant. Forty eight hours later, lysates were harvested and subjected to Western blotting with the indicated antibodies. The phosphorylation-dependent band shifts of both exogenously expressed and endogenous p107 were detected in phosphate affinity (phos-tag) gels. Experiments were performed in at least biological triplicates.
We tested the necessity of the L1-cleft interaction for UL97-mediated p107 or p130 phosphorylation with reciprocal mutations of the cleft domains of p107 and p130 as we did previously for Rb (32). Two mutants were analyzed for each protein. One set of previously studied alleles (p107-C846F (52) and p130-C894F (53)) mimic the Rb-C706F allele found in the small cell lung cancer H209 cell line (54, 55). Neither the p107-C846F (Fig. 3E) nor the p130-C894F (Fig. 3F) cleft mutant protein was efficiently phosphorylated by wild-type UL97. We generated a second set of cleft mutants (p107-N935F and p130-N1010F) corresponding to another previously designed Rb-N757F cleft mutant (56). Neither the p107-N935F (Fig. 3G) nor the p130-N1010F (Fig. 3H) cleft mutant protein was efficiently phosphorylated by wild-type UL97. We conclude that the UL97 L1 motif and the p107 and p130 clefts are required for UL97-mediated p107 and p130 phosphorylation.
The HP mutant showed reduced phosphorylation of p107 (Fig. 3A) but not p130 (Fig. 3B) in transfection assays. However, results with the HP mutant of UL97 (and with the quadruple mutant combination of L1, L2, L3, and HP motifs) must be interpreted with caution because this specific lesion (W368A) appears to attenuate overall kinase activity and viral replication (32). Therefore, to uncover any role for the HP-RXL interaction in UL97-mediated p107 or p130 phosphorylation, we determined the ability of wild-type UL97 to phosphorylate p107 or p130 alleles lacking RXL motifs, as we did previously for Rb (32). We generated epitope-tagged RXL deletion mutants of p107 (p107-ΔRXL) (57) and p130 (p130-ΔRXL) (58) and expressed them in Saos-2 cells by transient transfection. UL97 phosphorylated the RXL mutant p107 (Fig. 3, E and G) and p130 (Fig. 3H) proteins as well as their wild-type counterparts. Confirming previous results (57, 58), cyclin E showed reduced interaction with the RXL mutants of p107 and p130 compared with the wild-type proteins (Fig. 3I). Furthermore, the RXL mutants of p107 and p130 were not phosphorylated as completely as the wild-type proteins by cyclin E/CDK2 (Fig. 3J). Because the p107- and p130-RXL mutants used here show a reduced ability to interact physically and functionally with a hydrophobic patch-containing protein, we conclude the HP-RXL interaction is dispensable for UL97-mediated p107 or p130 phosphorylation.
UL97-mediated phosphorylation of p107 and p130 inactivate their ability to repress an E2F-responsive promoter
Exogenously expressed cyclin E inactivates exogenously expressed p107 or p130 in Saos-2 cells (20). We found that cotransfected UL97 was also capable of reversing the transcriptional repression of an E2F1 promoter reporter construct instituted by either p107 (Fig. 4A) or p130 (Fig. 4B) in Saos-2 cells. UL97 recovered p107-suppressed (Fig. 4C) or p130-suppressed (Fig. 4D) E2F1 reporter activity to a level indistinguishable from restoration by cyclin E/CDK2. Reporter rescue required the ability of UL97 to phosphorylate p107 or p130, as L1 or KD mutants failed to reverse the transcriptional repression mediated by p107 (Fig. 4A) or p130 (Fig. 4B). However, the L3 mutant that fully phosphorylates p107 and p130 (Fig. 3) rescued transcriptional repression (Fig. 4, A and B). We conclude that UL97 inactivates p107 and p130 by phosphorylation.
Figure 4.

UL97 relieves p107- or p130-mediated repression of an E2F-responsive promoter. A, Saos-2 cells were transfected with a luciferase reporter driven by the E2F1 promoter together with either an empty vector (−) or an expression plasmid for HA-tagged p107, and either an empty vector or an expression plasmid for the indicated allele of UL97. Lysates harvested 48 h after transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without p107 or UL97 (set at 100%). B, luciferase and Western blotting analyses were performed as in A except with HA-tagged p130. Luciferase activity is presented relative to the activity of the reporter without p130 or UL97 (set at 100%). C, luciferase and Western blotting analyses were performed as in A except cyclin E1/CDK2 (E/2) was included. Luciferase activity is presented relative to the activity of the reporter with UL97 (97) (set at 100%). D, luciferase and Western blotting analyses were performed as in B except cyclin E1/CDK2 was included. Luciferase activity is presented relative to the activity of the reporter with UL97 (set at 100%). Error bars denote the standard deviation of more than three biological replicates. *, p < 0.05; **, p < 0.01; n.s., not significant.
Rb-related p130 protein is required for efficient HCMV productive replication
UL97 also phosphorylates and inactivates Rb, but paradoxically, the Rb protein is required for efficient HCMV infection (59). Conversely, p107, which is also phosphorylated (Fig. 1) and inactivated (Fig. 4) by UL97, is not required for HCMV infection (60). Because p130 had yet to be tested, we monitored HCMV infectious progeny formation in cells in which p130 expression was stably reduced by shRNA-mediated interference. Similar to Rb, stable p130 knockdown cells produced fewer infectious progeny after 4 days of HCMV infection than did control cells transduced with a scrambled shRNA (Fig. 5). We have not tested longer time points but predict the observed decrease in replication would persist because it does in Rb-knockdown cells (60). Likewise, because a clinical strain virus also shows decreased replication in Rb-knockdown cells (59), we suspect one would also show defects in p130-knockdown cells, but we have not performed this experiment. Based on this result and previous data, we conclude that Rb and p130, but not p107, are required for efficient HCMV productive replication.
Figure 5.
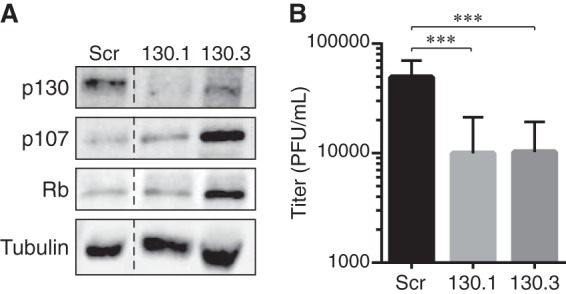
Stable p130 knockdown reduces HCMV productive replication. A, equal amounts of protein lysates from subconfluent, serum-starved NHDFs transduced with a retrovirus expressing a scrambled shRNA sequence (Scr) or either of two shRNAs targeting p130 (130.1 or 130.3) were analyzed by Western blotting with the indicated antibodies. Dashed line indicates the removal of irrelevant lanes and merging of the remaining images. B, serum-starved Scr, 130.1, or 130.2 cells were infected with AD169 at an m.o.i. of 1. Combined cell-free and cell-associated virus was collected 4 days postinfection, and the titers of virus were determined by standard plaque assay. Error bars represent the standard deviation of six biological replicates. ***, p < 0.0025.
UL97 forms a stable complex with p107 but not with p130
Because Rb and p130 are required for efficient HCMV infection but p107 is not, we explored whether the mechanism of UL97-mediated inactivation of p130 in the context of E2F-dependent gene expression was similar to that of Rb inactivation, and conversely whether UL97-mediated inactivation of p107 was somehow different from Rb inactivation. Our previous work (32) demonstrated that UL97 did not form a stable complex with Rb but did disrupt Rb-E2F complexes. Therefore, we examined UL97 binding to p107 and p130 and the ability of UL97 to disrupt p107-E2F and p130-E2F complexes.
With immunoprecipitation-Western blotting experiments from transfected cells, we detected a strong interaction between UL97 and p107 that required both the L1 motif of UL97 (Fig. 6A) and an intact cleft domain in p107 (Fig. 6B). Unlike cyclin E (Fig. 3I), the RXL motif of p107 was not required for interaction with UL97 (Fig. 6B). However, kinase activity was required as the UL97 point mutant lacking kinase activity failed to interact with p107 (Fig. 6, A and C), even when expressed to higher levels than the wild-type protein (Fig. 6C). We detected the UL97-p107 interaction by Western blotting after immunoprecipitation of p107 (Fig. 6, A and C) or UL97 (Fig. 6D) as well as after covalent capture of UL97 (Fig. 6B). p130 interacted weakly with UL97 in similar experiments (Fig. 6E). Mutating the UL97 L1 motif reduced this interaction (Fig. 6E). The UL97 point mutant lacking kinase activity failed to interact with p130 (Fig. 6E). By using the same antibody (to the HA epitope tag) to precipitate UL97 and the same antibody (FLAG) to detect either p107 or p130, we were able to compare the relative efficiency with which these two pocket proteins interacted with UL97, and we found that UL97 bound 3-fold more p107 than p130 (Fig. 6D). We conclude that UL97 associates strongly with p107 and weakly with p130 through L1-cleft interactions in a kinase activity-dependent manner.
