

Abstract
Members of the gammaproteobacterial Photorhabdus genus share mutualistic relationships with Heterorhabditis nematodes, and the pairs infect a wide swath of insect larvae. Photorhabdus species produce a family of stilbenes, with two major components being 3,5-dihydroxy-4-isopropyl-trans-stilbene (compound 1) and its stilbene epoxide (compound 2). This family of molecules harbors antimicrobial and immunosuppressive activities, and its pathway is responsible for producing a nematode “food signal” involved in nematode development. However, stilbene epoxidation biosynthesis and its biological roles remain unknown. Here, we identified an orphan protein (Plu2236) from Photorhabdus luminescens that catalyzes stilbene epoxidation. Structural, mutational, and biochemical analyses confirmed the enzyme adopts a fold common to FAD-dependent monooxygenases, contains a tightly bound FAD prosthetic group, and is required for the stereoselective epoxidation of compounds 1 and 2. The epoxidase gene was dispensable in a nematode-infective juvenile recovery assay, indicating the oxidized compound is not required for the food signal. The epoxide exhibited reduced cytotoxicity toward its producer, suggesting this may be a natural route for intracellular detoxification. In an insect infection model, we also observed two stilbene-derived metabolites that were dependent on the epoxidase. NMR, computational, and chemical degradation studies established their structures as new stilbene-l-proline conjugates, prolbenes A (compound 3) and B (compound 4). The prolbenes lacked immunosuppressive and antimicrobial activities compared with their stilbene substrates, suggesting a metabolite attenuation mechanism in the animal model. Collectively, our studies provide a structural view for stereoselective stilbene epoxidation and functionalization in an invertebrate animal infection model and provide new insights into stilbene cellular detoxification.
Keywords: crystal structure, Gram-negative bacteria, natural product, secondary metabolism, symbiosis
Introduction
Photorhabdus luminescens is an entomopathogenic gammaproteobacterium that artfully and simultaneously regulates mutualism and pathogenesis in two different invertebrate hosts. P. luminescens shares a mutualistic relationship with a specific nematode host, Heterorhabditis bacteriophora, and the pair is capable of infecting and killing a wide variety of insect larvae (1–3). A close relative, Photorhabdus asymbiotica, is also capable of causing soft tissue and bacteremic infections in humans, although it is thought that its nematode host dies during human infection (4). A developmentally arrested form of the nematode known as the infective juvenile (IJ)3 harbors Photorhabdus in its gut lumen and hunts for insect larvae in the soil. Upon successful penetration of an insect host, the nematode regurgitates Photorhabdus into the insect circulatory fluid (hemocoel). The bacteria replicate and produce immunosuppressants to commandeer the insect's innate immune system, insecticidals to aid in killing the insect, nematode development signals to promote Heterorhabditis to recover from its IJ developmental stage, and diverse antimicrobials to protect the newly acquired food source. The insects ultimately die by septicemia, whereas the nematodes prosper from feeding on the resulting Photorhabdus biomass. After multiple rounds of development and nutrient consumption, newly formed IJs with their partner bacteria emerge from the cadaver and hunt again.
Because of the multiple host-bacteria and microbe-bacteria interactions that occur during the multilateral symbiosis, the Photorhabdus genus has emerged as a rich source of biologically active small molecules (5, 6). Some of these pathways have been characterized. For example, Photorhabdus produces a variety of anthraquinone and indigoidine pigments (7–9). The genus also encodes isonitrile-functionalized metabolites that serve as potent nanomolar-level inhibitors of insect phenoloxidase, a central enzyme in the insect innate immune system (10, 11). α-Pyrone metabolites play a role in Photorhabdus quorum sensing (12). Lipids functionalized with urea, phurealipids, are produced to inhibit insect juvenile hormone epoxide hydrolase (13). Photorhabdus produces a spectrum of cytotoxic peptides, such as kolossin from a giant non-ribosomal peptide synthetase (14) and the highly potent proteasome inhibitor family, the glidobactins/lumimycins (15–17). Finally, Photorhabdus species are known to produce a variety of stilbene analogs, most of which harbor antibacterial, antifungal, and invertebrate immunosuppressive activities (18–23).
Stilbenes fall within the polyketide-phenylpropanoid class of natural products and are commonly found in plants. However, the first discovery of bacterial stilbenes, such as 3,5-dihydroxy-4-isopropyl-trans-stilbene (compound 1), was from Photorhabdus, representing a rarely reported chemical trait in bacteria (8, 24). Stilbene 1 has been shown to harbor antibiotic activities against a range of bacterial strains, including Staphylococcus aureus, Bacillus subtilis, and Micrococcus luteus (20, 24–27). The metabolite has been shown to have moderate inhibitory activity against phenoloxidase (18). Stilbene 1 also contributes to the bacterial “food signal,” an interkingdom-signaling pathway that contributes to the developmental recovery of the IJ stage of the nematode (21). More recently, a downstream stilbene metabolite derived from the epoxidation of 1, 2-isopropyl-5-(3-phenyl-oxiranyl)-benzene-1,3-diol (2), has been elucidated from greater waxmoth larvae (Galleria mellonella) infected by the Heterorhabditis-Photorhabdus pair (19). The metabolite was confirmed by synthesis, but its absolute configuration remained undefined. This stilbene epoxide also exhibited significant antimicrobial activities against a panel of microbes (19).
