

Abstract
Epstein-Barr virus-induced gene 3 (EBI3) is a subunit of the composite cytokines IL-27 and IL-35. Both have beneficial functions or effects in models of infectious and autoimmune diseases. This suggests that administration of EBI3 could be therapeutically useful by binding free p28 and p35 to generate IL-27 and IL-35. IL-27- and IL-35-independent functions of EBI3 could compromise its therapeutic uses. We therefore assessed the effects of EBI3 on cytokine receptor-expressing cells. We observed that EBI3 activates STAT3 and induces the proliferation of the IL-6-dependent B9 mouse plasmacytoma cell line. Analyses using blocking mAbs and Ba/F3 transfectants expressing gp130 indicate that EBI3 activity was linked to its capacity to mediate IL-6 trans-signaling, albeit less efficiently than soluble IL-6Rα. In line with this interpretation, co-immunoprecipitation and SPR experiments indicated that EBI3 binds IL-6. An important pro-inflammatory function of IL-6 trans-signaling is to activate blood vessel endothelial cells. We observed that EBI3 in combination with IL-6 could induce the expression of chemokines by human venal endothelial cells. Our results indicate that EBI3 can promote pro-inflammatory IL-6 functions by mediating trans-signaling. These unexpected observations suggest that use of EBI3 as a therapeutic biologic for autoimmune diseases will likely require co-administration of soluble gp130 to prevent the side effects associated with IL-6 trans-signaling. Together with previous studies that demonstrated activation of IL-6R by p28 (IL-30), new findings further suggest a complex interrelation between IL-27 and IL-6.
Keywords: cytokine, endothelial cell, inflammation, interleukin 6 (IL-6), interleukin 6 receptor (IL6R)
Introduction
Epstein-Barr virus-induced gene 3 (EBI3)2 encodes a 34-kDa soluble cytokine receptor that shares homologies with the p40 subunit of IL-12 and IL-23 and the non-signaling α chain receptors of the IL-6 cytokine family (ciliary neurotrophic factor and IL-11 receptors) (1). As indicated by its name, EBI3 was identified as a transcript induced in B cells by Epstein-Barr virus infection (2). EBI3 expression in peripheral blood cells can be triggered by mitogen activation (2). In vivo, EBI3 is present in interfollicular cells in tonsils, perifollicular cells in the spleen, and placental syncytial trophoblasts (3).
EBI3 pairs with IL-30 (p28) to form IL-27 (4), or, alternatively, with the p35 subunit of IL-12 to form IL-35 (5) (Fig. 1). Both IL-27 and IL-35 belong to the IL-6/IL-12 family of cytokines (6). IL-27 is mainly produced by activated dendritic cells and macrophages in response to Toll-like receptor ligands and pro-inflammatory cytokines (7). Its receptor comprises a unique IL-27Rα chain and the gp130 chain shared with members of the IL-6 cytokine family (Fig. 1) (8). Initially identified as a CD4+T cell activator (4), IL-27 can induce signaling in a large array of immune and non-immune cells, notably T, B, and myeloid cells. IL-27 has pleiotropic functions, including the regulation of inflammation in multiple models of infectious diseases (9). For example, it induces the secretion of IL-10 and limits inflammatory responses (10). It also inhibits Th17 CD4+ T cell differentiation and shows promising effects in EAE models of multiple sclerosis (11, 12). Like other cytokines of the IL-12 family, IL-27 has potent anti-tumor effect in mouse models of cancer (13).
Figure 1.
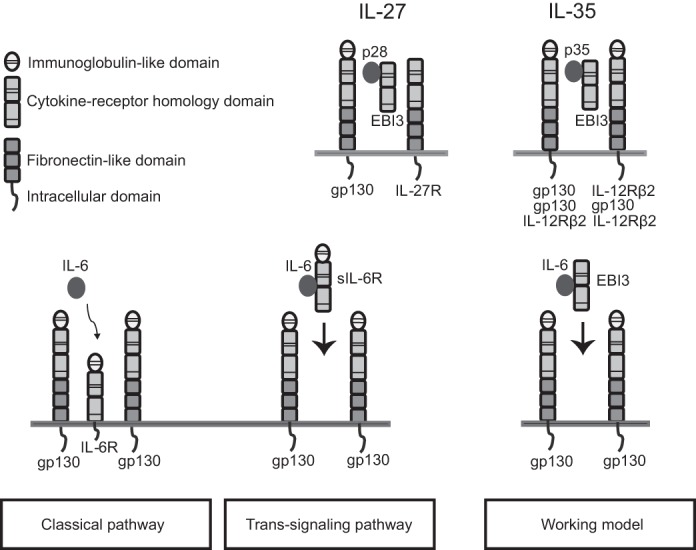
EBI3 participates in the composite cytokines IL-27 and IL-35 and could be implicated in IL-6 trans-signaling.
IL-35 is a composite cytokine produced by regulatory T cells (14). Like IL-27, IL-35 has anti-inflammatory roles in models of infectious diseases and EAE. IL-35, akin to IL-10 and TGF-β, promotes the differentiation of human and mouse induced T regulatory cells (iTR35) that play a key role in infection tolerance and contribute to tumor progression (15). IL-35 induces ex vivo conversion of mouse or human B cells into IL-35 or IL-10 producing B-cells (16). Studies using cells deficient in hematopoietin receptor chains revealed that IL-35 signals through unconventional receptors comprising IL-12Rβ2 or gp130 homo- or heterodimers (Fig. 1) (17).
EBI3 lacks the cysteine homologous to the residue forming the interchain disulfide bond in IL-12 and IL-23, indicating that cytokines involving EBI3 may be more unstable than their counterparts that utilize p40 (1). Furthermore, the p28 subunit of IL-27, IL-30, which can be secreted independently of EBI3 (18) is an IL-6 receptor (IL-6R) ligand with pro-inflammatory activities (19, 20). This suggests that injection of EBI3 or EBI3 derivatives such as EBI3-Fc fusion proteins could have therapeutic anti-inflammatory effect by favoring the formation of IL-27 and IL-35 complexes in vivo.
EBI3, like p40, can be secreted independent of its cytokine subunit partners (4, 21). Secreted p40 homodimers have been shown to bind IL-12R and to have specific biological activities (22–25). Therefore, we investigated whether EBI3 has functions outside IL-27 or IL-35 complexes and whether such functions could adversely affect the therapeutic use of recombinant EBI3 or EBI3 derivative in inflammatory or autoimmune diseases. We observed that EBI3 can activate gp130-expressing cells by mediating IL-6 trans-signaling (Fig. 1).
Results
mEBI3 activates the IL-6-dependent mouse plasmacytoma cell line B9
The B9 cell line is a mouse IL-6-dependent plasmacytoma widely used to measure mouse IL-6 biological activity (26). We previously generated a derivative of this cell line transfected with the IL-27Rα cDNA that responds to IL-27 (Fig. 2B, left panel) (20). We observed that the IL-27Rα-expressing B9 cells (B9-IL-27R) also respond in a dose-dependent manner to recombinant mEBI3 (Fig. 2B, right panel). To assess whether the observed effect reflects the capacity of mEBI3 to recruit and activate the IL-27Rα, we tested the effect of mEBI3 on the unmodified B9 cells that express only the IL-6Rα and therefore do not respond to IL-27 (Fig. 2A, left panel). Unexpectedly, these cells also proliferated in response to mEBI3 (Fig. 2A, right panel). Because mEBI3 can form a composite cytokine with the IL-12 p35 subunit (IL-35) shown to activate homodimers of the signal transducing subunit of gp130 (Fig. 1) (17), we assessed the expression of p35 mRNA by B9 cells by real-time RT-PCR. No expression could be detected, suggesting that the observed proliferation was not due to the formation of IL-35 by an interaction between the exogenous EBI3 and B9-secreted p35 (data not shown).
Figure 2.
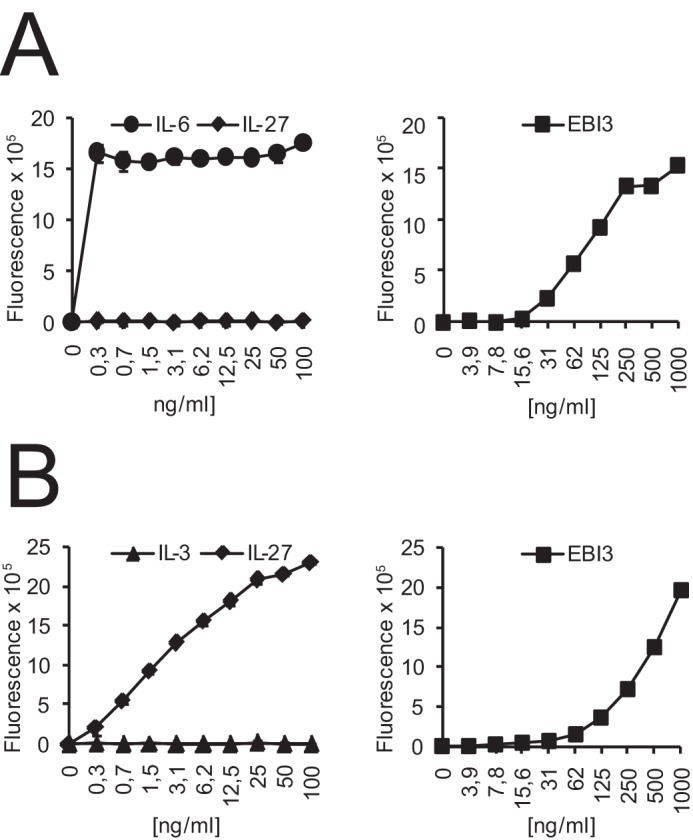
EBI3 activates IL-6-dependent mouse plasmacytoma cell line B9. Untransfected B9 cells (A) or B9 cells stably transfected with mouse IL-27Rα cDNA (B) were incubated with the indicated concentrations of mEBI3, IL-6, IL-3, or IL-27 for 72 h, and proliferation was measured by fluorescence using the AlamarBlue® colorimetric assay. Error bars indicate the S.D. of triplicate cultures. The data are representative of three independent experiments.