Figure 6.

UL97 does not disrupt all p107-E2F or p130-E2F complexes. A, Saos-2 cells were transfected with an expression plasmid for wild-type FLAG-tagged p107 together with an empty vector (−) or an expression plasmid for the indicated allele of UL97 or adenovirus E1a. 97, wild-type UL97; L1, L1 mutant; KD, kinase-dead mutant. Lysates harvested 48 h after transfection were subjected to immunoprecipitation (IP) with a p107 antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. h.c., heavy chain; C, rabbit IgG control. B, Saos-2 cells were transfected with an expression plasmid for the indicated p107 protein together with expression plasmids for HALO- and HA-tandem-tagged UL97 (HALO/HA-97). Lysates harvested 48 h after transfection were subjected to HALO tag capture and cleaved with TEV protease between the HALO and HA sequences to generate HA-tagged UL97 (HA-97). Input lysates and captured proteins were analyzed by Western blotting with the HA antibody. CM1, p107-C846F; CM2, p107-N935F; RXL, p107-ΔRXL. C, Saos-2 cells were transfected with a constant amount (1.6 μg) of an expression plasmid for wild-type FLAG-tagged p107 together with differing amounts of expression plasmids for either wild-type or kinase-dead UL97. Lysates were subjected to immunoprecipitation with a p107 antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. D, Saos-2 cells were transfected with an expression plasmid for FLAG-tagged p107 or p130 together with an empty vector (−) or an expression plasmid for HA-tagged UL97. Lysates harvested at 48 h after transfection were subjected to immunoprecipitation with the HA antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. The levels of FLAG-tagged p107 or p130 bound to HA-tagged UL97 were quantitated using ImageJ, and the values are presented below the panel. E, Saos-2 cells were transfected with an expression plasmid for wild-type FLAG-tagged p130 together with an empty vector (−) or an expression plasmid for the indicated allele of UL97 or adenovirus E1a. Lysates harvested 48 h after transfection were subjected to immunoprecipitation with a p130 antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. h.c., heavy chain; l.c., light chain; C, rabbit IgG control. F and G, quantification of the levels of bound E2F1 or E2F4 normalized to immunoprecipitated p107 or p130 in the absence (−) or presence of wild-type UL97 (F) or E1a (G) from A and E performed using ImageJ. Values are presented relative to the value in the absence of UL97 or E1a (set at 1). Error bars denote the standard deviation of more than three biological replicates. *, p < 0.05; **, p < 0.01; n.s., not significant. H, Saos-2 cells were transfected with a constant amount (1.8 μg) of an expression plasmid for FLAG-tagged p107 or Rb together with differing amounts of expression plasmids for UL97. Lysates harvested 48 h after transfection were subjected to immunoprecipitation with a p107 antibody or an Rb antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. C, rabbit IgG control. Arrow, E2F1-specific band; asterisk, nonspecific band. I and J, Saos-2 cells were transfected with an expression plasmid for FLAG-tagged p107 (I) or p130 (J) together with expression plasmids for cyclin E1/CDK2 (E/2) or HA-tagged UL97. Lysates harvested at 48 h after transfection were subjected to immunoprecipitation with a p107 (I) or p130 antibody (J). Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. K, quantification of the levels of bound E2F normalized to immunoprecipitated p107 or p130 from I and J performed using ImageJ. Values are presented relative to the value in the absence of cyclin E1/CDK2 (set at 1). Error bars denote the standard deviation of more than three biological replicates. *, p < 0.05. Experiments were performed in at least biological triplicates except those in C and D that were performed in biological duplicates.
Neither UL97 nor IE1 disrupts E2F interactions with p107 or p130
The textbook model for the inactivation of Rb family protein function (61) is that phosphorylation mediated by cellular CDKs or binding by viral oncoproteins disrupts their complexes with E2F transcription factors. Indeed, we previously found that UL97-mediated Rb phosphorylation disrupted its complexes with E2F1, E2F2, E2F3a, and E2F3b (32). However, UL97 was unable to disrupt p107 complexes with E2F1 or E2F4 (Fig. 6, A and F) whereas adenovirus E1a effectively disrupted them (Fig. 6, A and G). UL97 failed to disrupt p107-E2F1 complexes in the same experiment where lower levels of UL97 effectively disrupted Rb-E2F1 complexes (Fig. 6H). Likewise, UL97 was unable to disrupt p130 complexes with E2F4 (Fig. 6, E and F) but adenovirus E1a effectively disrupted them (Fig. 6, E and G). p130 complexes with E2F5 were not detected in our experiments in Saos-2 cells (Fig. 6E). In similar experiments, cyclin E/CDK2 showed a statistically significant ability to disrupt p107-E2F1, p107-E2F4, and p130-E2F4 complexes (Fig. 6, I–K). Although the magnitude of disruption by UL97 was in some cases comparable with cyclin E/CDK2, our results with UL97 never reached statistical significance (Fig. 6F). Therefore, we conclude that UL97-mediated inactivation of p107 and p130 does not occur through disruption of their complexes with the E2F proteins.
The HCMV IE1 protein reversed p107-mediated repression of the E2F-responsive adenovirus E2 promoter during cotransfection/reporter assays in C33A cells (34) indicating that, like UL97, IE1 can inactivate p107. We confirmed the ability of IE1 to inactivate p107 by demonstrating IE1-mediated reversal of p107-instituted repression of a reporter driven by the E2F1 promoter in Saos-2 cells (Fig. 7A). We also demonstrated IE1-mediated inactivation of p130 with a similar assay (Fig. 7B). Despite reports of in vitro kinase activity for the IE1 protein (49), we found no evidence that IE1 induced the phosphorylation of p107 or p130 in vivo (Fig. 7C). However, we did detect a stable interaction between IE1 and both p107 (Fig. 7D) and p130 (Fig. 7E). Like UL97-mediated inactivation of p107 and p130, IE1-mediated inactivation of p107 or p130 did not occur through the disruption of p107-E2F1 (Fig. 7, D and F), p107-E2F4 (Fig. 7, D and F), or p130-E2F4 (Fig. 7, E and F) complexes. Our work mirrors a previous study (62) that detected an interaction between IE1 and p107 that did not dissociate E2F4. We conclude that IE1 binds and inactivates p107 and p130 without disrupting their complexes with E2F proteins.
Figure 7.

IE1 relieves p107- or p130-mediated repression of an E2F-responsive promoter. A, Saos-2 cells were transfected with a luciferase reporter driven by the E2F1 promoter together with either an empty vector (−) or an expression plasmid for HA-tagged p107, and either an empty vector or an expression plasmid for IE1. Lysates harvested 48 h after transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without p107 or IE1 (set at 100%). B, luciferase assay and Western blotting analyses were performed as in A except with HA-tagged p130. Luciferase activity is presented relative to the activity of the reporter without p130 or IE1 (set at 100%). Error bars denote the standard deviation of more than three biological replicates. *, p ≤0.05; n.s., not significant. C, Saos-2 cells were transfected with an expression plasmid for HA-p107 or HA-p130 together with either an empty vector (EV) or an expression plasmid for HA-tagged IE1 or V5-tagged UL97. Lysates harvested 48 h after transfection were analyzed by Western blotting with the indicated antibodies. D, Saos-2 cells were transfected with an expression plasmid for HA-tagged p107 and either an empty vector (−) or an expression plasmid for IE1. Lysates harvested 48 h after transfection were subjected to immunoprecipitation with a p107 antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. E, Saos-2 cells were transfected with an expression plasmid for HA-tagged p130 and either an empty vector (−) or an expression plasmid for IE1. Lysates harvested 48 h after transfection were subjected to immunoprecipitation with a p130 antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. F, quantification of the levels of bound E2F normalized to immunoprecipitated p107 or p130 from D and E performed using ImageJ. Values are presented relative to the value in the absence of IE1 (set at 1). Error bars denote the standard deviation of biological triplicates. n.s., not significant.