In this study, we characterized Plu2236, an orphan FAD-dependent monooxygenase (FMO), by X-ray crystallography, which revealed an overall fold common to FMOs with a tightly bound FAD prosthetic group. Through bacterial genetics (Δplu2236) and in vitro biochemical reconstitution of Plu2236, we show that this enzyme is a stilbene epoxidase and is required to stereoselectively convert 1 to 2. By defining this enzymatic stilbene epoxidation process, we were able to further examine the developmental and biological effects of the P. luminescens stilbene epoxide in two animal models, nematode IJ recovery (H. bacteriophora) and insect pathogenicity (G. mellonella). In the invertebrate animal infection model, we observed two new stilbene-derived metabolites that were dependent on the epoxidase. Characterization of these metabolites led to new stilbene-l-proline conjugates, prolbenes A (3) and B (4), and the completion of the absolute configurational assignment of stilbene epoxide (2).
l-Proline is one of the dominant amino acids in the insect circulatory fluid and represents a nutrient signal in P. luminescens that stimulates the production of select secondary metabolic pathways in the bacterium (10). The chemical features of the circulatory fluid and the electrophilicity of the stilbene epoxide promote l-proline functionalization and, correspondingly, attenuation of the stilbene epoxide antimicrobial activities. Finally, we show that stilbene epoxidation reduces bacterial host cytotoxicity and likely contributes to an enzymatic stilbene detoxification strategy in P. luminescens.
Results
X-ray crystallographic characterization of an orphan FMO
The crystal structure of an orphan FMO from P. luminescens TT01 was determined in the trigonal P3221 space group with one molecule in the asymmetric unit (Fig. 1A and Table 1). Analysis of the crystal structure using PISA (28) does not suggest any stable multimeric assembly of the P. luminescens FMO (PlFMO). The structure of the FMO reveals a typical two domain fold (29, 30) with the smaller NADP-binding domain situated on top of a larger FAD-binding domain with a long helix (residues 354–385) near the C terminus stabilizing their interactions (Fig. 1A). The NADP-binding domain (residues 76–89 and 178–300) contains a seven-stranded β-sheet packed against three helices. The larger FAD-binding domain consists of a four-stranded parallel β-sheet packed between a three-stranded antiparallel β-sheet and a group of six α-helices. A Rossmann fold, common to the nucleotide-binding proteins, made up of the β1-α1-β2 motif containing the characteristic GXGXXG sequence is present at the N terminus of the FAD-binding domain. In PlFMO, this sequence corresponds to GAGIGG between residues 7 and 12 (Fig. 1C; supplemental Fig. S1). The tightly bound prosthetic group FAD is well defined in the electron density maps (Fig. 1B) and makes extensive interactions with PlFMO, including 18 hydrogen bonds (Fig. 1C). A search for structural homologs of PlFMO identified Bacteroides thetaiotaomicron TetX (BtTetX) among others with a z-score of 35.6 (30). A structural superposition of BtTetX onto PlFMO reveals a well conserved fold (Fig. 1D; supplemental Fig. S1) with a root mean-square-deviation of 3.0 Å over 345 Cα atomic pairs despite low sequence identity of 23%. Also, the FAD of PlFMO is bound in the IN conformation, where isoalloxazine and adenine rings are distal to each other, as observed in the case of BtTetX (30).
Figure 1.

X-ray crystallographic structure of FAD-dependent epoxidase. A, schematic representation of overall fold of PlFMO with the FAD and NADPH domains shown in salmon and cyan, respectively, along with the long helix α11, stabilizing the FAD and NADPH domains, shown in red. The β-strands, α-helices, and 310-helices are marked sequentially in black, blue, and pink numbers, respectively. B, mFo − DFc omit-map shown at a contour level of 2.5σ along with the final refined model of FAD. C, LIGPLOT representation, created using the PDBsum (53) server, showing both hydrogen bonding and hydrophobic interactions between the FAD and PlFMO. D, superposition of PlFMO (PDB code 4HB9) and BtTetX (PDB code 2XDO) structures using the DALI server (54). A schematic representation of PlFMO is colored as in D and that of BtTetX is colored in gray. All images were created using the PyMOL molecular graphics system, version 1.5 Schrödinger, LLC.
Table 1.
Crystallographic statistics
| Data collection | PlFMO |
| PDB code | 4HB9 |
| Space group | P3221 |
| Unit cell dimensions (Å) | a = b = 71.008 c = 157.910 |
| Matthew's coefficient (Å3/Da) | 2.52 |
| Solvent content (%) | 51 |
| Wavelength (Å) | 0.9790 |
| Resolution (Å) | 50.00–1.93 (1.96–1.93)a |
| No. of unique reflections | 35,439 (1728) |
| Completeness (%) | 100.00 (99.3) |
| Rsymm (%) | 10.2 (77.2) |
| Multiplicity | 19.2 (18.3) |
| 〈I/σ(I)〉 | 30.5 (5.4) |
| Refinement | |
| No. of reflections | 35,385 |
| No. of reflections in test set | 1765 |
| R-factor (%) | 17.5 |
| Rfree (%) | 21.8 |
| Root mean square deviations from ideal values | |
| Bond length (Å) | 0.010 |
| Bond angles (°) | 1.444 |
| Ramachandran plot | |
| MolProbity (55) residues in | |
| Favored region (%) | 97.4 |
| Allowed region (%) | 100.0 |
a Values in parentheses correspond to the highest-resolution shell.