The mEBI3-induced B9 cell proliferation can be inhibited by anti-IL-6 and anti-gp130 mAbs
B9 cells express low levels of IL-6 mRNA and proliferate in response to soluble IL-6Rα in a dose-dependent manner (26). This proliferation is believed to reflect trans-signaling induced by the formation of a soluble IL-6Rα·IL-6 complex (10, 26). We therefore investigated whether mEBI3 can substitute for IL-6Rα and promote B9 proliferation by trans-signaling (Fig. 1). Because this signaling would imply the formation of a complex between recombinant mEBI3 and B9-secreted IL-6 followed by the recruitment and activation of gp130 by this initial complex (27), we first assessed the effect of blocking anti-IL-6 and anti-gp130 mAbs on EBI3-induced proliferation. Isotype-matched rat IgG was used as negative control. A marked inhibition of the mEBI3-induced B9 proliferation was observed by blocking either the gp130 receptor (Fig. 3A) or the B9-secreted IL-6 (Fig. 3B). These results indicate that both IL-6 and gp130 were involved in B9 proliferative response, supporting the hypothesis that mEBI3 could substitute for IL-6Rα in the activation of gp130 by trans-signaling (Fig. 1).
Figure 3.

EBI3-induced B9 cell proliferation can be inhibited by anti-IL-6, anti-gp130, and 25F10, a mAb that block IL-6 trans-signaling. Untransfected B9 cells were incubated with mEBI3 (500 ng/ml), IL-6 (0.01 ng/ml), or IL-27 (100 ng/ml) with or without anti-gp130 (A), anti-IL-6 (B), 25F10 (C), or anti-IL-6R (D) mAbs. Proliferation was measured as described in the legend to Fig. 2. Error bars indicate the S.D. of triplicate cultures. Statistical differences between IL-6 ± mAbs and EBI3 ± mAbs were determined using Student's t test. ***, p < 0.001; **, p < 0.01. The data are representative of three independent experiments.
The mEBI3-induced B9 cell proliferation can be inhibited by the trans-signaling inhibitory mAb 25F10
We further explored the mechanism involved in the activation of B9 by mEBI3 using the mAb 25F10. This mAb, which was generated at NovImmune using the IL-6·IL-6Rα complex as immunogen, can prevent IL-6 trans-signaling (28). The addition of mAb 25F10 inhibited B9-IL-27R cell proliferation induced by IL-6 but not by IL-27, included as control (Fig. 3C). Interestingly, 25F10 mAb also inhibited the proliferation of B9-IL-27R cells induced by mEBI3 (Fig. 3C). To investigate whether the observed proliferation was IL-6R-independent, we assessed the effect of blocking anti-IL-6Rα mAb on IL-6, EBI3, or IL-27-induced proliferation using the B9-IL-27R transfectants: although IL-6-induced proliferation was inhibited, either EBI3 or IL-27 proliferations were unaffected in the presence of blocking anti-IL-6R mAb (Fig. 3D). Collectively, these results suggest that mEBI3 can form a trans-signaling complex with IL-6 and that the EBI3·IL-6 and IL-6Rα·IL-6 complexes are sufficiently similar for recognition by the same mAb. In line with this hypothesis, the epitope recognized by the mAb 25F10 has been mapped near Glu-261 in the D3 mIL-6Rα domain (28). This residue diverge between mouse, human, and rat IL-6α (28) or between IL-6Rα and EBI3 (28) but precedes the conserved Phe-262 (29).
mEBI3 activates STAT3 phosphorylation in B9 and CD4 T cells, and this phosphorylation is blocked by anti-IL-6 mAb
We analyzed whether induction of B9 cell proliferation was paralleled by activation of the JAK/STAT pathway. Although 15 min of stimulation with IL-6 induced a clear STAT3 tyrosine phosphorylation, no signal was observed in response to mEBI3 (Fig. 4A). Because activation of B9 by a trans-signaling mechanism involving an EBI3·IL-6 complex would require a secretion of IL-6 in the B9 culture medium unlikely to occur in 15 min, we tested the effect of longer incubation. Interestingly, overnight incubation with mEBI3 resulted in a STAT3 phosphorylation detectable by flow cytometry (Fig. 4A) or Western blot (Fig. 4C) that could be prevented by the inclusion of a blocking anti-IL-6 mAb (Fig. 4, A and C). B9 transfectants expressing IL-27Rα were used as control for the specificity of the blocking anti-IL-6 mAb (Fig. 4, B and C). Induction of STAT3 phosphorylation could be observed with EBI3 concentrations ranging from 0.5 to 2 μg/ml (Fig. 4C). These results indicate that mEBI3 can induce the JAK/STAT signaling pathway in B9 cells and confirm the implication of IL-6 in the activation of B9 cells. To assess whether mEBI3 could also induce STAT3 phosphorylation in primary mouse cells, we stimulated mouse splenocytes with IL-6, IL-27, or mEBI3 for 15 min or 16 h and analyzed STAT3 phosphorylation in CD4 T cells by flow cytometry (Fig. 5). Whereas no STAT3 activation could be detected in cells incubated with mEBI3 for 15 min, a detectable signal was observed at 16 h. This signal could be prevented by the inclusion of a blocking anti-IL-6 mAb (Fig. 5), suggesting that it was induced by a complex formed between the recombinant mEBI3 and IL-6 secreted by the splenocytes. To investigate whether the observed EBI3-induced STAT3 phosphorylation was IL-6R-independent, we assessed the effect of blocking anti-IL-6Rα mAb inclusion during the splenocyte stimulation. Although the IL-6-induced STAT3 phosphorylation was blocked by anti-IL-6R mAb, it did not affect the EBI3 and IL-27-induced STAT3 phosphorylation (Fig. 5B). These results indicate that EBI3 can activate the JAK/STAT pathway in CD4 T cells independently of the canonical IL-6/IL-6R signaling and suggest that EBI3 can form an active complex with IL-6.
Figure 4.

EBI3 activates STAT3 phosphorylation in B9 cells, and this phosphorylation is IL-6-dependent. A, B9 cells were stimulated with mEBI3 (1 μg/ml), IL-27 (100 ng/ml), or IL-6 (100 ng/ml) for 15 min or 16 h with or without anti-IL-6 mAb. B, B9 cells stably transfected with mouse IL-27Rα cDNA were stimulated with IL-27 (100 ng/ml) for 15 min with or without anti-IL-6 mAb. The cells were fixed, permeabilized, and stained with FITC-labeled anti-pSTAT3. Fluorescence was assessed by flow cytometry. The filled histograms indicate unstimulated cells. Mean fluorescence intensity values of the unstimulated (upper left corners) and stimulated cells (upper right corners) are indicated in the histograms. C, B9 cells were stimulated with mEBI3 (0.5, 1, or 2 μg/ml) for 16 h, IL-27 (100 ng/ml), or IL-6 (100 ng/ml) for 15 min with or without anti-IL-6 mAb. D, B9 cells stably transfected with mouse IL-27Rα cDNA were stimulated with IL-27 (100 ng/ml) for 15 min with or without anti-IL-6 mAb. Cell lysates were analyzed by Western blot using anti-pSTAT3 or anti-STAT3 followed by anti-rabbit IgG-HRP. The data are representative of three independent experiments.
Figure 5.