HCMV infection fails to disrupt the transcriptional repressive complexes assembled by p107 or p130
Our inability to detect UL97- or IE1-mediated disruption of p130-E2F complexes during transfection assays (Figs. 6 and 7) is consistent with the previous finding that the p130-E2F4 complex can be captured from HCMV-infected cells (46). We confirmed that p130-E2F4 complexes are stable in serum-starved cells infected with HCMV (Fig. 8A, lane 3) at a multiplicity of infection (m.o.i. = 3) capable of expressing UL97 in most of the cells (Fig. 8B) and that p130-E2F4 complexes are disrupted by serum stimulation of uninfected cells (Fig. 8A, lane 6). Thus in a setting more relevant than transfection assays, UL97 still fails to disrupt p130-E2F4 complexes.
Figure 8.

HCMV infection does not disrupt p130-E2F4-MuvB complexes. A, serum-starved HFFs were mock-infected (M) or infected with wild-type HCMV (V) or the UL97 L1 mutant virus (L1) at an m.o.i. of 3. At 2 h post-infection DMSO or 10 μm maribavir (MBV: +) was added. Lysates harvested 45 h post-infection were subjected to immunoprecipitation (IP) with p130, LIN54, or LIN9 antibodies or normal rabbit IgG (C) as a control. Uninfected serum-stimulated cells (S) were incubated with 15% FBS for 18 h before harvesting. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. pS672, p130 phospho-Ser-672 specific antibody. Lanes with immunoprecipitates are numbered below. B, serum-starved HFFs were infected with HCMV as in A. At 45 h post-infection cells were subjected to immunostaining with a UL97 antibody and Hoechst treatment to counterstain DNA. C, U-2 OS cells were transfected with an expression plasmid for FLAG-tagged LIN52 together with an expression plasmid for HA-tagged UL97. Lysates harvested 48 h after transfection were subjected to immunoprecipitation with the FLAG antibody (F) or normal mouse IgG (C) as a control. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. D, Saos-2 cells were transfected with expression plasmids for wild-type FLAG-tagged p107 and wild-type E2F4 together with an empty vector (−) or an expression plasmid for UL97. Lysates harvested 48 h after transfection were subjected to immunoprecipitation with an E2F4 antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. C, rabbit IgG control. E, Saos-2 cells were transfected as in D except wild-type FLAG-tagged p130 was included. Lysates harvested 48 h after transfection were subjected to immunoprecipitation with an E2F4 or p130 phospho-Ser-672-specific antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. C, rabbit IgG control. Lanes with immunoprecipitates are numbered below. F, Saos-2 cells were transfected with a luciferase reporter driven by the E2F1 promoter together with either an empty vector (−) or an expression plasmid for p130ΔCDK4 (Δ) and either an empty vector or a wild-type UL97 expression plasmid. Lysates harvested 48 h after transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without p130ΔCDK4 or UL97 (set at 100%). Error bars denote the standard deviation of more than three biological replicates. **, p < 0.01. G, luciferase and Western blotting analyses were performed as in F except expression plasmids for cyclin E1/CDK2 (E/2) were included. H, Saos-2 cells were transfected with an expression plasmid for FLAG-tagged wild-type p130 (WT) or p130ΔCDK4 (Δ) together with expression plasmids for cyclin E1/CDK2 (E/2). Lysates harvested 48 h after transfection were subjected to immunoprecipitation with a FLAG antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. C, mouse IgG control. I, quantification of the levels of bound E2F4 normalized to immunoprecipitated FLAG-tagged p130ΔCDK4 (Δ) from H performed using ImageJ. Values are presented relative to the value in the absence of cyclin E1/CDK2 (set at 1). The error bar denotes the standard deviation of biological triplicate samples. *, p < 0.05. J, input lysates from A were analyzed by Western blotting with the indicated antibodies. Experiments were performed in at least biological triplicates except those in D and E that were performed in biological duplicates.
In addition to E2F proteins, transcriptionally repressive associations assembled by p107 and p130 contain components of the MuvB complex, including LIN9, LIN52, and LIN54 (15, 16). Like complexes with E2F4, serum stimulation was able to disrupt p130-LIN54 (Fig. 8A, lanes 6 and 11) and p130-LIN9 complexes (Fig. 8A, lanes 6 and 16). However, in HCMV-infected cells p130 coprecipitated with LIN54 (Fig. 8A, lanes 3 and 8) or LIN9 (Fig. 8A, lanes 3 and 13). Similarly, both p130 and p107 were found in stable association with the ectopically expressed MuvB component LIN52 in the presence of UL97 in U-2 OS cells (Fig. 8C). We conclude that UL97 fails to disrupt p107-MuvB and p130-MuvB complexes. Although we did not directly test IE1, results indicate that p130-E2F4-MuvB complexes are stable within infected cells that express IE1 (Fig. 8, A and J). Thus we predict that, like UL97, IE1 is unable to disrupt complexes between p107 or p130 and MuvB components in the context of infection.
Comparing the electrophoretic mobility of p130 precipitated by LIN54 or LIN9 in mock (Fig. 8A, lanes 7 and 12) or HCMV-infected cells (Fig. 8A, lanes 8 and 13), it appears that a phosphorylated (slower migrating) form of p130 associates with these MuvB components during productive viral replication. However, Ser-672-phosphorylated p130 was no longer able to bind LIN54 (Fig. 8A, lane 8) or LIN9 (Fig. 8A, lane 13) during HCMV infection. Similarly, in UL97-transfected cells, E2F4 bound slower-migrating phosphorylated forms of p107 (Fig. 8D) and p130 (Fig. 8E, lane 3) but not Ser-672-phosphorylated p130 (Fig. 8E, lane 5). Therefore, it appears that p130 (and likely p107) participates in at least two unique complexes with E2F4 and MuvB in HCMV-infected cells, one that is disrupted by UL97-mediated phosphorylation of p130 Ser-672, and one that is maintained despite UL97-mediated phosphorylation of p130 residues other than Ser-672.
We reasoned that disruption of p130-E2F4-MuvB complexes by UL97-mediated phosphorylation of Ser-672 (with probable simultaneous phosphorylation of other residues) was likely to inactivate the transcriptional repressive function of p130. Therefore, we asked whether the other complex that contains E2F4, MuvB, and p130 phosphorylated at residues other than Ser-672, and that is co-present with UL97, is capable of repressing an E2F-driven reporter. The p130ΔCDK4 protein (T401A/S627A/S1035A) repressed the E2F1 promoter in Saos-2 cells (Fig. 8F) but was inactivated by UL97 (Fig. 8F). We conclude that, despite the inability of UL97 to disrupt p130-E2F4-MuvB complexes in which Ser-672 is not phosphorylated, UL97 can still functionally inactivate this stable complex. Furthermore, we conclude that disruption of p130 complexes via Ser-672 phosphorylation is mediated by UL97 but is not necessary for UL97-mediated p130 inactivation. Although we cannot test p107 because an antibody specific for the phosphorylated form of the homologous residue (Ser-650) is unavailable, we hypothesize that a similar mechanism would explain the ability of UL97 to inactivate p107 (see Fig. 9). Interestingly, cyclin E/CDK2 was able to both functionally inactivate p130ΔCDK4 (Fig. 8G) and disrupt its association with E2F4 (Fig. 8, H and I). This suggests that, like with UL97, p130 Ser-672 phosphorylation is not required for inactivation, but unlike UL97, complex disruption may be required for cyclin E/CDK2 to inactivate p130.
Figure 9.
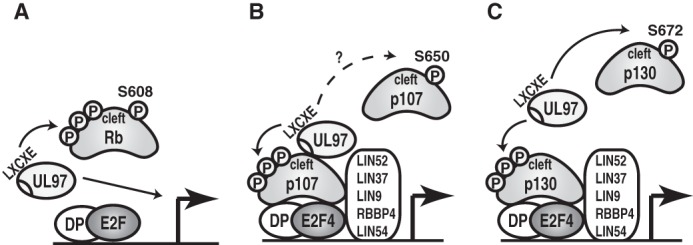
Models for Rb, p107, or p130 inactivation by UL97. A, UL97 rescues Rb-mediated repression of E2F-responsive gene expression by both phosphorylation-mediated (P) disruption of Rb-E2F complexes as well as through an undefined L1 LXCXE-dependent mechanism. B, UL97 associates with and phosphorylates p107 to rescue p107-mediated repression of E2F-responsive gene expression in a UL97-L1- and p107-cleft-dependent manner but does not disrupt p107-E2F or p107-MuvB complexes. Whether UL97 disrupts some p107 complexes by phosphorylating p107 at Ser-650 (homologous to Rb Ser-608 and p130 Ser-672) is not known (?). C, UL97 phosphorylates p130 to rescue p130-mediated repression of E2F-responsive gene expression in a UL97-L1- and p130-cleft-dependent manner but does not disrupt p130-E2F or p130-MuvB complexes. UL97 phosphorylates p130 Ser-672 disrupting p130-E2F4 complexes, but Ser-672 phosphorylation is not necessary for p130 inactivation by UL97.