Deletion of plu2236 destroys bacterial stilbene epoxidation in liquid cultures
The protein sequence and X-ray structure of Plu2236 support an undefined monooxygenation reaction. To determine the specific function(s) of this orphan FMO, we cleanly deleted plu2236 in wild type P. luminescens TT01 using allelic-exchange mutagenesis. LC/MS comparisons of organic extracts from liquid cultures of wild type and Δplu2236 strains, along with organic extracts from insect larvae infected by these bacteria, revealed that the production of a major peak (m/z [M + H]+ = 271) was completely abrogated in the mutant strain relative to the wild type strain (Fig. 2, A and B). The UV-visible, mass spectral, and chromatographic properties of this peak were consistent with stilbene epoxide (2) (Fig. 2C). The planar structure of 2 from P. luminescens was first reported as an antimicrobial metabolite from an insect model infected by the nematode carrying P. luminescens (19). We isolated this peak from a larger-scale wild type culture, and NMR analysis confirmed the planar structural assignment (supplemental Fig. S2). We also isolated and confirmed the structure of its precursor stilbene (1, m/z [M + H]+ = 255) by NMR from the wild type culture (Fig. 2C; supplemental Fig. S3) (23). Stilbene 1 represents the observed epoxidase substrate, and as expected, it accumulates in the plu2236 mutant strain (Fig. 2A). These experiments demonstrate that Plu2236 is an FAD-dependent stilbene epoxidase responsible for converting 1 to 2. The stilbene biosynthetic gene “cluster” in P. luminescens is genomically fragmented, with mini-gene clusters localized to at least four distinct regions of the genome (21). The stilbene epoxidase gene (plu2236) is adjacent to stlA (plu2234), a phenylalanine ammonia lyase that converts phenylalanine to cinnamic acid, initiating phenylpropanoid/stilbene biosynthesis.
Figure 2.

HPLC/MS analysis for production of stilbene 1 and stilbene epoxide 2. A, in P. luminescens culture broth (top, P. luminescens WT; bottom, Δplu2236). B, in G. mellonella insect infected with P. luminescens (top, P. luminescens WT-infected G. mellonella; bottom, Δplu2236-infected G. mellonella). The 10 mg of crude ethyl acetate-soluble materials were dissolved in 200 μl of methanol, and 10 μl of samples were subsequently injected for HPLC/MS analysis. The m/z 255 and m/z 271 correspond to protonated molecular ions of 1 and 2 in the positive ion mode, respectively. Peak intensity of the metabolites was determined by extracted ion counts in the same scale of y axis. Shown in C are the chemical structures of the P. luminescens metabolites, stilbene (1), and stilbene epoxide (2), respectively.
In vitro reconstitution of Plu2236