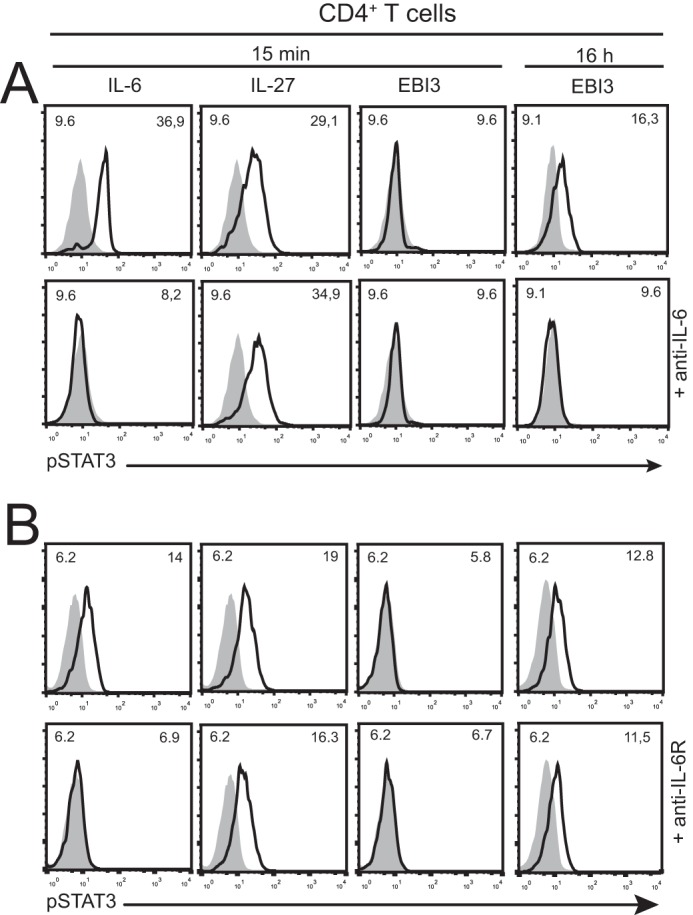
EBI3 activates STAT3 phosphorylation in CD4+ T cells, and this phosphorylation is blocked by anti-IL-6 mAb. A, mouse splenocytes were stimulated with mEBI3 (1 μg/ml), IL-27 (100 ng/ml), or IL-6 (10 ng/ml) for 15 min or 16 h with or without anti-IL-6 mAb. B, mouse splenocytes were stimulated with mEBI3 (1 μg/ml), IL-27 (100 ng/ml), or IL-6 (10 ng/ml) for 15 min or 16 h with or without anti-IL-6R mAb. The cells were fixed, permeabilized, and stained with APC-labeled anti-CD4 and FITC-labeled anti-pSTAT3. Fluorescence was measured by flow cytometry. Histograms represent the fluorescence of the cells gated for CD4 expression. The filled histograms indicate unstimulated cells. Mean fluorescence intensity values of the controls (upper left corners) and stimulated cells (upper right corners) are indicated in the histograms. The data are representative of three independent experiments.
EBI3 forms a secreted complex with IL-6
Next, we evaluated the binding between EBI3 and IL-6 by co-immunoprecipitation (Fig. 6). We generated stable HEK-293 transfectants expressing IL-6, IL-27 (i.e. co-expressing EBI3 and IL-30), or a combination of EBI3 and IL-6. Both EBI3 and IL-6 were detected in respective transfectant cell culture medium (Fig. 6A). When EBI3 was immunoprecipitated with a specific mAb, co-immunoprecipitation of IL-6 could be detected (Fig. 6A, bottom panel). As expected, no signal for IL-6 was observed when the co-immunoprecipitation assays were performed using medium containing IL-6 alone or IL-27 (Fig. 6A, bottom panel). To investigate whether both mouse and human EBI3 could bind IL-6, His-Biotin-tagged mouse or human EBI3 was incubated with unconjugated mouse or human IL-6, and the mix was subjected to IMAC. Co-immunoprecipitation of EBI3 and IL-6 could be detected using HRP-labeled streptavidin to reveal EBI3 and anti-IL-6 to detect IL-6 (Fig. 6B). As expected, no signal was observed when co-immunoprecipitation assays were performed using medium containing only IL-6 (Fig. 6B). These results indicate that both mouse and human EBI3 can bind IL-6.
Figure 6.
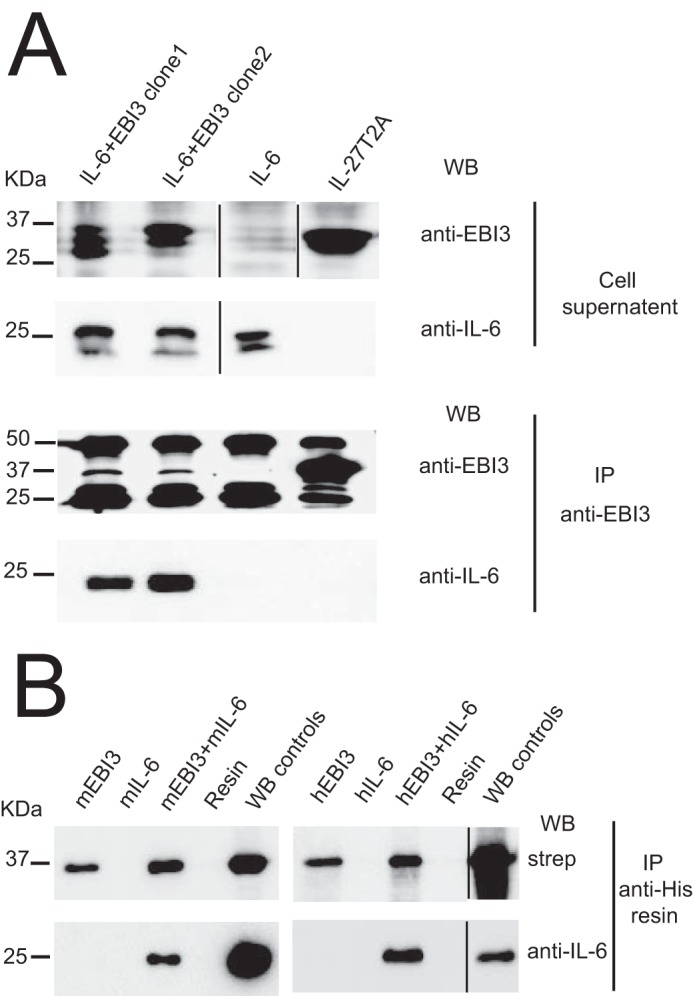
EBI3 forms a secreted complex with IL-6. A, HEK-239 Flp-InTM cells were transfected with expression vectors coding for mEBI3-T2A-IL-6 (clone nos. 1 and 2), mIL-27T2A, and mIL-6. Proteins were immunoprecipitated (IP) from cellular supernatants using anti-mEBI3 mAb and anti-rat IgG-agarose resin. Signals for mEBI3 (35 kDa) and mIL-6 (25 kDa) were revealed by successive incubation with anti-mEBI3 and biotinylated anti-mIL-6 Abs followed by anti-rat IgG-HRP and streptavidin-HRP, respectively. Signals at 50 and 25 kDa represent background because of the detection of the Ig heavy and light chains eluted from rat IgG-agarose by the secondary antibody used for EBI3 detection. B, His-biotin-tagged recombinant mouse or human EBI3 and unconjugated mouse or human IL-6 were subjected to IMAC. Nickel-nitrilotriacetic acid (Ni-NTA) resin incubated without the addition of EBI3 and IL-6 was used as negative control (resin lanes). EBI3 (100 ng) and IL-6 (20 ng) were used as positive detection controls. Signals were revealed with either streptavidin-HRP or anti-IL-6 followed by HRP-conjugated anti-IgG. WB, Western blotting.
IL-6 binding to EBI3 could be detected by SPR
We further proved binding of EBI3 with IL-6 with SPR measurements. EBI3 was covalently immobilized on the surface of the SPR chip and incubated with IL-6. A shift of ∼3.5 nm was observed with IL-6 (Fig. 7, A and D). When EBI3 immobilized on the SPR chip was incubated with identical concentrations of IL-11, a background shift of 0.8 nm was observed (Fig. 7, C and D). When equal concentrations of soluble G-CSFR were immobilized on the surface by the same manner, a background shift of 0.8 nm was observed with IL-6 (Fig. 7, B and D). The significantly larger SPR response for EBI3-IL-6 confirms the selectivity of interaction between these proteins. We then used SPR to estimate the dissociation constant (KD) of the EBI3·IL-6 complex, which was measured at 4.1 μm (Fig. 7E). These data further indicate that EBI3 can bind IL-6.
Figure 7.