We further investigated how UL97-mediated phosphorylation might inactivate p107 and p130 without disrupting their complexes with E2Fs or MuvB. In certain cell types, p107- and p130-assembled complexes contain the chromatin-modifying HDAC1 protein or the chromatin-remodeling BRG1 protein (11). However, we failed to detect p107 or p130 association with either HDAC1 or BRG1 in human fibroblasts (Fig. 8A) or Saos-2 cells (Fig. 6, A and E). Therefore, UL97-mediated inactivation of p107 and p130 is unlikely to occur through or to require disruption of their binding to HDAC1 or BRG1. We conclude that UL97 inactivates p107 and p130 through a unique mechanism that does not require the complete disruption of the transcriptionally repressive complexes assembled by either p107 or p130.
Despite the inability to disrupt the transcriptionally repressive complexes assembled by p107 and p130, HCMV infection induces the accumulation of proteins encoded by E2F-regulated genes such as p107, E2F1, Rb, MCM7, cyclin E1, and cyclin B1 (Fig. 8J). Serum stimulation (Fig. 8J) of mock-infected serum-starved cells also induces the accumulation of these proteins, as well as that of cyclin A2, cyclin D1, and cyclin D2, which are not induced by HCMV infection (63–65). Maribavir, a small molecule inhibitor of UL97, attenuated the accumulation of the protein products of the tested E2F-regulated genes, except for cyclin E1 (Fig. 8J), indicating UL97 kinase activity is required for most E2F-responsive gene expression in HCMV-infected cells. The L1 LXCXE motif required for UL97-mediated p107 and p130 phosphorylation (Fig. 3) and important for UL97-mediated Rb phosphorylation (32) was not required for the accumulation of the protein products of the tested E2F-regulated genes (Fig. 8J) (32), likely because of complementation by IE1, which independently inactivates p107 (Fig. 7A) (34) and p130 (Fig. 7B), and it cooperates with the L1 mutant UL97 (L1m) to inactivate Rb (32).
In summary, we describe novel ways in which the HCMV v-CDK UL97 inactivates Rb (Fig. 9A), p107 (Fig. 9B), and p130 (Fig. 9C). Similarities include the requirement for the L1 motif and catalytic activity for UL97 and the cleft domain of the Rb family members. Differences include whether or not associations with E2F proteins are disrupted and whether or not a stable complex between UL97 and the Rb family member is detected.
Discussion
To understand the physiological function of a kinase, it is imperative to know its substrates. Global methods have been utilized to identify potential substrates of the HCMV-encoded v-CDK UL97, including the use of protein arrays (66) and phosphoproteomics (67). Still, novel, important, and confirmed substrates emerge through the classic approach of testing likely candidates such as Rb (48, 68), lamin A/C (69), and HDAC1 (70). Through this method, we have identified the Rb-related p107 and p130 proteins as UL97 substrates. Neither p107 nor p130 were detected by more advanced global methods (66, 67), reaffirming that such approaches are not comprehensive and that individual candidate interrogation is still viable and valuable.
Our previous work (32) demonstrated that UL97 uses its first LXCXE motif and the LXCXE-binding cleft in Rb to mediate Rb phosphorylation, the disruption of Rb complexes with E2F1, E2F2, E2F3a, and E2F3b, and the reversal of Rb-mediated repression of E2F-responsive promoter reporters (Fig. 9A). Interestingly, we could not correlate UL97-mediated Rb inactivation with E2F dissociation as the UL97-L1 mutant was competent at complex disruption but failed to reverse Rb-mediated reporter repression (32). This represents a mechanism for Rb inactivation and for the stimulation of E2F-dependent transcription that is distinct from those mediated by cellular cyclin-CDK complexes (17) or other viral proteins (26, 29, 35, 36). Here we show that UL97 also inactivates p107 and p130 by seemingly novel mechanisms.
UL97 phosphorylated p107 through a reaction that required the L1 motif of UL97 and the p107 cleft (Fig. 3). UL97-mediated p107 phosphorylation reversed the ability of p107 to suppress an E2F-responsive reporter (Fig. 4), yet it failed to disrupt p107-E2F1, p107-E2F4 (Fig. 6), or p107-MuvB (Fig. 8) complexes. UL97 did however form a stable complex with p107 that required both the L1 motif of UL97 and the kinase activity of UL97 (Figs. 6 and 9B). Inactivation of p130 by UL97 was similar but not identical (Fig. 9C). UL97 phosphorylated p130 through a reaction that required the L1 motif of UL97 and the p130 cleft (Fig. 3). UL97-mediated p130 phosphorylation reversed the ability of p130 to suppress an E2F-responsive reporter (Fig. 4), yet failed to disrupt all p130-E2F4 complexes or p130-MuvB complexes (Figs. 6 and 8). Unlike p107, p130 bound but did not form a stable complex with UL97 (Fig. 6). In contrast to UL97, cyclin E/CDK2-mediated phosphorylation of p107 or p130 disrupted their transcriptionally repressive complexes.
Our immunoprecipitation assays utilize a ratio of UL97 to p130 that is 2-fold higher than that used in our luciferase assays. Despite increasing the UL97/p130 ratio over that which is required to inactivate p130 (Fig. 4B), UL97 still fails to disrupt p130-E2F4 complexes (Fig. 6E). Furthermore, the levels of UL97 achieved by high multiplicity infection of most of the cells in a culture (Fig. 8, A and B) also fail to disrupt p130-E2F complexes. Therefore, we consider it unlikely that a higher level expression of UL97 would lead to complex disruption, and we conclude that UL97 inactivates p130 (and by extension p107) without disrupting their complexes with E2F proteins.
Ser-672 on p130 is analogous to Ser-608 on Rb and Ser-650 on p107. Phosphorylation of those sites weakens the interactions between Rb or p107 and E2F proteins (21, 24). We found that when UL97 phosphorylated p130 on Ser-672, p130 failed to interact with E2F4 (Fig. 8). We have not tested the ability of UL97 to phosphorylate p107 on Ser-650, but we hypothesize that such a phosphorylation event occurs and disrupts p107-E2F complexes (Fig. 9B). Interestingly, UL97 phosphorylated a mutant p130 protein with an unphosphorylatable residue at position 672 (Fig. 1) and reversed the ability of this mutant protein to suppress an E2F reporter (Fig. 8). Thus, UL97 must generate at least two differentially phosphorylated forms of p130 that are inactivated in different ways (one by phosphorylation at Ser-672 and complex disruption and one not). This appears to agree with the recent revelation that Rb exists as multiple different mono-phosphorylated forms during the G1 phase (71). More work is needed to determine whether UL97 mono-phosphorylates the Rb family proteins and to define the physiological roles of mono-phosphorylated Rb family proteins in uninfected cells and perhaps during HCMV infection.
The reasons for the diversity of mechanisms used by UL97 to inactivate Rb, p107, and p130 (Fig. 9) and for the mechanistic divergence from those used by other cellular and viral proteins are unknown, but they may be related to either the different roles that the Rb family proteins play during HCMV infection or the different cellular (tumor suppressor) functions of the Rb family proteins. The hypophosphorylated forms of Rb and p130 are degraded by pp71 (36), an HCMV protein delivered directly to cells upon infection by virions (72). The subsequent phosphorylation of Rb and p130 by UL97 appears to protect them from pp71-mediated degradation and helps to maintain their presence in HCMV-infected cells while at the same time inactivating their ability to repress E2F-responsive transcription. The artificial depletion of Rb (59) or p130 (Fig. 5) impairs productive HCMV infection. Therefore, perhaps the Rb and p130 residues phosphorylated by UL97 are selected to simultaneously neutralize pp71-mediated degradation and release E2F repression, yet maintain the functions of Rb and p130 that HCMV requires.