To support the function of PlFMO in a cell-free system, we overexpressed and purified N-terminal His6-tagged Plu2236 (supplemental Fig. S4). In vitro protein biochemical experiments were conducted using 200 μm isolated stilbene substrate 1, 100 μm NADH or NADPH, and 5 μm of the FAD-containing enzyme (Fig. 3A). After 30 min, the reaction mixtures were extracted with an equal volume of ethyl acetate; the organic fraction was dried, and the products were analyzed by high-resolution ESI-QTOF-MS. Extracted ion chromatogram analyses of the in vitro reactions showed conversion of stilbene substrate 1 to stilbene epoxide 2 in the presence of supplemented NADPH or NADH (Fig. 3, B and C). NADPH was preferred over NADH. These in vitro protein biochemical experiments demonstrate that PlFMO is necessary and sufficient for the stereoselective epoxidation reaction.
Figure 3.
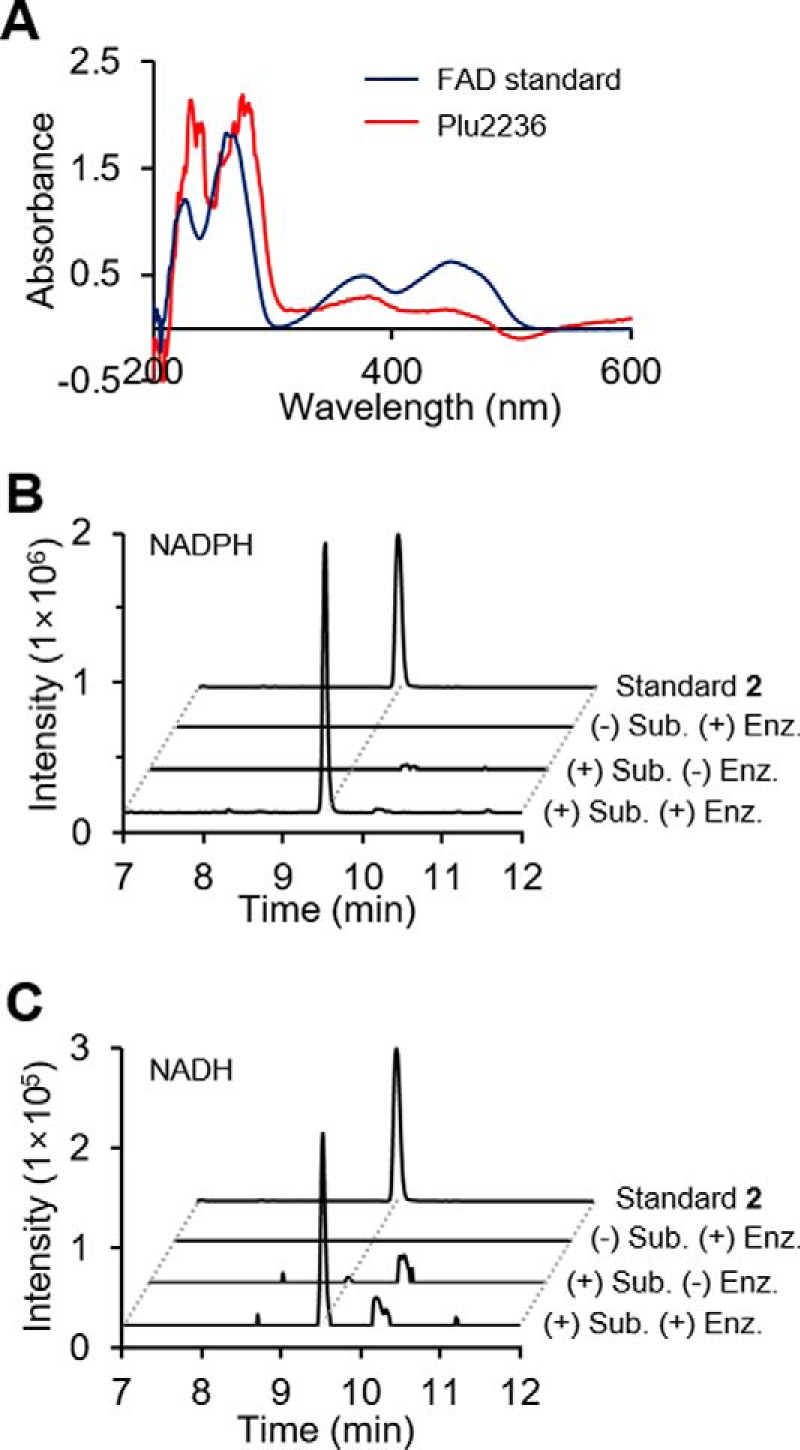
In vitro reconstitution of PlFMO. A, UV-visible spectra of purified PlFMO (red) and a free FAD standard (blue). B and C, in vitro production of the stilbene epoxide (2) in the presence of NADPH (B) or NADH (C), respectively. Intensity of the peak shown in B and C was determined by ion extraction and is shown on the same scale, with m/z 271.1334 corresponding to stilbene epoxide (2) within a 10 ppm window, and intensity of standard 2 indicates 2 × 107 counts. All experiments were prepared in triplicate. HPLC/MS data were analyzed using a gradient from 40 to 100% aqueous acetonitrile containing 0.1% formic acid over 15 min with a 0.7 ml/min solvent flow rate.
Discovery of new epoxidase-dependent stilbenes in an invertebrate animal infection model
To assess stilbene production in the animal model, we first analyzed organic extracts from wild type P. luminescens TT01 in a waxmoth larval (G. mellonella) infection model compared with non-infected control larvae. Larvae were frozen, ground, and extracted over a time course (6, 12, 24, 36, and 48 h) using several organic solvents, including n-hexane, dichloromethane, ethyl acetate, and n-butanol. C18-reversed-phase LC/MS analysis demonstrated that stilbene (1, tR = 23.5 min) and stilbene epoxide (2, tR = 22.5 min) could readily be detected in the ethyl acetate extracts of Photorhabdus-infected larvae (supplemental Fig. S5). The stilbenes were detectable after 24 h and were maximally observed at 48 h in the time-dependent analysis (Fig. 4B, inset). During the course of this analysis, we observed the production of two additional distinct peaks (tR = 14.2 and 15.3 min) that were dependent on the stilbene epoxidase and shared similar production characteristics as the stilbenes (Fig. 4). However, these additional peaks exhibited similar UV-visible spectra (λmax 210 nm) that were distinct from the stilbenes and consistent with strong phenyl chromophores (supplemental Fig. S6). High-resolution ESI-QTOF-MS data analysis suggested that these molecules were novel stilbene-derived isomers with the chemical formula C22H27NO5 (supplemental Fig. S7).
Figure 4.