EBI3-IL-6 interaction assessed by SPR. A and B, EBI3 (A) or G-CSFR (B) was immobilized via EDC/NHS on 50-mm-thick gold surface coated with a peptide monolayer as indicated under “Experimental procedures.” Conditions of binding and washing were indicated. C, the sensorgrams for the binding of IL-6 on EBI3 and G-CSFR clearly show the preferential interaction of EBI3 and IL-6. PBS (pH 7.4) rinsing is also shown. D, SPR sensorgrams showing the greater response for the EBI3 + IL-6 binding partners, while controls with EBI3 modified SPR chip had a significantly lower response to IL-11 than EBI3 + IL-6, or a G-CSFR modified SPR chip exposed to IL- 6. E, dose-response curve for IL-6 with an EBI3 modified SPR chip. The concentration of KD is shown as the dotted line in the figure. The data are representative of three independent experiments.
mEBI3-IL-6 activates gp130-expressing Ba/F3 cells
Recombinant fusion proteins between sIL-6Ra·IL-6 (“hyper-IL-6”) in which the complex is stabilized by a flexible linker have been widely used to demonstrate that the sIL-6Ra·IL-6 complex can activate cells expressing only gp130 (“trans-signaling”) (Fig. 1) and to investigate the role of IL-6 trans-signaling (30). We therefore assessed whether a structurally analogous fusion between EBI3 and IL-6 could, like hyper-IL-6, activate gp130-expressing Ba/F3 cells. Ba/F3 was chosen because this IL-3-dependent cell line does not express IL-6Rα, can be rendered sensitive to other cytokines by transfection with the relevant receptor cDNA, and has been widely used to study type I cytokine receptors (31). As expected, expressions of gp130 rendered Ba/F3 cells sensitive to hyper-IL-6, as well as to IL-27, because Ba/F3 spontaneously express IL-27Rα (32) (Fig. 8A, left panel). No proliferation in response to either IL-6 (Fig. 8A, left panel) or EBI3 alone could be detected (Fig. 8A, right panel), confirming that these transfectants do not constitutively express either IL-6 or IL-6Rα and are therefore suitable for investigating the trans-signaling capacity of the EBI3-IL-6 fusion. A clear proliferation in response to the EBI3-IL-6 fusion protein was observed (Fig. 8A, right panel). The EBI3-IL-6 fusion protein could also stimulate the activation of STAT3 (Fig. 8B). The up-regulation of STAT3 phosphorylation induced by 30 min stimulations with 1 μg/ml of EBI3-IL-6 was, however, substantially lower than that induced by hyper-IL-6 at 100 ng/ml. Altogether, these results indicate that EBI3 can mediate IL-6 trans-signaling, albeit less potently than sIL-6Rα.
Figure 8.
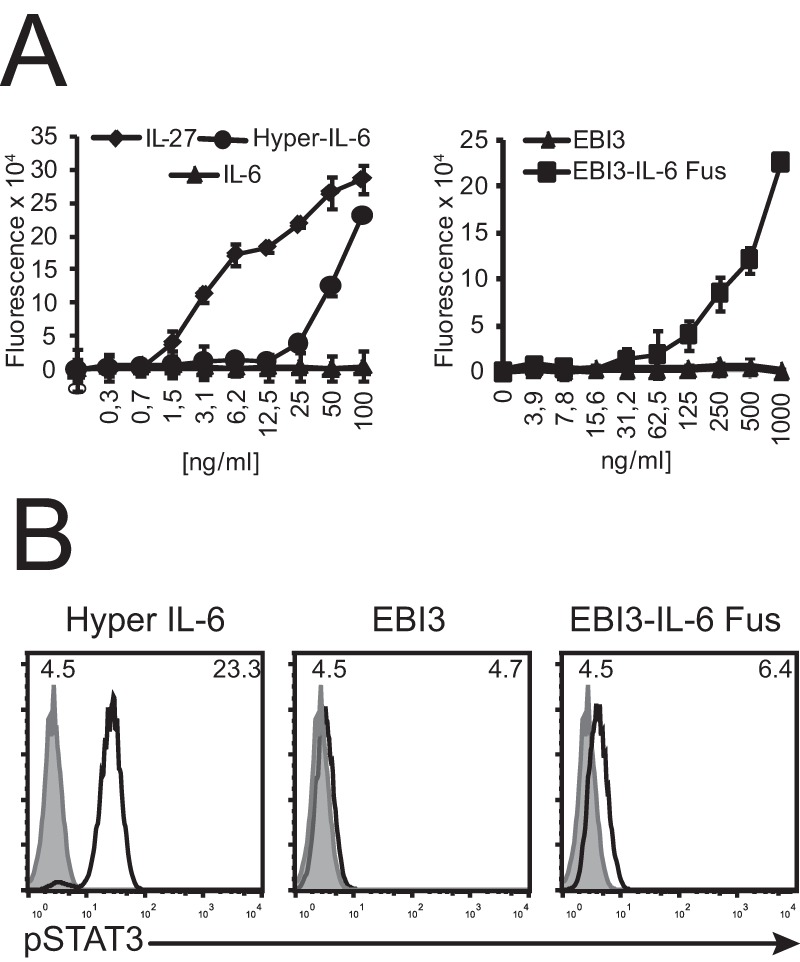
EBI3-IL-6 fusion protein activates gp130-expressing Ba/F3 cells. A, BAF/3 cells stably transfected with mouse gp130 cDNA were incubated with the indicated concentrations of mEBI3, IL-6, IL-27, mEBI3-IL-6 fusion (EBI3-IL-6 Fus), or hyper-IL-6 for 72 h. Proliferation was measured as described in the legend to Fig. 1. The error bars indicate the S.D. of triplicate cultures. B, BAF/3-gp130 were stimulated with mEBI3 (1 μg/ml), mEBI3-IL-6 fusion (1 μg/ml), or hyper-IL-6 (100 ng/ml) for 30 min. The cells were fixed, permeabilized, and stained with FITC-labeled anti-pSTAT3. Fluorescence was measured by flow cytometry. The filled histograms indicate unstimulated cells. Mean fluorescence intensity values of the non-stimulated (upper left corners) and stimulated cells (upper right corners) are indicated in the histograms. The data are representative of three independent experiments.
EBI3-IL-6 binds immobilized gp130
We covalently immobilized human soluble gp130 on the surface of the SPR chip and incubated it with hEBI3-IL6 fusion or hEBI3. Although the scarcity of the purified protein limited the concentration that could be used to a maximum ∼55 times lower than that used for the IL-6 binding to EBI3, a smaller, albeit significant shift of ∼0.5 nm was observed (Fig. 9, A and C), whereas no signal was detected with EBI3, indicating that hEBI3-IL-6 binds gp130 (Fig. 9, B and C).
Figure 9.

EBI3-IL-6 fusion interaction with soluble gp130 assessed by SPR. Soluble gp130 was immobilized via EDC/NHS on 50-mm-thick gold surface coated with a peptide monolayer as indicated under “Experimental procedures.” The sensorgram for EBI3-IL-6 (A and C) or EBI3 (B and C) are indicated. PBS (pH 7.4 or 4.5) rinsing is also shown. The decrease of the SPR signal observed with the injection of EBI3 is due to the different refractive index of the solution. The data are representative of three independent experiments.
Induction of chemokine production by EBI3-mediated IL-6 trans-signaling
Previous reports indicate that IL-6-induced chemokine production by HUVEC is strictly mediated by trans-signaling and that it can be induced using combinations of IL-6 and sIL-6Rα (33). Therefore, we used this system to investigate whether EBI3 could mediate trans-signaling on human cells. When HUVEC were stimulated for 4 h in the presence of sIL-6Rα and IL-6, a strong up-regulation of MCP-1 and MCP-3 mRNA (Fig. 10A) or MCP-1 and MCP-2 (Fig. 10C) production could be observed. Interestingly, up-regulation of MCP-1 and MCP-3 mRNA levels or production was also induced by the conjunction of hIL-6 and hEBI3, albeit less efficiently (Fig. 10, A and C). As expected, no chemokine mRNA or protein level up-regulation was detected when cells were stimulated with hIL-6 alone, confirming that trans-signaling (Fig. 1) was required for the induction of chemokine production by HUVEC. Up-regulation of MCP-1 and MCP-3 mRNA levels could be detected with EBI3 concentrations between 0.5 and 2 μg/ml (Fig. 10B). These results indicate that EBI3 can mediate IL-6 trans-signaling in human cells.
Figure 10.

The combination of IL-6 and EBI3 induces chemokine expression and production in HUVEC in a dose-dependent manner, and this expression is blocked by anti-gp130 mAb. A, cells were stimulated for 4 h with EBI3 (1 μg/ml) and sIL-6R (100 ng/ml) with or without IL-6 (10 ng/ml). Gene expression was assessed by real-time RT-PCR. B, cells were stimulated for 4 h with IL-6 (10 ng/ml) and an increasing concentrations of EBI3. Gene expression was assessed by real-time RT-PCR. C and D, cells were stimulated for 24 h with EBI3 (2 μg/ml) and IL-6 (10 ng/ml) or with EBI3-IL-6 recombinant fusion protein (200 ng/ml) with or without anti-gp130 mAb. Chemokine levels were measured by ELISA. Statistical differences between sIL-6R ± IL-6 and EBI3 ± IL-6 were determined using a Student's t test. ***, p < 0.001; *, p < 0.05. The error bars in the figures represent S.D. of triplicate cultures. The data are representative of two independent experiments.
EBI3-mediated IL-6 trans-signaling can be induced by EBI3-IL-6 fusion protein and inhibited by anti-gp130 mAb
We next observed that the up-regulation of MCP-1 and MCP-3 secretion can also be induced by the conjunction of hIL-6 and hEBI3 (Fig. 10C) or the hEBI3-IL-6 fusion (Fig. 10D) and that the responses induced by the combinations of sIL-6Ra and IL-6, of EBI3 and IL-6 or hEBI3-IL-6 can be inhibited by the inclusion of anti-gp130 mAb (Fig. 10, C and D). These results indicate that EBI3 can mediate IL-6 trans-signaling through gp130.