The artificial depletion of p107 did not inhibit HCMV productive replication (60). Unlike Rb and p130 (6, 8), p107 is not a strong tumor suppressor (73), and therefore it may affect processes less important for controlling both oncogenesis and viral infection. However, the experiments demonstrating the non-essential nature of p107 for HCMV productive replication were performed in G0 cells arrested by serum withdrawal where p107 is not initially expressed but is induced by HCMV infection. It remains to be determined whether p107 is required for HCMV infections of S or G2 phase cells, as well as how the Rb family proteins, presumably in their phosphorylated forms, contribute positively to HCMV infection. This positive contribution seemingly extends beyond the classic paradigm of driving infected cells into the S phase to support the replication of viral DNA genomes by promoting the expression of the genes encoding DNA replication proteins and nucleotide biosynthetic enzymes. This is because HCMV encodes most of its own DNA replication proteins (26), and because deoxyribonucleotides are limiting in HCMV-infected cells even when the Rb family proteins are phosphorylated (74).
A unique feature of all the HCMV proteins that interact with the Rb family members is that they do not lead to overt cellular transformation, whether expressed alone or together during viral infection. Rb-inactivating proteins from other viruses such as adenovirus E1a, SV40 T antigen, and papillomavirus E7 are all highly oncogenic. Thus the different mechanisms that HCMV UL97 uses to inactivate Rb in comparison with viral oncoproteins may be one of the reasons why HCMV is not a classic tumor virus, although it does appear to have a complex relationship with human cancers that is continuing to emerge (39, 43–45). Understanding the differences between Rb protein inactivation by viral oncoproteins and UL97 may reveal the key functions of Rb, p107, and p130 that protect cells from transformation, perhaps leading to more targeted and better tolerated cancer chemotherapies.
Experimental procedures
Cells and viruses
Saos-2 (containing truncated Rb) (75), U-2 OS, HEK293T, primary human foreskin fibroblasts (HFF), and primary normal human dermal fibroblasts (NHDF) (Clonetics) cells were grown and maintained at 37 °C in Dulbecco's modified Eagle's medium (DMEM, Sigma) supplemented with 10% fetal bovine serum (FBS, Sigma), 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.292 mg/ml glutamine (PSG, Sigma). All infections were performed under serum-starved conditions. For serum starvation, HFF cells were incubated in DMEM with 0.1% FBS for 48 h. For serum stimulation, serum-starved HFF cells were incubated in DMEM with 15% serum for 18 h. The HCMV strain used was AD169. HCMV recombinants with UL97 deleted (Δ97), with individual UL97 LXCXE motif disruptions (L1 mutant (C151G), RC295, the L2 mutant (C428G), RC312, the L3 mutant (C693G), RC316), or with all three UL97 LXCXE motifs disrupted (triple mutant, T3260) were described previously (32, 68, 76). To inhibit UL97 kinase activity in HCMV-infected cells, maribavir (Acme Bioscience, A4028; 10 μm) was added 2 h postinfection. Transduction of NHDF cells with retroviruses expressing shRNAs was performed as described previously (77). Scrambled shRNA (78) or p130 shRNA sequences (130.1 and 130.2) (79) were previously described. Virus titers measuring replication in p130 knockdown NHDF cells were quantitated by plaque assay on non-transduced NHDF cells.
Plasmids
Expression plasmids have been described for the following: HA-tagged alleles for the herpesviral kinases (25); V5-tagged UL97 and kinase-dead derivative (80); pHAp107 (81); pHAp130 (82); pE2F1-Luc(−242) (83); pCGN- and pSG5-based HA-tagged UL97 L1m (C151G), L2m (C428G), L3m (C693G), Tri (L1m + L2m + L3m), HPm (W368A), Quad (Tri + HPm), pSG5-IE1, and pCMV-FLAG-Rb (32); pGEX3X-p130 (322–1139 amino acids) (84); Rc/cyclin A2 and Rc/cyclin E1 (85); Rc/cyclin D2 (86); CMV-CDK4 (87); p130ΔCDK4 and p130-PM19A (50); pCMV-E1a (88); LIN52-V5-pEF6 (16); and pcDNA3-E2F4 (89). pCDK2-HA was purchased from Addgene (catalog no. 1884). pFC14a-HA-UL97 WT and KD were constructed by inserting a PCR product amplified from pCGN-UL97 WT or KD into the AsiSI and XhoI sites of pFC14a (Promega) using the In-Fusion HD cloning kit (Clontech). pCMV-FLAG-p107, pCMV-FLAG-p130, pCMV-FLAG-p130ΔCDK4, and pCMV-FLAG-LIN52 were constructed by inserting PCR products amplified from pHAp107, pHAp130, p130ΔCDK4, or LIN52-V5-pEF6 into HindIII and XbaI sites in p3xFLAG-CMV-10 (Sigma) using the In-Fusion HD cloning kit. pSG5-HAp107 and pSG5-HAp130 were constructed by inserting PCR products amplified from pHAp107 or pHAp130 into BamHI sites in pSG5 using the In-Fusion HD cloning kit. pSG5-based plasmids were used during cotransfection experiments with IE1. Expression plasmids for mutant alleles were generated by standard PCR mutagenesis techniques. They are as follows: two distinct cleft mutants of pHAp107 (N935F or C846F); alanine exchange mutant of RXL motif of pHAp107 (657KRRL660 to AAAA; pHAp107-ΔRXL); two distinct cleft mutants of pHAp130 (N1010F or C894F); and an RXL motif deletion of pHAp130 (680RRL682 deletion; pHAp130-ΔRXL). All alleles generated by PCR have been confirmed by sequencing. The sequences of primers for mutagenesis are available upon request.
Western blottings and antibodies
p107- or p130-transfected Saos-2 cells were suspended in lysis buffer (20 mm Tris-HCl (pH 7.4), 0.5% Triton X-100, 300 mm NaCl, 1 mm EDTA, 0.1% SDS, 25 mm NaF, 25 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 mm β-glycerophosphate) and incubated on ice for 40 min followed by centrifugation as described previously (90). HCMV-infected fibroblasts were lysed in an SDS solution (1% SDS, 2% β-mercaptoethanol) by boiling for 10 min followed by vortexing as described previously (48). Equal amounts of proteins were separated by 7.5 or 10% SDS-PAGE and transferred onto nitrocellulose membranes. Protein bands were quantified with the Odyssey Fc Imager and the Image Studio version 2.1.10 software (LI-COR). Phos-tag reagent (47) was purchased from the NARD Institute (AAL-107) and used according to the manufacturer's instructions. λ protein phosphatase treatment was performed as reported previously (32). Primary antibodies were purchased from Abcam (cyclin A2, catalogue no. ab32498; p130-Ser(P)-672, ab76255); Abgent (p130-Ser(P)-672, AJ1683b); Ambion (GAPDH, AM4300); BD Transduction Laboratories (CDK2, 610146; Rb2/p130, 610262); Bethyl Laboratories (E2F4, A302–133A for immunoprecipitation and A302-134A for Western blotting; LIN9, A300-BL2981; LIN54, A303-799A); Calbiochem (adenovirus 2 E1a, DP11); Cell Signaling Technology (E2F1, 3742; Rb, 9309); Covance (HA, MMS-101P); Invitrogen (V5, R960-25); Millipore (HDAC1, 05-100); Santa Cruz Biotechnology (BRG-1, sc-17796; CDK4, sc-23896; cyclin B1, sc-245; cyclin D1, sc-718; cyclin D2, sc-181; cyclin E, sc-247; E2F4, sc-866; E2F5, sc-1083; p107, sc-318; and p130, sc-317), Sigma (FLAG M2, F1804; α-tubulin, T9026); and Virusys (UL44, CA006-100). Antibodies against HCMV IE1 (1B12) and UL97 have been previously described (51, 91). Mouse TrueBlot Ultra (eBioscience, 18-8817) and rabbit TrueBlot (18-8816) were used as secondary antibodies for Western blotting of immunoprecipitation samples.
In vivo phosphorylation assays
Saos-2 cells were transfected with expression plasmids for wild-type or mutated p107 or p130 together with wild-type or mutated HCMV UL97 or various cyclin/CDK pairs using TransIT-2020 (Mirus) as described previously (25). At 48 h post-transfection, the transfected cells were harvested and subjected to Western blotting analysis.