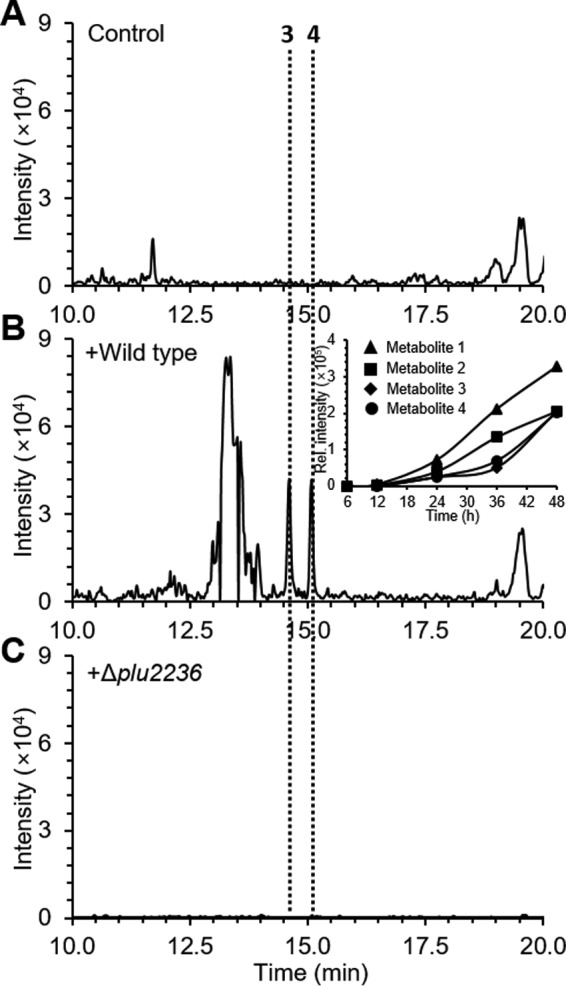
HPLC/MS was used for the detection of prolbene metabolites 3 and 4 from insect model G. mellonella larvae infected with P. luminescens WT (B) and Δplu2236 (C). Control indicates non-infected insect larvae (A). Peaks were extracted with m/z 385 ion signal corresponding to positively charged ion of the metabolites 3 and 4. Inset in B shows time-dependent production behavior of metabolites 1-4. The relative intensity of the metabolites in the inset was determined by integration values of peaks extracted with corresponding m/z.
Isolation and characterization of new prolbenes A and B
To isolate the new stilbene-derived metabolites detected in the invertebrate animal infection model, we cultivated wild type P. luminescens in a hemolymph mimetic medium (HMM) that is based on the concentrations of the 20 proteinogenic amino acids present in G. mellonella circulatory fluid (10, 31, 32). Ethyl acetate extracts from initial 5-ml cultures were monitored by LC/MS, and the unknown target peaks were successfully produced under the P. luminescens culture conditions (supplemental Fig. S8). A subsequent 16 liters of HMM culture was initiated, and after 48 h of aerobic cultivation at 30 °C, the cleared supernatant was extracted twice with equal portions of ethyl acetate (32 liters total), yielding 2.3 g of crude material. Multiple reversed-phase HPLC fractionations of the organic materials allowed us to homogeneously isolate the two molecules, 3 (tR = 18.9 min, 1.5 mg) and 4 (tR = 20.5 min, 1.8 mg). Their planar chemical structures were proposed based on the interpretation of their 1D and 2D NMR (gCOSY, gHSQC, and gHMBC) spectra (supplemental Figs. S9–S16).
Compound (3) was isolated as an amorphous solid with the molecular formula C22H27NO5 based on its HR-ESI-QTOF-MS spectrum (observed [M + H]+ at m/z 386.1964; calculated 386.1967) (Fig. 5C). Analysis of the 1H NMR spectral data (Table 2; supplemental Fig. S9) suggested the presence of two separate spin systems, including an isopropyl stilbene scaffold and a proline group, which was consistent with the COSY NMR spectral data (Fig. 5A; supplemental Fig. S10). COSY cross-peaks from H-10/H-14 (2H, δH 7.59, m) to H-12 (1H, δH 7.16, t, J = 7.2 Hz), and from a methine H-15 (1H, δH 4.23, m) to the two methyl groups H-16 and H-17 indicated the presence of a mono-substituted benzene ring and an isopropyl group, respectively. The 1H NMR spectrum coupled with HSQC spectrum (supplemental Fig. S11) also showed the resonance of a singlet peak (2H, δH 6.93, m) in the double bond region, and key HMBC correlations sharing two hydroxylated quaternary carbons C-1/C-5 (δC 157.5) and an upfield-shifted C-2 resonance (δC 106.6) from H-2/H-4 and H-15 allowed us to construct a 2-isopropyl-1,3-diol-type of phenyl group (supplemental Fig. S12). The connectivity of the two separated rings of compound 3 was established by combinatorial analysis of COSY and HMBC NMR data (Fig. 5A). The COSY NMR spectrum showed a three-bond correlation between two methines H-7 (1H, δH 5.62, d, J = 3.7 Hz) and H-8 (1H, δH 4.26, brs), suggesting the presence of ethane-1,2-diol-like single bond. Establishment of the 1,2-diphenylethane scaffold was achieved from HMBC correlations from H-7 to C-2/C-4 and C-9 (δC 138.7) and from H-8 to C-10/C-14 and C-3 (δC 142.1). The proline group of 3 was constructed based on analysis of the COSY and HMBC NMR correlations (Fig. 5A). The 1H NMR showed the resonance of an α-proton H-2′ (1H, δH 3.92, dd, J = 9.5, 3.8 Hz) and sequential COSY cross-peaks from H-2′ to a nitrogen-bearing methylene H-5′ (2H, δH 3.19, t, J = 7.1 Hz, δH 2.59, dd, J = 15.6, 8.4 Hz) established the pyrrolidine scaffold of proline. Carboxylic acid group attachment at C-2′ (δC 65.3) was permitted by HMBC correlations from H-2′ and H-3′ (2H, δH 2.20, m, δH 2.06, m) to C-1′ (δC 178.6). Finally, the full planar structure of 3 was unambiguously constructed by the key HMBC correlations from H-2′ and H-5′ in the proline group to C-8 (δC 75.4), indicating that the two partial structures were connected via a C–N bond (Fig. 5A). As such, the chemical structure of 3 was assigned as (2-(3,5-dihydroxy-4-isopropylphenyl)-2-hydroxy-1-phenylethyl)proline.