Discussion
IL-6 is a cytokine that elicits a broad range of immune and acute phase responses with both pro- and anti-inflammatory functions. The dual role of this cytokine, at least in part, depends on the nature of the signaling pathway induced: IL-6 “classical” cis-signaling involving membrane IL-6Rα is believed to maintain body homeostasis, whereas IL-6 trans-signaling via the soluble IL-6 receptor mediates most pro-inflammatory responses (34, 35). We and others observed that IL-30, the p28 subunit of IL-27, has functions unrelated to its capability to form a complex with EBI3: it can activate the IL-6 receptor by cis- or trans-signaling and can trigger the differentiation of B cells in plasmocytes (19, 20, 36). That EBI3 also has roles unrelated to IL-27 is suggested by publications indicating its presence in tissues negative for p28: no significant p28 expression was detected in EBI3-secreting Hodgkin lymphoma-derived cell lines, in Epstein-Barr virus-transformed B cell lines (37), or in EBI3-expressing tumor-infiltrating dendritic cells (37). Although the report did not rule out that these cells express p35, the authors suggested that EBI3 regulates immune response independently from its association to p28. In accordance with this hypothesis, EBI3 is involved in transplantation tolerance induced by tolerogenic dendritic cells in mice through a mechanism involving IFN-γ expression by double-negative T cells in the absence of both p35 and p28 partners (38). Furthermore, EBI3 expression was shown to be up-regulated in response to IL-1α and TNF-α in human intestinal epithelial cells, suggesting a potential role for EBI3 in the early host response to inflammation with no observed expression of p28 and lack of coordination with p35 (39).
Studies investigating the role of EBI3 were mostly based on the use of EBI3-deficient mouse model (40–42). While testing its biological activities in vitro, we observed that EBI3 supports the proliferation of the IL-6-responsive mouse plasmacytoma cell line B9. This cell line is IL-27-unresponsive (20), indicating that EBI3 could have IL-27Rα-independent regulatory roles. Previous studies revealed that sIL-6Rα induces B9 cell proliferation through a trans-signaling pathway involving the formation of a complex between minute amounts of IL-6 secreted by the plasmacytoma cell line and exogenous sIL-6Rα (26). Our analysis of the activation of B9 cells by EBI3 indicated that it could be blocked by either anti-gp130 or anti-IL-6 mAbs and that induction of STAT3 phosphorylation required a long stimulation time. This suggests that proliferation was induced through an analogous trans-signaling triggered by an alternative complex formed by the B9-released IL-6 and recombinant EBI3. Also in agreement with this hypothesis, we observed in co-immunoprecipitation studies that EBI3 binds IL-6 when the two proteins are co-expressed or mixed in vitro or in SPR biding assays. To further test the ability of EBI3 to mediate IL-6 trans-signaling, we produced an EBI3-IL-6 fusion protein structurally similar to the hyper-IL-6 used to study IL-6 trans-signaling (30). This fusion protein could induce the activation of Ba/F3 cells transfected with gp130, albeit less potently than hyper-IL-6. We investigated the effect of EBI3 on primary splenocytes and observed that it could induce an IL-6-dependent STAT3 activation in the CD4 T-cells.
A physiologically important pro-inflammatory role of IL-6 trans-signaling is to activate IL-6Rα negative endothelial cells (43). HUVEC have been used as a model to assess the effect of IL-6 trans-signaling in human endothelial cells (33). We observed that EBI3 could, in conjunction with IL-6, induce MCP-1 and MCP-3 gene expression in HUVEC, indicating that EBI3 can mediate IL-6 trans-signaling in human cells and suggesting that the EBI3·IL-6 complex could be involved in the recruitment of leukocytes at sites of inflammation.
Our results imply that EBI3 could have either pro- or anti-inflammatory effects on gp130-expressing cells depending on its binding to the cytokine partner subunits p35, IL-30 (p28), or IL-6. The effects of EBI3 on IL-6 trans-signaling were observed with EBI3 concentrations between 500 ng and 2 μg/ml, much higher than those detected in peripheral blood (3). These concentrations are, however, close to those detected in sera during the last months of pregnancy (117–446 ng/ml) (3), suggesting that EBI3 could contribute to the role of IL-6 in preterm delivery (44), believed to involve trans-signaling (45). These data and previous observations that IL-30 can activate the IL-6 receptor by cis- or trans-signaling (18–20) further demonstrate a high level of complexity and interchangeability in this family of cytokines and cytokine receptors (46). Our observations suggest caution in using EBI3 derivatives as potential therapeutic agents to favor the generation or stabilization of IL-27 and IL-35 through complexing unbound IL-30 or p35. Further studies should indicate whether this novel EBI3 activity contributes to IL-6 trans-signaling in autoimmune and inflammatory diseases models.
Experimental procedures
Cells and reagents
B9-IL-27Rα and Ba/F3-gp130 cells were generated as described previously (20, 47). The cells were grown in RPMI 1640 (Wisent Bio Products, St-Bruno, Canada) supplemented with 10% FBS, 10 mm HEPES, 2 mm l-glutamine, 1% nonessential amino acids, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from Wisent) in the presence of IL-6 (10 ng/ml; R&D Systems, Cedarlane) or IL-3 (10 ng/ml; PeproTech Inc., Cedarlane) and hygromycin (1 mg/ml) for B9-IL-27Rα and Ba/F3-gp130, respectively. High FiveTM insect cells were cultured in Express Five® medium (Invitrogen, ThermoFisher Scientific) supplemented with 20 mm l-glutamine at 27 °C. Flp-In-293 cells were expanded in DMEM (Wisent Bio Products) supplemented with 10% FBS. HUVEC were cultured in M-199 medium (ThermoFisher Scientific) supplemented with 20% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml endothelial cell growth supplement, and 100 μg/ml heparin in 0.1% collagen precoated dishes. Recombinant mIL-27, hIL-6, hIL-11, hsIL-6Rα, human soluble G-CSFR, and human soluble gp130 were purchased from R&D Systems (Cedarlane). The following primary antibodies were used: anti-His mAb (Qiagen), anti-mEBI3 (clone no. DNT27; eBioscience, Cedarlane), anti-mIL-6 (clone no. MP5-20F3; R&D Systems), biotinylated polyclonal anti-mIL-6 (R&D Systems), and anti-hIL-6 (clone no. 6708; R&D Systems). The following HRP-conjugated antibodies were used: anti-mouse IgG (GE Healthcare Biosciences, Cedarlane), anti-rat IgG, and anti-goat IgG (R&D Systems). HRP-labeled streptavidin (GE Healthcare Biosciences) was used to detect biotinylated proteins and antibodies. The following antibodies were used at 5 μg/ml to block the IL-6- and gp130-dependent activities: anti-mgp130 (clone no. 125623; R&D Systems) and anti-mIL-6 (Biolegend, Cedarlane). For flow cytometry assay, FITC-labeled anti-phospho-STAT3 mAb and APC-labeled anti-phospho-CD4 mAb were obtained from BD Biosciences.
Experimental animals
All procedures conformed to the Canadian Council on Animal Care guidelines and were approved by the Animal Ethics Committee of the Université de Montréal. 6–8-week-old female C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME).
Expression and purification of mEBI3, mEBI3-IL-6, and mhyper-IL-6 recombinant proteins
Derivatives of the pIB/V5-His vector coding for a carboxy-His6-tagged mEBI3 or mEBI3-IL-6 and mIL-6-sIL-6R (hyper-IL-6) fusion proteins linked by GGG(SGGGG)3SHHHHHH sequence were generated using synthetic DNA (GeneArt, ThermoFisher Scientific) and standard molecular biology methods and then transfected in High FiveTM insect cells (ThermoFisher Scientific) according to the supplier's instructions. Transfected cells were selected using blasticidin S hydrochloride (100 μg/ml). Batches of 500 ml of transfected cell culture medium were concentrated 25 times with Vivaflow® 200 cross-flow cassettes (Sartorius Inc., Mississauga, Canada) in the presence of 0.5 mm PMSF and purified by immobilized metal-affinity chromatography (IMAC) using nickel-nitrilotriacetic acid-agarose beads (Qiagen) under the conditions suggested by the manufacturer. Purified proteins were dialyzed two times against PBS, sterile-filtered, quantified, and analyzed by Western blot for the presence of mEBI3 and mIL-6 using anti-His5 mAb followed by anti-mouse HRP-conjugated IgG. Biological activity of mEBI3 was assessed using a B9 plasmacytoma proliferation assay (26).
Expression and purification of bacterial EBI3
Synthetic (GenArt) cDNA coding for His6/Avitag-tagged mouse and human mature EBI3 were subcloned in the expression plasmid pET24d (Novagen). Expression of EBI3 was performed in Escherichia coli BL21 (DE3) (Novagen) as described (48). EBI3 was purified from bacterial inclusion bodies by IMAC under the denaturing conditions suggested by Qiagen. EBI3 was refolded by subsequent dialysis against 20 mm glycine (pH 3.0), 0.2 mm oxidized glutathione, and 2 mm reduced glutathione and decreasing urea concentrations. EBI3 was sterile-filtered and quantified by SDS-PAGE and Coomassie Blue staining using BSA as standard. Biological activity was assessed using a B9 plasmacytoma proliferation assay (26).