In vitro kinase assays
Wild-type and kinase-dead UL97 proteins were purified by transfecting 15-cm plates of HEK293T cells that were at 60% confluence with 32 μg of pFC14a-UL97 WT or pFC14a-UL97 KD using polyethyleneimine (PEI) at 1 mg/ml. Cells were harvested at 48 h in purification buffer (1 mm DTT, 0.005% IGEPAL CA-630, and PBS) containing a protease inhibitor mixture (Promega) and lysed by sonication. Lysates were cleared by centrifugation and then incubated overnight with HaloLink resin (Promega) to bind the HaloTag on the kinase. After three washes, TEV protease was used to release HA-UL97 WT or HA-UL97 KD from the resin. The eluted kinases were stored in purification buffer with 15% glycerol at −80 °C. To generate the substrates for the autoradiography-based in vitro kinase assays, Saos-2 cells (1.6 × 106/100-mm plate) were transfected with 1.8 μg of pHAp107 or pHAp130 using TransIT-2020. After 48 h, cells were suspended with modified CSK buffer (100 mm Pipes (pH 6.8); 500 mm NaCl; 300 mm sucrose; 1 mm EGTA; 1 mm MgCl2; 0.1% Triton X-100; 10 μg/ml pepstatin A; 25 μg/ml leupeptin; 1 mm PMSF; 25 mm NaF; 10 mm β-glycerophosphate), collected by centrifugation, and then diluted with an equal volume of ET gel buffer (50 mm Tris (pH 7.4), 0.1% Triton X-100; 1 mm EDTA). The lysates were subjected to immunoprecipitation with anti-p107 or p130 antibodies (2 μg) and protein A/G-magnetic beads (Thermo Fisher Scientific). The beads were washed four times with ET gel buffer with 250 mm NaCl and once with ET gel buffer with 150 mm NaCl. To generate the substrate for the Western blotting-based in vitro kinase assays, GST-p130 was purified from Escherichia coli strain Rosetta2(DE3) (Novagen) transformed with pGEX3X-p130 (322–1139 amino acids) as described previously (90). For the kinase reactions, all substrates were washed one final time with kinase buffer (50 mm Tris (pH 8); 5 mm β-glycerophosphate; 10 mm MgCl2; 2 mm DTT) and resuspended in the kinase reaction containing ∼250 ng of kinase; 75 μm ATP; 1 μCi of [γ-32P]ATP; 50 mm Tris (pH 8); 5 mm β-glycerophosphate; 10 mm MgCl2; and 2 mm DTT. The reactions were incubated at 37 °C for 30 min, stopped with SDS-PAGE loading buffer, and boiled for 10 min. Samples were run on a 6% SDS-PAGE, transferred to a nitrocellulose membrane, and exposed to film for autoradiography. Western blotting was then performed on the membrane to verify the presence of the kinases and p107 using an HA antibody and the presence of p130 using a p130-specific antibody. For detection of phosphorylated Ser-672 on p130, a kinase reaction was performed as described above except in the presence of unlabeled ATP (1 mm) and bacterially purified GST-p130 as a substrate. Western blotting was then performed using a p130 antibody and a phospho-Ser-672-specific antibody.
Luciferase assays
Saos-2 cells (2.5 × 105/6-well) were transfected with expression plasmids for pHAp107 (0.25 μg) or pHAp130 (0.7 μg) together with wild-type/mutated HCMV UL97 (1 μg for cotransfection with p107; 0.7 or 1 μg for cotransfection with p130), or Rc/cyclin E1 (0.5 μg) and pCDK2-HA (0.5 μg), and pE2F1-Luc(−242) (0.02 μg) using TransIT-2020. Luciferase assays were performed as described previously (32).
Immunoprecipitations
Saos-2 cells (1.6–1.8 × 106/100-mm) were transfected with pCMV-FLAG-p107, pCMV-FLAG-p130, pSG5-HAp107, or pSG5-HAp130 (1.6 μg) together with a pCGN vector carrying wild-type/mutated HCMV UL97, pCMV-E1a (3.2 μg) or Rc/cyclin E1 (1.6 μg) and pCDK2-HA (1.6 μg), or a pSG5 vector carrying UL97 or IE1 (3.2 μg) using TransIT-2020. After 48 h, cells were suspended with modified CSK buffer with 150 mm NaCl and centrifuged. The extracts were immunoprecipitated with anti-FLAG, HA, p107, p130, phospho-Ser-672 p130 or E2F4 antibodies (2 μg) or normal mouse IgG or normal rabbit IgG (2 μg) as controls, and protein G-Sepharose beads or protein A-Sepharose beads (GE Healthcare), or protein A/G-magnetic beads. Beads were washed three times with ET gel buffer with 150 mm NaCl and eluted with SDS gel loading buffer. For covalent capture with HaloLink resin, Saos-2 cells were transfected with expression plasmids of HA-tagged p107 alleles together with pFC14a-UL97 WT and lysed as described above. The extracts were incubated with HaloLink resin for 3 h. After three washes, TEV protease was used for release. U-2 OS cells (1.5 × 106/100-mm) were transfected with pCMV-FLAG-LIN52 (1.6 μg) together with pCGN-based HA-UL97 (3.2 μg) using TransIT-2020. After 48 h, the immunoprecipitation was performed using anti-FLAG antibody. In the context of infection, HFF cells (7 × 105/100-mm) were serum-starved for 48 h and infected with AD169 or L1 mutant UL97 virus at an m.o.i. of 3. After 2 h, cells were treated with 10 μm maribavir or DMSO as a vehicle and subjected to immunoprecipitation with p130, LIN54, or LIN9 antibodies at 45 h postinfection. Serum-stimulated cells were incubated with 15% FBS for 18 h before harvesting. Immunoprecipitation including anti-Rb antibody was performed as described previously (32). Western blotting quantitation was performed with ImageJ software. Statistical analyses utilized a two-tailed unpaired Student's t test.
Immunofluorescence analysis
HCMV-infected HFF cells were fixed with 2% paraformaldehyde and stained as described previously (25, 92) with a UL97 primary antibody and a secondary antibody conjugated to Alexa Fluor 594 (Molecular Probes). The images were captured using an Olympus Fluoview FV 1000 confocal microscope and software. The specificity of anti-UL97 antibody was confirmed using mock-infected HFF cells.
Author contributions
S. I. and R. F. K. designed the experiments. S. I. performed the experiments. A. C. U. produced the purified UL97 kinases and performed in vitro kinase assays in Fig. 1, F and G. H. R. V. performed the p130 knockdown experiments in Fig. 5. S. I. and R. F. K. wrote the paper with comments from all authors.
Acknowledgments
We thank our laboratory managers Phil Balandyk and Diccon Fiore for expert technical assistance and the members of our laboratory for helpful discussions. We thank the following investigators for providing research materials: Jiri Bartek, Sunwen Chou, Don Coen, Doug Cress, Jim DeCapprio, Philip Hinds, Larisa Litovchick, Joe Nevins, Mark Prichard, Sander van den Heuvel, and Eileen White.
This work was supported in part by National Institutes of Health Grants R01-AI080675 (to R. F. K.) and P01-CA022443 (to Paul Lambert). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Rb
- retinoblastoma
- HFF
- human foreskin fibroblast
- NHDF
- normal human dermal fibroblast
- HCMV
- human cytomegalovirus
- CDK
- cyclin-dependent kinase
- HP
- hydrophobic patch
- EBV
- Epstein-Barr virus
- m.o.i.
- multiplicity of infection
- KSHV
- Kaposi's sarcoma-associated herpesvirus
- TEV
- tobacco etch virus.