Figure 5.
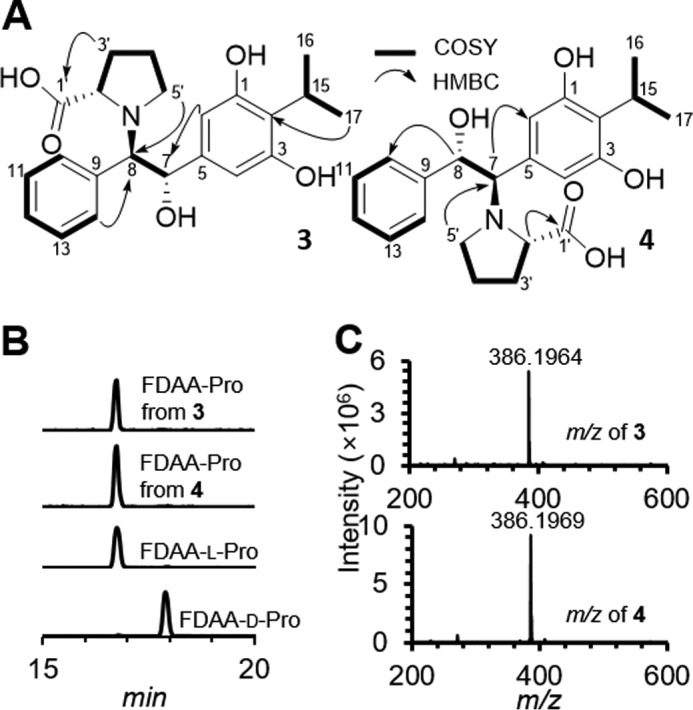
Structural characterization of prolbene metabolites 3 and 4. Shown in A are key 2D NMR correlations deduced from the interpretation of the 1H-1H COSY (bold) and 1H-13C HMBC (arrow) NMR spectra of compounds 3 and 4. B, Marfey's analysis: FDAA derivatives of proline were analyzed by HPLC/MS with reversed-phase C18 column, and HPLC/MS was established by extracted ion method with corresponding m/z 368 of derivatives. C, observed HR-ESI-QTOFMS spectra of 3 (top) and 4 (bottom).
Table 2.
1H and 13C NMR spectral data of compounds 3 and 4 in pyridine-d5 (δH 8.73, δC 149.9)
The chemical structures of the compounds 3 and 4 are shown in Figs. 5A and 7.
| Position | Compound 3 |
Compound 4 |
||||
|---|---|---|---|---|---|---|
| δH mult (J, Hz)a | δCb | δH mult (J, Hz) a | δCb | |||
| 1 | 157.5 | C | 157.5 | C | ||
| 2 | 6.93 s | 106.6 | CH | 7.12 s | 110.8 | CH |
| 3 | 142.1 | C | 133.8 | C | ||
| 4 | 6.93 s | 106.6 | CH | 7.12 s | 110.8 | CH |
| 5 | 157.5 | C | 157.5 | C | ||
| 6 | 120.0 | C | 121.4 | C | ||
| 7 | 5.62d (3.7) | 74.7 | CH | 4.52d (4.2) | 73.8 | CH |
| 8 | 4.26 brs | 75.4 | CH | 5.77d (4.2) | 74.3 | CH |
| 9 | 138.7 | C | 143.3 | C | ||
| 10 | 7.59 mc | 131.2 | CH | 7.55d (7.6) | 127.5 | CH |
| 11 | 7.21 mc | 127.5 | CH | 7.20t (7.6) | 128.2 | CH |
| 12 | 7.16t (7.2) | 127.3 | CH | 7.11 mc | 127.4 | CH |
| 13 | 7.21 mc | 127.5 | CH | 7.20t (7.6) | 128.2 | CH |
| 14 | 7.59 mc | 131.2 | CH | 7.55d (7.6) | 127.5 | CH |
| 15 | 4.23 mc | 25.2 | CH | 4.25 dt (14.2, 7.1) | 25.3 | CH |
| 16 | 1.74d (7.0) | 21.6 | CH3 | 1.75d (7.0) | 21.5 | CH3 |
| 17 | 1.73d (7.0) | 21.6 | CH3 | 1.74d (7.0) | 21.5 | CH3 |
| 1′ | 178.6 | C | 175.9 | C | ||
| 2′ | 3.92 dd (9.5, 3.8) | 65.3 | CH | 4.29 dd (9.5, 4.6) | 65.5 | CH |
| 3′ | 2.20 m | 30.8 | CH2 | 2.23 ddd (16.3, 8.1, 3.9) | 29.9 | CH2 |
| 3′ | 2.06 m | 2.11 m | ||||
| 4′ | 1.72 m | 24.1 | CH2 | 1.74 mc | 23.9 | CH2 |
| 4′ | 1.50 | 1.55 m | ||||
| 5′ | 3.19t (7.1) | 52.7 | CH2 | 3.52 m | 52.4 | CH2 |
| 5′ | 2.59 dd (15.6, 8.4) | 3.01 dd (16.8, 9.6) | ||||
a These were assigned by 1H NMR at 600 MHz.
b These were assigned by HSQC and HMBC NMR.
c The coupling constants cannot be assigned because of overlapping signals.