Expression and purification of hEBI3-IL-6 fusion protein
A synthetic pcDNA3.4 derivatives coding for a human EBI3–6H-(SGGG)3S-IL-6 fusion protein (GenArt) was transiently transfected in HEK 293T cells (ATCC, Cedarlane) using polyethylenimine (49). Transfected cells were maintained in Opti-MEMTM (ThermoFisher Scientific) for 5 days. Cell culture medium was concentrated by ultrafiltration. EBI3-IL-6 was purified by IMAC, sterile-filtered, quantified, and analyzed by Western blot for the presence of hEBI3 and hIL-6. Biological activity of the fusion protein was assessed using a B9 plasmacytoma proliferation assay (26).
Proliferation assays
Untransfected B9 cells, B9-IL-27Rα, or Ba/F3-gp130 cells were incubated for 72 h in triplicate with the indicated dilutions of recombinant proteins (19). For inhibitory studies, IL-6 or gp130 targeting antibodies (5 μg/ml) or the anti-IL6·IL-6Rα complex 25F10 mAb (10 μg/ml; NovImmune, Plan-Les-Ouates, Switzerland) were added as indicated. Proliferation was measured using a fluorometric assay (AlamarBlue®; AbDSerotec, Cedarlane). Mean unstimulated cell fluorescence background was subtracted from the values obtained with the stimulated cells.
Measurement of STAT3 activation by flow cytometry
Untransfected B9 cells, B9-IL-27Rα, or Ba/F3-gp130 cells were serum- and cytokine-starved for 16 h. B9, B9-IL-27Rα, Ba/F3-gp130, or C57BL/6 mouse primary splenocytes (106 cells/condition) were activated with cytokines in the presence or absence of anti-IL-6 mAb or anti-IL-6R mAb for 15 min or 16 h at 37 °C. The cells were fixed, permeabilized, and stained with FITC-labeled anti-phospho-STAT3 (Tyr-705) and APC-labeled anti-CD4 mAbs as described (19). Fluorescence was assessed using a FACSCalibur (BD Biosciences). The data were analyzed using the FlowJo software (Tree Star Inc., Ashland, OR).
Measurement of STAT3 activation by Western blot
Untransfected B9 cells or B9-IL-27Rα cells were serum- and cytokine-starved for 16 h. The cells were then activated with cytokines in the presence or absence of anti-IL-6 mAb for 15 min or 16 h at 37 °C. The cells were lysed in 1% Triton X-100, 10% glycerol, 50 mm NaCl, 50 mm HEPES, 5 mm EDTA (pH 7.3). Protein levels were quantified using a bicinchoninic acid assay. Aliquots of lysates containing 20 μg of proteins were analyzed by Western blotting for total and phosphorylated STAT3 using specific Abs (Cell Signaling Technology) followed by HRP-labeled anti-rabbit IgG.
Generation of stable HEK-293 transfectants co-expressing mEBI3 and mIL-6
pcDNA5 derivatives coding for mIL-6, mEBI3, mIL-30 (p28), or the combination of these proteins were generated using synthetic cDNAs (GeneArt) and standard molecular biology techniques. For the bicistronic pcDNA5 vectors, the cDNA coding for mEBI3 and mIL-6 or mEBI3 and p28 (IL-27) were separated by a sequence coding for a T2A “self-cleaving peptide” (50). Stable Flp-InTM-293 transfectants were generated and selected following ThermoFisher Scientific protocol.
Co-immunoprecipitations and Western blotting
Flp-In-293 transfectants expressing mIL-6, mIL-27 T2A, or mEBI3-T2A-IL-6 were expanded to confluence in DMEM, 10% FBS. Confluent cells were maintained in Opti-MEMTM medium for 72 h. Supernatants (20 ml) were concentrated using Amicon Ultra-15 centrifugal filters (EMD-Millipore), mEBI3 was immunoprecipitated using 1 μg of rat anti-mEBI3 (clone no. DNT27; eBioscience) and 50 μl of agarose-conjugated anti-rat IgG antibody (Rockland, Cedarlane) for 16 h at 4 °C. The immunoaffinity-purified proteins were washed three times with cold PBS and eluted with 0.1 m glycine HCl (pH 2.5). Eluted fractions were analyzed by Western blotting using anti-mEBI3 followed by HRP-conjugated anti-rat IgG or biotinylated anti-mIL-6 followed by HRP-conjugated streptavidin. For co-immunoprecipitation of mouse or human EBI3 and IL-6, 1 μg of 6-His/Biotin-tagged mEBI3 and 100 ng of mIL-6 (R&D Systems) or 5 μg of 6-His/Biotin-tagged hEBI3 and 500 ng of hIL-6 (R&D Systems) were co-immunoprecipitated using His-select® HF nickel affinity gel (Sigma-Aldrich) for 16 h at 4 °C in 1 ml of binding buffer (50 mm NaH2PO4, 300 mm NaCl, 10 mm immidazol, pH 8). The immunoaffinity-purified proteins were washed three times with cold buffer containing (50 mm NaH2PO4, 300 mm NaCl, 20 mm imidazole, pH 8) and eluted with (50 mm NaH2PO4, 300 mm NaCl, 500 mm immidazol, pH 8). Eluted fractions were analyzed by Western blotting using streptavidin-HRP for EBI3 detection or polyclonal anti-mouse or human IL-6 (R&D Systems) followed by HRP-labeled anti-IgG for IL-6 detection.
Surface plasmon resonance binding assay
Binding of IL-6 and IL-11 on EBI3 or G-CSFR was evaluated with surface plasmon resonance (SPR). All SPR measurements were acquired with a home-built instrument described elsewhere (51). To perform the binding assays, the SPR chip was functionalized with a peptide monolayer consisting of 3-mercaptopropyl-LHDLHD-OH(3-MPA-LHDLHD-OH) in conditions previously reported (52). Immobilizations of EBI3 and G-CSFR were realized using a peptide cross-linking reaction. First, a baseline was acquired for 5 min in acidic water (pH 3) adjusted with hydrochloric acid or PBS adjusted to pH 4.5 with hydrochloric acid. Then a 1:1 solution of 35 mm ethyl-(dimethylaminopropyl) carbodiimide and 110 mm N-hydroxysuccinimide was injected for 5 min on the sensors to activate the carboxylic acid functions of the peptide monolayer immobilized at the surface of the SPR chip. The SPR chip was then washed with acidic water or PBS (pH 4.5) for 3 min. EBI3 or G-CSFR was diluted to 204 nm in acidic water or PBS (pH 4.5) and reacted with the SPR chip for 15 min. Subsequently, the EBI3 or G-CSFR solution was washed with acidic water or PBS (pH 4.5) for 3 min. Then a 1 m ethanolamine solution (pH 8.5) was added for 5 min to block any remaining activated site on the sensor. Ethanolamine solution was washed with PBS (pH 7.4) for 3 min, and then PBS with 0.1% BSA was injected for 5 min to further block any remaining active sites on the SPR chip. The surface was further washed with PBS (pH 7.4) for 3 min. A solution of IL-6 or IL-11 at 680 nm prepared in PBS (pH 7.4) was injected for 20 min before finally rinsing the sensor surface with PBS (pH 7.4) for 5 min.
Determination of IL-6-EBI3 dissociation constant
The dissociation constant (KD) of IL-6 and EBI3 was determined using experimental SPR data. The sensor surface was modified with EBI3 as described above. IL-6 solutions of concentrations ranging from 50 nm to 50 μm were then exposed to the sensor surface for 5 min and rinsed with PBS (pH 7.4). The shift of the SPR band for each concentration was used to determine the KD using the Langmuir isotherm model with Matlab (Natick, MA) and the curve fitting toolbox. The equation used in Matlab is ΔλSPR = [C/(KD + C)] * ΔλSPR,max, and experimental data were fitted with a non-linear least-squares method. In the equation, ΔλSPR is the SPR shift for each concentration, C is the IL-6 concentration, KD is the dissociation constant (KD), and ΔλSPR,max is the maximum SPR shift calculated with the model.
Binding assay of the EBI3-IL-6 fusion protein to gp130
The binding of EBI3-IL-6 to gp130 was assessed by SPR. The sensor was coated with the peptide monolayer, in which the carboxylic acid groups immobilized at the surface of the SPR chip were activated and washed as described above. Then a 5 μg/ml solution of soluble gp130 in PBS (pH 7.4) was reacted with the SPR chip for 20 min. The gp130 solution was washed with PBS (pH 7.4) for 1 min and with PBS + 0.1% BSA for 5 min. Then a 1 m ethanolamine solution (pH 8.5) was added for 5 min to block any remaining activated site on the sensor. Ethanolamine solution was washed with PBS + 0.1% BSA for 5 min. A 90 nm solution of either EBI3-IL-6 or EBI3 was injected for 15 min. Finally, the sensor surface was rinsed with PBS + 0.1% BSA for 5 min. EBI3-IL-6 and EBI3 solutions were prepared in PBS at pH 7.4 or pH 4.5, respectively.