References
- 1. Friend S. H., Bernards R., Rogelj S., Weinberg R. A., Rapaport J. M., Albert D. M., and Dryja T. P. (1986) A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323, 643–646 [DOI] [PubMed] [Google Scholar]
- 2. Ewen M. E., Xing Y. G., Lawrence J. B., and Livingston D. M. (1991) Molecular cloning, chromosomal mapping, and expression of the cDNA for p107, a retinoblastoma gene product-related protein. Cell 66, 1155–1164 [DOI] [PubMed] [Google Scholar]
- 3. Mayol X., Graña X., Baldi A., Sang N., Hu Q., and Giordano A. (1993) Cloning of a new member of the retinoblastoma gene family (pRb2) which binds to the E1A transforming domain. Oncogene 8, 2561–2566 [PubMed] [Google Scholar]
- 4. Burkhart D. L., and Sage J. (2008) Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weinberg R. A. (1995) The retinoblastoma protein and cell cycle control. Cell 81, 323–330 [DOI] [PubMed] [Google Scholar]
- 6. Helin K., Holm K., Niebuhr A., Eiberg H., Tommerup N., Hougaard S., Poulsen H. S., Spang-Thomsen M., and Norgaard P. (1997) Loss of the retinoblastoma protein-related p130 protein in small cell lung carcinoma. Proc. Natl. Acad. Sci. U.S.A. 94, 6933–6938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paggi M. G., and Giordano A. (2001) Who is the boss in the retinoblastoma family? The point of view of Rb2/p130, the little brother. Cancer Res. 61, 4651–4654 [PubMed] [Google Scholar]
- 8. Claudio P. P., Howard C. M., Pacilio C., Cinti C., Romano G., Minimo C., Maraldi N. M., Minna J. D., Gelbert L., Leoncini L., Tosi G. M., Hicheli P., Caputi M., Giordano G. G., and Giordano A. (2000) Mutations in the retinoblastoma-related gene RB2/p130 in lung tumors and suppression of tumor growth in vivo by retrovirus-mediated gene transfer. Cancer Res. 60, 372–382 [PubMed] [Google Scholar]
- 9. Ho V. M., Schaffer B. E., Karnezis A. N., Park K. S., and Sage J. (2009) The retinoblastoma gene Rb and its family member p130 suppress lung adenocarcinoma induced by oncogenic K-Ras. Oncogene 28, 1393–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaffer B. E., Park K. S., Yiu G., Conklin J. F., Lin C., Burkhart D. L., Karnezis A. N., Sweet-Cordero E. A., and Sage J. (2010) Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 70, 3877–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Classon M., and Dyson N. (2001) p107 and p130: versatile proteins with interesting pockets. Exp. Cell Res. 264, 135–147 [DOI] [PubMed] [Google Scholar]
- 12. Nevins J. R. (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 9, 585–593 [PubMed] [Google Scholar]
- 13. Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 14. Henley S. A., and Dick F. A. (2012) The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Litovchick L., Sadasivam S., Florens L., Zhu X., Swanson S. K., Velmurugan S., Chen R., Washburn M. P., Liu X. S., and DeCaprio J. A. (2007) Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 26, 539–551 [DOI] [PubMed] [Google Scholar]
- 16. Guiley K. Z., Liban T. J., Felthousen J. G., Ramanan P., Litovchick L., and Rubin S. M. (2015) Structural mechanisms of DREAM complex assembly and regulation. Genes Dev. 29, 961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adams P. D. (2001) Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim. Biophys. Acta 1471, M123–133 [DOI] [PubMed] [Google Scholar]
- 18. Dick F. A., and Rubin S. M. (2013) Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams P. D., Li X., Sellers W. R., Baker K. B., Leng X., Harper J. W., Taya Y., and Kaelin W. G. Jr. (1999) Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol. Cell. Biol. 19, 1068–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ashizawa S., Nishizawa H., Yamada M., Higashi H., Kondo T., Ozawa H., Kakita A., and Hatakeyama M. (2001) Collective inhibition of pRB family proteins by phosphorylation in cells with p16INK4a loss or cyclin E overexpression. J. Biol. Chem. 276, 11362–11370 [DOI] [PubMed] [Google Scholar]
- 21. Rubin S. M. (2013) Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 38, 12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burke J. R., Deshong A. J., Pelton J. G., and Rubin S. M. (2010) Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J. Biol. Chem. 285, 16286–16293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burke J. R., Hura G. L., and Rubin S. M. (2012) Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 26, 1156–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liban T. J., Thwaites M. J., Dick F. A., and Rubin S. M. (2016) Structural conservation and E2F binding specificity within the retinoblastoma pocket protein family. J. Mol. Biol. 428, 3960–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuny C. V., Chinchilla K., Culbertson M. R., and Kalejta R. F. (2010) Cyclin-dependent kinase-like function is shared by the β- and γ-subset of the conserved herpesvirus protein kinases. PLoS Pathog. 6, e1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hume A. J., and Kalejta R. F. (2009) Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div. 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee C., and Cho Y. (2002) Interactions of SV40 large T antigen and other viral proteins with retinoblastoma tumour suppressor. Rev. Med. Virol. 12, 81–92 [DOI] [PubMed] [Google Scholar]
- 28. Felsani A., Mileo A. M., and Paggi M. G. (2006) Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene 25, 5277–5285 [DOI] [PubMed] [Google Scholar]
- 29. Helt A. M., and Galloway D. A. (2003) Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24, 159–169 [DOI] [PubMed] [Google Scholar]
- 30. Chellappan S., Kraus V. B., Kroger B., Munger K., Howley P. M., Phelps W. C., and Nevins J. R. (1992) Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. U.S.A. 89, 4549–4553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee J. O., Russo A. A., and Pavletich N. P. (1998) Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 391, 859–865 [DOI] [PubMed] [Google Scholar]
- 32. Iwahori S., Hakki M., Chou S., and Kalejta R. F. (2015) Molecular determinants for the inactivation of the retinoblastoma tumor suppressor by the viral cyclin-dependent kinase UL97. J. Biol. Chem. 290, 19666–19680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gill R. B., Frederick S. L., Hartline C. B., Chou S., and Prichard M. N. (2009) Conserved retinoblastoma protein-binding motif in human cytomegalovirus UL97 kinase minimally impacts viral replication but affects susceptibility to maribavir. Virol. J. 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Poma E. E., Kowalik T. F., Zhu L., Sinclair J. H., and Huang E. S. (1996) The human cytomegalovirus IE1–72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J. Virol. 70, 7867–7877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kalejta R. F., and Shenk T. (2003) Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. U.S.A. 100, 3263–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalejta R. F., Bechtel J. T., and Shenk T. (2003) Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23, 1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hagemeier C., Caswell R., Hayhurst G., Sinclair J., and Kouzarides T. (1994) Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. EMBO J. 13, 2897–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fortunato E. A., Sommer M. H., Yoder K., and Spector D. H. (1997) Identification of domains within the human cytomegalovirus major immediate-early 86-kilodalton protein and the retinoblastoma protein required for physical and functional interaction with each other. J. Virol. 71, 8176–8185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ranganathan P., Clark P. A., Kuo J. S., Salamat M. S., and Kalejta R. F. (2012) Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J. Virol. 86, 854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cobbs C. S., Harkins L., Samanta M., Gillespie G. Y., Bharara S., King P. H., Nabors L. B., Cobbs C. G., and Britt W. J. (2002) Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 62, 3347–3350 [PubMed] [Google Scholar]
- 41. Harkins L. E., Matlaf L. A., Soroceanu L., Klemm K., Britt W. J., Wang W., Bland K. I., and Cobbs C. S. (2010) Detection of human cytomegalovirus in normal and neoplastic breast epithelium. Herpesviridae 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taher C., de Boniface J., Mohammad A. A., Religa P., Hartman J., Yaiw K. C., Frisell J., Rahbar A., and Söderberg-Naucler C. (2013) High prevalence of human cytomegalovirus proteins and nucleic acids in primary breast cancer and metastatic sentinel lymph nodes. PLoS ONE 8, e56795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soroceanu L., and Cobbs C. S. (2011) Is HCMV a tumor promoter? Virus Res. 157, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dziurzynski K., Chang S. M., Heimberger A. B., Kalejta R. F., McGregor Dallas S. R., Smit M., Soroceanu L., Cobbs C. S., and HCMV and Gliomas Symposium (2012) Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 14, 246–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herbein G., and Kumar A. (2014) The oncogenic potential of human cytomegalovirus and breast cancer. Front. Oncol. 4, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McElroy A. K., Dwarakanath R. S., and Spector D. H. (2000) Dysregulation of cyclin E gene expression in human cytomegalovirus-infected cells requires viral early gene expression and is associated with changes in the Rb-related protein p130. J. Virol. 74, 4192–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., and Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 48. Hume A. J., Finkel J. S., Kamil J. P., Coen D. M., Culbertson M. R., and Kalejta R. F. (2008) Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320, 797–799 [DOI] [PubMed] [Google Scholar]