Compound 4 was also isolated as an amorphous solid. Its molecular formula was determined to be C22H27NO5 (observed [M + H]+ m/z 386.1969, calculated 386.1967) based on HR-ESI-QTOF-MS data (Fig. 5C), and its planar structure was deduced by 1D and 2D NMR experiments (supplemental Figs. S13–S16). Briefly, similar to 3, the 1H NMR spectrum of 4 indicated the presence of a stilbene scaffold and a proline group. However, the HMBC correlation from H-2′ (1H, δH 4.29, dd, J = 9.5, 4.6 Hz) and H-5′ (2H, δH 3.52, m, δH 3.01, dd, J = 16.8, 9.6 Hz) to C-7 (δC 73.8) permitted the substitution of the proline group at C-7 in the stilbene scaffold (Fig. 5A). Therefore, the structure of 4 was established as (1-(3,5-dihydroxy-4-isopropylphenyl)-2-hydroxy-2-phenylethyl)proline.
The absolute configurations of the prolbenes were assigned based on chemical degradation, ECD, and computational experiments (Figs. 5B and 6, A and B). Prolbenes A (3) and B (4) were both determined to harbor l-proline moieties based on acid hydrolysis, amino acid derivatization, and LC/MS analyses compared with l- and d-proline standards (Marfey's analyses (Fig. 5B)). Given the l-proline moieties, establishment of the remaining absolute configurations at C-7 and C-8 in compounds 3 and 4 was first supported by ECD spectroscopy and quantum-chemical ECD calculations (Fig. 6A). Prior to the ECD simulations, advanced conformational searches of the two possible diastereomer sets ((A: 7S, 8R) and (B: 7R, 8S) for 3; and (A: 7R, 8S) and (B: 7S, 8R) for 4) yielded 15 and 22; and 16 and 21 conformers within 10 kJ/mol of a relative energy level in the MMFF94 force field (supplemental Tables S1 and S2), respectively. The geometries of the conformers were further optimized at the B3LYP/6–31+G(d) level of theory (supplemental Tables S3–S6). The resulting conformers were subjected to ECD calculations utilizing the identical theory level, and the simulated ECD spectra were Boltzmann-averaged based on the Gibbs free energy of each conformer. The experimental ECD spectra of both 3 and 4 showed positive Cotton effects at 220 nm and broad negative Cotton effects at 290 nm (Fig. 6A). Comparisons of the experimental spectra with the computed ECD spectra were informative; however, they were not identical, presumably due to the presence of weak chromophores in 3 and 4. Thus, we also employed the gauge-including atomic orbital (GIAO) NMR chemical shift calculations supported by DP4 analysis (33). These conformers were calculated at the B3LYP/6–31G(d,p) level of theory, and the chemical shifts were calculated and Boltzmann-averaged at 298.15 K from the GIAO magnetic shielding tensor values (supplemental Tables S1 and S2) (33). Comparison of experimental NMR data of 3 and 4 with computed NMR chemical shift values of the diastereomer sets A and B (supplemental Table S7), followed by DP4 analysis, illuminated that diastereomer A (7S, 8R) of 3 and (7R, 8S) of 4 showed DP4 probabilities of 95.1 and 100% given that both 1H and 13C NMR chemical shift values were considered (Fig. 6B and supplemental Figs. S17 and S18). These experiments were consistent with the ECD spectra. Thus, the absolute configurations of C-7 and C-8 in 3 and 4 were assigned as 7S, 8R and 7R, 8S, respectively. Correspondingly, these experiments allowed us to assign the absolute configurations of C-7 and C-8 in stilbene epoxide 2 as 7S, 8S, respectively, demonstrating Plu2236's enzymatic stereoselectivity (Figs. 6B and 7).
Figure 6.

Absolute configuration determination of prolbene metabolites 3 and 4. A, calculated ECD spectra of 3 (top) and 4 (bottom) in the gas phase at the B3LYP/6–31+G(d) level and its experimental ECD in methanol (black). B, DP4 probabilities obtained from the two possible diastereomers A (left) and B (right) of prolbenes 3 and 4. NMR chemical shift calculations were performed using the Gaussian 09 package (Gaussian Inc.) in B3LYP/6–31G(d,p) theory level.
Figure 7.

Proposed biosynthesis of stilbene epoxidation and l-proline functionalization in P. luminescens.