Human endothelial cell culture
Confluent HUVEC in their fifth passage were kept for 4 h in M-199 plus 1% BSA. The medium was replaced with M-199 containing 1% BSA and 5 μg/ml polymixin B in the presence or absence of IL-6 (10 ng/ml), sIL-6Rα (100 ng/ml), EBI3 (0,1–2 μg/ml), EBI3-IL-6 recombinant fusion protein (200 ng/ml), or a combination of either sIL-6Rα-IL-6 or EBI3-IL-6 for 4 h at 37 °C prior to RNA extraction or 24 h for measuring chemokines levels in the cell culture medium. For inhibition studies, blocking anti-gp130 Ab or isotype control (R&D Systems) were used at 10 μg/ml.
Analysis of MCP-1 and MCP-3 expression by RT-PCR
Expression of MCP-1 (NCBI accession no. IR5047) and MCP-3 (NCBI accession no. IR5048.2) was analyzed by real-time RT-PCR. Briefly, RNA was extracted using EZ-10 column total RNA mini-prep kits (Bio Basic Inc., Markham, Canada). RNA integrity was assessed using an Agilent Bioanalyzer at the genomic platform of the Institute for Research in Immunology and Cancer (Montreal, Canada). Random-primed single-stranded cDNA was then synthesized using Moloney murine leukemia virus reverse transcriptase (ThermoFisher Scientific) and 0.5 μg of total RNA. MCP-1 and MCP-3 cDNA levels were assessed using TaqMan probes and a TaqMan Fast qPCR MasterMix (Applied Biosystems; Thermo Fisher Scientific) at the Institute for Research in Immunology and Cancer genomic platform.
Analysis of MCP-1 and MCP-3 levels by ELISA
MCP-1 and MCP-3 concentrations were measured using human ELISA kits according to the instructions of the supplier (Abcam Inc., Cedarlane).
Statistical analysis
The error bars represent means ± S.D. of triplicate cultures. Statistical analysis was performed using Student's unpaired t test. p < 0.05 was considered statistically significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Author contributions
S. C. performed most of the experiments. J. L. C. conducted the experiments using SPR. The paper was written by S. C., J.-F. G., J.-F. M., and M. S. with contributions of all other authors. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank the Institute for Research in Immunology and Cancer genomic platform for the DNA sequencing and the real-time PCR analysis. We are grateful to the group of Dr. Jean-Philippe Gratton (Département de Pharmacologie, Université de Montréal) for the gift of HUVEC and for advice and help with the HUVEC experiments. We thank S. Sénéchal (Flow cytometry platform, Université de Montréal) for help with flow cytometry.
This work was supported by Canadian Institutes of Health Research Grant MOP-57832 (to J.-F. G.) and Natural Science and Engineering Research Council of Canada Grant 412305-2011 (to J.-F. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- EBI3
- Epstein-Barr virus-induced gene 3
- APC
- allophycocyanin
- G-CSFR
- granulocyte colony-stimulating factor receptor
- gp
- glycoprotein
- h
- human
- m
- mouse
- R
- receptor
- s
- soluble
- SPR
- surface plasmon resonance
- HUVEC
- human venal endothelial cells
- IMAC
- immobilized metal-affinity chromatography
- EDC/NHS
- N-ethyl-N′-(3-dimethyl-aminopropyl)-carbodiimide/N-hydroxysuccinimide.
References
- 1. Jones L. L., and Vignali D. A. (2011) Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol. Res. 51, 5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Devergne O., Hummel M., Koeppen H., Le Beau M. M., Nathanson E. C., Kieff E., and Birkenbach M. (1996) A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J. Virol. 70, 1143–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Devergne O., Coulomb-L'Herminé A., Capel F., Moussa M., and Capron F. (2001) Expression of Epstein-Barr virus-induced gene 3, an interleukin-12 p40-related molecule, throughout human pregnancy: involvement of syncytiotrophoblasts and extravillous trophoblasts. Am. J. Pathol. 159, 1763–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pflanz S., Timans J. C., Cheung J., Rosales R., Kanzler H., Gilbert J., Hibbert L., Churakova T., Travis M., Vaisberg E., Blumenschein W. M., Mattson J. D., Wagner J. L., To W., Zurawski S., et al. (2002) IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity 16, 779–790 [DOI] [PubMed] [Google Scholar]
- 5. Devergne O., Birkenbach M., and Kieff E. (1997) Epstein-Barr virus-induced gene 3 and the p35 subunit of interleukin 12 form a novel heterodimeric hematopoietin. Proc. Natl. Acad. Sci. U.S.A. 94, 12041–12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vignali D. A., and Kuchroo V. K. (2012) IL-12 family cytokines: immunological playmakers. Nat. Immunol. 13, 722–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pirhonen J., Sirén J., Julkunen I., and Matikainen S. (2007) IFN-α regulates Toll-like receptor-mediated IL-27 gene expression in human macrophages. J. Leukoc. Biol. 82, 1185–1192 [DOI] [PubMed] [Google Scholar]
- 8. Pflanz S., Hibbert L., Mattson J., Rosales R., Vaisberg E., Bazan J. F., Phillips J. H., McClanahan T. K., de Waal Malefyt R., and Kastelein R. A. (2004) WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J. Immunol. 172, 2225–2231 [DOI] [PubMed] [Google Scholar]
- 9. Bosmann M., and Ward P. A. (2013) Modulation of inflammation by interleukin-27. J. Leukoc. Biol. 94, 1159–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Freitas do Rosário A. P., Lamb T., Spence P., Stephens R., Lang A., Roers A., Muller W., O'Garra A., and Langhorne J. (2012) IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: a critical mechanism for protection from severe immunopathology during malaria infection. J. Immunol. 188, 1178–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stumhofer J. S., Laurence A., Wilson E. H., Huang E., Tato C. M., Johnson L. M., Villarino A. V., Huang Q., Yoshimura A., Sehy D., Saris C. J., O'Shea J. J., Hennighausen L., Ernst M., and Hunter C. A. (2006) Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat. Immunol. 7, 937–945 [DOI] [PubMed] [Google Scholar]
- 12. Batten M., Li J., Yi S., Kljavin N. M., Danilenko D. M., Lucas S., Lee J., de Sauvage F. J., and Ghilardi N. (2006) Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat. Immunol. 7, 929–936 [DOI] [PubMed] [Google Scholar]
- 13. Liu Z., Yu J., Carson W. E 3rd, Bai X. F. (2013) The role of IL-27 in the induction of anti-tumor cytotoxic T lymphocyte response. Am. J. Transl. Res. 5, 470–480 [PMC free article] [PubMed] [Google Scholar]
- 14. Li X., Mai J., Virtue A., Yin Y., Gong R., Sha X., Gutchigian S., Frisch A., Hodge I., Jiang X., Wang H., and Yang X. F. (2012) IL-35 is a novel responsive anti-inflammatory cytokine: a new system of categorizing anti-inflammatory cytokines. PLoS One 7, e33628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collison L. W., Chaturvedi V., Henderson A. L., Giacomin P. R., Guy C., Bankoti J., Finkelstein D., Forbes K., Workman C. J., Brown S. A., Rehg J. E., Jones M. L., Ni H. T., Artis D., et al. (2010) IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 11, 1093–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang R. X., Yu C. R., Dambuza I. M., Mahdi R. M., Dolinska M. B., Sergeev Y. V., Wingfield P. T., Kim S. H., and Egwuagu C. E. (2014) Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 20, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Collison L. W., Delgoffe G. M., Guy C. S., Vignali K. M., Chaturvedi V., Fairweather D., Satoskar A. R., Garcia K. C., Hunter C. A., Drake C. G., Murray P. J., and Vignali D. A. (2012) The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 13, 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stumhofer J. S., Tait E. D., Quinn W. J. 3rd, Hosken N., Spudy B., Goenka R., Fielding C. A., O'Hara A. C., Chen Y., Jones M. L., Saris C. J., Rose-John S., Cua D. J., Jones S. A., Elloso M. M., et al. (2010) A role for IL-27p28 as an antagonist of gp130-mediated signaling. Nat. Immunol. 11, 1119–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crabé S., Guay-Giroux A., Tormo A. J., Duluc D., Lissilaa R., Guilhot F., Mavoungou-Bigouagou U., Lefouili F., Cognet I., Ferlin W., Elson G., Jeannin P., and Gauchat J. F. (2009) The IL-27 p28 subunit binds cytokine-like factor 1 to form a cytokine regulating NK and T cell activities requiring IL-6R for signaling. J. Immunol. 183, 7692–7702 [DOI] [PubMed] [Google Scholar]
- 20. Tormo A. J., Meliani Y., Beaupré L. A., Sharma M., Fritz J. H., Elson G., Crabé S., and Gauchat J. F. (2013) The composite cytokine p28/cytokine-like factor 1 sustains B cell proliferation and promotes plasma cell differentiation. J. Immunol. 191, 1657–1665 [DOI] [PubMed] [Google Scholar]