- 49. Pajovic S., Wong E. L., Black A. R., and Azizkhan J. C. (1997) Identification of a viral kinase that phosphorylates specific E2Fs and pocket proteins. Mol. Cell. Biol. 17, 6459–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hansen K., Farkas T., Lukas J., Holm K., Rönnstrand L., and Bartek J. (2001) Phosphorylation-dependent and -independent functions of p130 cooperate to evoke a sustained G1 block. EMBO J. 20, 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. He Z., He Y. S., Kim Y., Chu L., Ohmstede C., Biron K. K., and Coen D. M. (1997) The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J. Virol. 71, 405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu L., Enders G., Lees J. A., Beijersbergen R. L., Bernards R., and Harlow E. (1995) The pRB-related protein p107 contains two growth suppression domains: independent interactions with E2F and cyclin/cdk complexes. EMBO J. 14, 1904–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meloni A. R., Smith E. J., and Nevins J. R. (1999) A mechanism for Rb/p130-mediated transcription repression involving recruitment of the CtBP corepressor. Proc. Natl. Acad. Sci. U.S.A. 96, 9574–9579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bignon Y. J., Shew J. Y., Rappolee D., Naylor S. L., Lee E. Y., Schnier J., and Lee W. H. (1990) A single Cys706 to Phe substitution in the retinoblastoma protein causes the loss of binding to SV40 T antigen. Cell Growth Differ. 1, 647–651 [PubMed] [Google Scholar]
- 55. Kaye F. J., Kratzke R. A., Gerster J. L., and Horowitz J. M. (1990) A single amino acid substitution results in a retinoblastoma protein defective in phosphorylation and oncoprotein binding. Proc. Natl. Acad. Sci. U.S.A. 87, 6922–6926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen T. T., and Wang J. Y. (2000) Establishment of irreversible growth arrest in myogenic differentiation requires the RB LXCXE-binding function. Mol. Cell. Biol. 20, 5571–5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Leng X., Noble M., Adams P. D., Qin J., and Harper J. W. (2002) Reversal of growth suppression by p107 via direct phosphorylation by cyclin D1/cyclin-dependent kinase 4. Mol. Cell. Biol. 22, 2242–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lacy S., and Whyte P. (1997) Identification of a p130 domain mediating interactions with cyclin A/cdk 2 and cyclin E/cdk 2 complexes. Oncogene 14, 2395–2406 [DOI] [PubMed] [Google Scholar]
- 59. VanDeusen H. R., and Kalejta R. F. (2015) The retinoblastoma tumor suppressor promotes efficient human cytomegalovirus lytic replication. J. Virol. 89, 5012–5021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. VanDeusen H. R., and Kalejta R. F. (2015) Deficiencies in cellular processes modulated by the retinoblastoma protein do not account for reduced human cytomegalovirus replication in its absence. J. Virol. 89, 11965–11974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dyson N. J. (2016) RB1: a prototype tumor suppressor and an enigma. Genes Dev. 30, 1492–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Johnson R. A., Yurochko A. D., Poma E. E., Zhu L., and Huang E. S. (1999) Domain mapping of the human cytomegalovirus IE1–72 and cellular p107 protein-protein interaction and the possible functional consequences. J. Gen. Virol. 80, 1293–1303 [DOI] [PubMed] [Google Scholar]
- 63. Salvant B. S., Fortunato E. A., and Spector D. H. (1998) Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J. Virol. 72, 3729–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bresnahan W. A., Boldogh I., Thompson E. A., and Albrecht T. (1996) Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology 224, 150–160 [DOI] [PubMed] [Google Scholar]
- 65. Wiebusch L., and Hagemeier C. (2001) The human cytomegalovirus immediate early 2 protein dissociates cellular DNA synthesis from cyclin-dependent kinase activation. EMBO J. 20, 1086–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li R., Zhu J., Xie Z., Liao G., Liu J., Chen M. R., Hu S., Woodard C., Lin J., Taverna S. D., Desai P., Ambinder R. F., Hayward G. S., Qian J., Zhu H., and Hayward S. D. (2011) Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10, 390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oberstein A., Perlman D. H., Shenk T., and Terry L. J. (2015) Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome. Proteomics 15, 2006–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Prichard M. N., Sztul E., Daily S. L., Perry A. L., Frederick S. L., Gill R. B., Hartline C. B., Streblow D. N., Varnum S. M., Smith R. D., and Kern E. R. (2008) Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J. Virol. 82, 5054–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hamirally S., Kamil J. P., Ndassa-Colday Y. M., Lin A. J., Jahng W. J., Baek M. C., Noton S., Silva L. A., Simpson-Holley M., Knipe D. M., Golan D. E., Marto J. A., and Coen D. M. (2009) Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 5, e1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bigley T. M., Reitsma J. M., Mirza S. P., and Terhune S. S. (2013) Human cytomegalovirus pUL97 regulates the viral major immediate early promoter by phosphorylation-mediated disruption of histone deacetylase 1 binding. J. Virol. 87, 7393–7408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Narasimha A. M., Kaulich M., Shapiro G. S., Choi Y. J., Sicinski P., and Dowdy S. F. (2014) Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 3, e02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Penkert R. R., and Kalejta R. F. (2012) Tale of a tegument transactivator: the past, present and future of human CMV pp71. Future Virol. 7, 855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wirt S. E., and Sage J. (2010) p107 in the public eye: an Rb understudy and more. Cell Div. 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kuny C. V., and Kalejta R. F. (2016) Human cytomegalovirus can procure deoxyribonucleotides for viral DNA replication in the absence of retinoblastoma protein phosphorylation. J. Virol. 90, 8634–8643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shew J. Y., Lin B. T., Chen P. L., Tseng B. Y., Yang-Feng T. L., and Lee W. H. (1990) C-terminal truncation of the retinoblastoma gene product leads to functional inactivation. Proc. Natl. Acad. Sci. U.S.A. 87, 6–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Prichard M. N., Gao N., Jairath S., Mulamba G., Krosky P., Coen D. M., Parker B. O., and Pari G. S. (1999) A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 73, 5663–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Saffert R. T., and Kalejta R. F. (2007) Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81, 9109–9120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pasque V., Gillich A., Garrett N., and Gurdon J. B. (2011) Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J. 30, 2373–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ueno T., Sasaki K., Yoshida S., Kajitani N., Satsuka A., Nakamura H., and Sakai H. (2006) Molecular mechanisms of hyperplasia induction by human papillomavirus E7. Oncogene 25, 4155–4164 [DOI] [PubMed] [Google Scholar]
- 80. Prichard M. N., Britt W. J., Daily S. L., Hartline C. B., and Kern E. R. (2005) Human cytomegalovirus UL97 Kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J. Virol. 79, 15494–15502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhu L., van den Heuvel S., Helin K., Fattaey A., Ewen M., Livingston D., Dyson N., and Harlow E. (1993) Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev. 7, 1111–1125 [DOI] [PubMed] [Google Scholar]
- 82. Vairo G., Livingston D. M., and Ginsberg D. (1995) Functional interaction between E2F-4 and p130: evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev. 9, 869–881 [DOI] [PubMed] [Google Scholar]
- 83. Johnson D. G., Ohtani K., and Nevins J. R. (1994) Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 8, 1514–1525 [DOI] [PubMed] [Google Scholar]
- 84. Dong F., Cress W. D. Jr, Agrawal D., and Pledger W. J. (1998) The role of cyclin D3-dependent kinase in the phosphorylation of p130 in mouse BALB/c 3T3 fibroblasts. J. Biol. Chem. 273, 6190–6195 [DOI] [PubMed] [Google Scholar]
- 85. Hinds P. W., Mittnacht S., Dulic V., Arnold A., Reed S. I., and Weinberg R. A. (1992) Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70, 993–1006 [DOI] [PubMed] [Google Scholar]
- 86. Baker G. L., Landis M. W., and Hinds P. W. (2005) Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle 4, 330–338 [PubMed] [Google Scholar]
- 87. van den Heuvel S., and Harlow E. (1993) Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262, 2050–2054 [DOI] [PubMed] [Google Scholar]
- 88. White E., Cipriani R., Sabbatini P., and Denton A. (1991) Adenovirus E1B 19-kilodalton protein overcomes the cytotoxicity of E1A proteins. J. Virol. 65, 2968–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sardet C., Vidal M., Cobrinik D., Geng Y., Onufryk C., Chen A., and Weinberg R. A. (1995) E2F-4 and E2F-5, two members of the E2F family, are expressed in the early phases of the cell cycle. Proc. Natl. Acad. Sci. U.S.A. 92, 2403–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Iwahori S., Shirata N., Kawaguchi Y., Weller S. K., Sato Y., Kudoh A., Nakayama S., Isomura H., and Tsurumi T. (2007) Enhanced phosphorylation of transcription factor Sp1 in response to herpes simplex virus type 1 infection is dependent on the ataxia telangiectasia-mutated protein. J. Virol. 81, 9653–9664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhu H., Shen Y., and Shenk T. (1995) Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69, 7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Iwahori S., Yasui Y., Kudoh A., Sato Y., Nakayama S., Murata T., Isomura H., and Tsurumi T. (2008) Identification of phosphorylation sites on transcription factor Sp1 in response to DNA damage and its accumulation at damaged sites. Cell. Signal. 20, 1795–1803 [DOI] [PubMed] [Google Scholar]