- 21. Jones L. L., Chaturvedi V., Uyttenhove C., Van Snick J., and Vignali D. A. (2012) Distinct subunit pairing criteria within the heterodimeric IL-12 cytokine family. Mol. Immunol. 51, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jana M., Dasgupta S., Pal U., and Pahan K. (2009) IL-12 p40 homodimer, the so-called biologically inactive molecule, induces nitric oxide synthase in microglia via IL-12Rβ1. Glia 57, 1553–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brahmachari S., and Pahan K. (2008) Role of cytokine p40 family in multiple sclerosis. Minerva Med. 99, 105–118 [PMC free article] [PubMed] [Google Scholar]
- 24. Shimozato O., Ugai S., Chiyo M., Takenobu H., Nagakawa H., Wada A., Kawamura K., Yamamoto H., and Tagawa M. (2006) The secreted form of the p40 subunit of interleukin (IL)-12 inhibits IL-23 functions and abrogates IL-23-mediated antitumour effects. Immunology 117, 22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mondal S., Roy A., and Pahan K. (2009) Functional blocking monoclonal antibodies against IL-12p40 homodimer inhibit adoptive transfer of experimental allergic encephalomyelitis. J. Immunol. 182, 5013–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diamant M., Hansen M. B., Rieneck K., Svenson M., Yasukawa K., and Bendtzen K. (1994) Stimulation of the B9 hybridoma cell line by soluble interleukin-6 receptors. J. Immunol. Methods 173, 229–235 [DOI] [PubMed] [Google Scholar]
- 27. Thibault V., Richards C. D., Botelho F., and Gauldie J. (1997) Antibodies to rat soluble IL-6 receptor stimulate B9 hybridoma cell proliferation. FEBS Lett. 408, 182–186 [DOI] [PubMed] [Google Scholar]
- 28. Lacroix M., Rousseau F., Guilhot F., Malinge P., Magistrelli G., Herren S., Jones S. A., Jones G. W., Scheller J., Lissilaa R., Kosco-Vilbois M., Johnson Z., Buatois V., and Ferlin W. (2015) Novel insights into interleukin 6 (IL-6) cis- and trans-signaling pathways by differentially manipulating the assembly of the IL-6 signaling complex. J. Biol. Chem. 290, 26943–26953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rousseau F., Basset L., Froger J., Dinguirard N., Chevalier S., and Gascan H. (2010) IL-27 structural analysis demonstrates similarities with ciliary neurotrophic factor (CNTF) and leads to the identification of antagonistic variants. Proc. Natl. Acad. Sci. U.S.A. 107, 19420–19425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peters M., Blinn G., Solem F., Fischer M., Meyer zum Büschenfelde K. H., and Rose-John S. (1998) In vivo and in vitro activities of the gp130-stimulating designer cytokine hyper-IL-6. J. Immunol. 161, 3575–3581 [PubMed] [Google Scholar]
- 31. Fischer M., Goldschmitt J., Peschel C., Brakenhoff J. P., Kallen K. J., Wollmer A., Grötzinger J., and Rose-John S. (1997) I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 15, 142–145 [DOI] [PubMed] [Google Scholar]
- 32. Sprecher C. A., Grant F. J., Baumgartner J. W., Presnell S. R., Schrader S. K., Yamagiwa T., Whitmore T. E., O'Hara P. J., and Foster D. F. (1998) Cloning and characterization of a novel class I cytokine receptor. Biochem. Biophys. Res. Commun. 246, 82–90 [DOI] [PubMed] [Google Scholar]
- 33. Romano M., Sironi M., Toniatti C., Polentarutti N., Fruscella P., Ghezzi P., Faggioni R., Luini W., van Hinsbergh V., Sozzani S., Bussolino F., Poli V., Ciliberto G., and Mantovani A. (1997) Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 6, 315–325 [DOI] [PubMed] [Google Scholar]
- 34. Scheller J., Chalaris A., Schmidt-Arras D., and Rose-John S. (2011) The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 1813, 878–888 [DOI] [PubMed] [Google Scholar]
- 35. Rose-John S. (2012) IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 8, 1237–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Garbers C., Spudy B., Aparicio-Siegmund S., Waetzig G. H., Sommer J., Hölscher C., Rose-John S., Grötzinger J., Lorenzen I., and Scheller J. (2013) An interleukin-6 receptor-dependent molecular switch mediates signal transduction of the IL-27 cytokine subunit p28 (IL-30) via a gp130 protein receptor homodimer. J. Biol. Chem. 288, 4346–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Larousserie F., Bardel E., Pflanz S., Arnulf B., Lome-Maldonado C., Hermine O., Brégeaud L., Perennec M., Brousse N., Kastelein R., and Devergne O. (2005) Analysis of interleukin-27 (EBI3/p28) expression in Epstein-Barr virus- and human T-cell leukemia virus type 1-associated lymphomas: heterogeneous expression of EBI3 subunit by tumoral cells. Am. J. Pathol. 166, 1217–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hill M., Thebault P., Segovia M., Louvet C., Bériou G., Tilly G., Merieau E., Anegon I., Chiffoleau E., and Cuturi M. C. (2011) Cell therapy with autologous tolerogenic dendritic cells induces allograft tolerance through interferon-γ and Epstein-Barr virus-induced gene 3. Am. J. Transplant 11, 2036–2045 [DOI] [PubMed] [Google Scholar]
- 39. Maaser C., Egan L. J., Birkenbach M. P., Eckmann L., and Kagnoff M. F. (2004) Expression of Epstein-Barr virus-induced gene 3 and other interleukin-12-related molecules by human intestinal epithelium. Immunology 112, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chung Y., Yamazaki T., Kim B. S., Zhang Y., Reynolds J. M., Martinez G. J., Chang S. H., Lim H., Birkenbach M., and Dong C. (2013) Epstein Barr virus-induced 3 (EBI3) together with IL-12 negatively regulates T helper 17-mediated immunity to Listeria monocytogenes infection. PLoS Pathog. 9, e1003628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu J. Q., Liu Z., Zhang X., Shi Y., Talebian F., Carl J. W. Jr, Yu C., Shi F. D., Whitacre C. C., Trgovcich J., and Bai X. F. (2012) Increased Th17 and regulatory T cell responses in EBV-induced gene 3-deficient mice lead to marginally enhanced development of autoimmune encephalomyelitis. J. Immunol. 188, 3099–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nieuwenhuis E. E., Neurath M. F., Corazza N., Iijima H., Trgovcich J., Wirtz S., Glickman J., Bailey D., Yoshida M., Galle P. R., Kronenberg M., Birkenbach M., and Blumberg R. S. (2002) Disruption of T helper 2-immune responses in Epstein-Barr virus-induced gene 3-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 99, 16951–16956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hou T., Tieu B. C., Ray S., Recinos Iii A., Cui R., Tilton R. G., and Brasier A. R. (2008) Roles of IL-6-gp130 signaling in vascular inflammation. Curr. Cardiol. Rev. 4, 179–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chiesa C., Pacifico L., Natale F., Hofer N., Osborn J. F., and Resch B. (2015) Fetal and early neonatal interleukin-6 response. Cytokine 76, 1–12 [DOI] [PubMed] [Google Scholar]
- 45. Lee S. Y., Buhimschi I. A., Dulay A. T., Ali U. A., Zhao G., Abdel-Razeq S. S., Bahtiyar M. O., Thung S. F., Funai E. F., and Buhimschi C. S. (2011) IL-6 trans-signaling system in intra-amniotic inflammation, preterm birth, and preterm premature rupture of the membranes. J. Immunol. 186, 3226–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garbers C., Hermanns H. M., Schaper F., Müller-Newen G., Grötzinger J., Rose-John S., and Scheller J. (2012) Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev. 23, 85–97 [DOI] [PubMed] [Google Scholar]
- 47. Tormo A. J., Beaupré L. A., Elson G., Crabé S., and Gauchat J. F. (2013) A polyglutamic acid motif confers IL-27 hydroxyapatite and bone-binding properties. J. Immunol. 190, 2931–2937 [DOI] [PubMed] [Google Scholar]
- 48. Cognet I., Guilhot F., Chevalier S., Guay-Giroux A., Bert A., Elson G. C., Gascan H., and Gauchat J. F. (2004) Expression of biologically active mouse ciliary neutrophic factor (CNTF) and soluble CNTFRα in Escherichia coli and characterization of their functional specificities. Eur. Cytokine Netw. 15, 255–262 [PubMed] [Google Scholar]
- 49. Longo P. A., Kavran J. M., Kim M. S., and Leahy D. J. (2013) Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol. 529, 227–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Szymczak A. L., Workman C. J., Wang Y., Vignali K. M., Dilioglou S., Vanin E. F., and Vignali D. A. (2004) Correction of multi-gene deficiency in vivo using a single “self-cleaving” 2A peptide-based retroviral vector. Nat. Biotechnol. 22, 589–594 [DOI] [PubMed] [Google Scholar]
- 51. Zhao S. S., Bukar N., Toulouse J. L., Pelechacz D., Robitaille R., Pelletier J. N., and Masson J. F. (2015) Miniature multi-channel SPR instrument for methotrexate monitoring in clinical samples. Biosens. Bioelectron 64, 664–670 [DOI] [PubMed] [Google Scholar]
- 52. Ratel M., Provencher-Girard A., Zhao S. S., Breault-Turcot J., Labrecque-Carbonneau J., Branca M., Pelletier J. N., Schmitzer A. R., and Masson J. F. (2013) Imidazolium-based ionic liquid surfaces for biosensing. Anal. Chem. 85, 5770–5777 [DOI] [PubMed] [Google Scholar]