Biological activity studies associated with stilbene epoxidation and functionalization
Importantly, stilbenes from Photorhabdus strains have been associated with multiple activities, including antibacterial, antifungal, insect innate immunosuppressive (phenoloxidase inhibition), and nematode IJ recovery activities (18, 19, 21, 25). We first tested the minimal inhibitory concentrations (MICs) of stilbenes 1-4 for antibacterial activities against two bacteria, Gram-positive B. subtilis and Gram-negative Escherichia coli. Stilbene 1 showed strong inhibition against B. subtilis and E. coli, with MIC values of 3.125 and 12.5 μg/ml, respectively (supplemental Table S8). Similarly, stilbene epoxide 2 exhibited significant antibacterial activities (MIC values; 12.5 μg/ml for B. subtilis and 25 μg/ml for E. coli). Prolbenes A (3) and B (4) were inactive against the bacteria at a maximum concentration of 200 μg/ml (supplemental Table S8). We then tested their antifungal activities against a model yeast strain. Stilbenes 1 and 2 showed MIC values of 12.5 and 50 μg/ml, respectively, against Saccharomyces cerevisiae, and prolbenes 3 and 4 were inactive (up to 200 μg/ml) (supplemental Table S8). We also tested their in vitro immunosuppressive activities against a model tyrosinase. Although stilbenes had been reported to have micromolar-level activities against an insect phenoloxidase (18), stilbenes 1 and 2 revealed only very weak tyrosinase inhibitory activities (8.6 and 4.5% inhibition, respectively) at the concentration of 100 μm, whereas 3 and 4 were inactive at the same dose (supplemental Fig. S19). Next, we examined the plu2236 mutant strain in the G. mellonella infection model compared with wild type P. luminescens. Both strains were fully virulent killing all animals within 48 h, indicating that the epoxide is not required for pathogenicity. Because of functional redundancy of virulence factors, we also tested the purified stilbenes 1 and 2 in the same infection model. Both stilbenes failed to result in insect death within 5 days, indicating minimal insect cytotoxicity. Previously, stilbene 1 was shown to chemically complement a stilbene-negative mutant in IJ recovery, and it was unclear whether epoxidation was required for this food signal (21). Consequently, we tested the role of the epoxidase (Δplu2236) in an IJ recovery assay relative to wild type. The recovery rates of IJs on two independently prepared plu2236 mutant strains were 65 and 76%, which were comparable with the wild type control (73%) (supplemental Fig. S20). These nematode development assays demonstrate that plu2236 is dispensable for the Heterorhabditis food signal. Consistent with this notion, we found that the dual insect-human pathogen P. asymbiotica, which also shares a mutualistic relationship with Heterorhabditis nematodes (34), produced stilbene 1 as expected, but stilbene epoxide 2 was not detected under identical HMM-based cultivation conditions (supplemental Fig. S21). We finally tested the growth inhibitory effects of 1 and 2 against the producing host P. luminescens. We examined the wild type strain, a knock-out strain of the LysR family transcriptional repressor hexA, which is known to regulate stilbene production (22, 35), and the new stilbene epoxidase plu2236 mutant. Minimal inhibitory concentration results revealed that both metabolites 1 and 2 effectively inhibited the growth of their producing strains at high supplemental metabolite concentrations (up to 100 μg/ml), whereas stilbene epoxide 2 was much less toxic compared with stilbene 1 (Fig. 8). Collectively, these studies support that enzymatic stilbene epoxidation maintains virulence-associated activities (i.e. antibiosis) and contributes to bacterial stilbene detoxification during the multipartite lifestyle of P. luminescens.
Figure 8.

Growth inhibitory comparison of stilbene 1 and stilbene epoxide 2. Producing strains P. luminescens wild type (A), Δplu2236 (B), and ΔhexA (C) were used for bacterial host cytotoxicity measurements of the stilbene metabolites 1 and 2. Optical density (A600) was measured and collected over three different time points (12, 24, and 48 h in triplicate).
Discussion
Although stilbenes represent common phenylpropanoid metabolites in plants, their production in bacteria is only very rarely reported. Distinct from the type III polyketide synthase (PKS) biosynthesis of stilbenes in plants (36), Photorhabdus strains convergently evolved a type II PKS route to the stilbene scaffolds (37). Interestingly, evidence suggests that these bacterial molecules proceed through cyclohexanedione intermediates (37), a family of molecules linked to plant pheromone signaling (38). The Photorhabdus stilbenes are known to harbor extraordinarily diverse bioactivities, including antibacterial, antifungal, insect immunosuppressive, and nematode developmental activities (18, 20, 21). Despite the important ecological functions of Photorhabdus stilbenes in their multipartite life cycle, the mechanism of bacterial stilbene epoxidation and the biological roles of the resulting stilbene epoxides have remained unknown. In this study, X-ray crystallography was used to characterize the structure of the orphan FMO Plu2236. Genetic inactivation of plu2236, comparative metabolite analysis of the major stilbene metabolites in wild type cultures (1 and 2), and in vitro reconstitution of the enzyme demonstrated that this FMO is a stilbene epoxidase. Stereochemical determination of stilbene epoxide 2, which was genetically dependent on PlFMO, and its l-proline-functionalized products (3 and 4) observed in an invertebrate animal infection model allowed us to define the stereoselectivity of the epoxidase.
Formation of prolbenes A (3) and B (4) in the in vivo waxmoth infection model demonstrated a route for stilbene functionalization in insects. The epoxide represents an electrophilic moiety, and consistent with its electrophilicity, previous studies have identified hydrolytically ring-opened products in bacterial cultures (22). It is likely that the epoxide reacts with other small molecule or macromolecule nucleophiles. Insect circulatory fluid (hemolymph) is rich in free amino acids with individual amino acids detected in the millimolar level (10). The three most abundant amino acids in the waxmoth G. mellonella hemolymph are l-glutamine (100 mm), l-proline (73 mm), and l-alanine (30 mm) (10). We attribute in vivo prolbene production to the electrophilicity of the stilbene epoxide, the high concentration of l-proline in the hemolymph, and a higher nucleophilicity of proline's secondary amine relative to the primary amines in the other proteinogenic amino acids. l-Proline functionalization occurs spontaneously in cell-free medium (supplemental Fig. S22). Although the stilbenes harbored antimicrobial and immunosuppressive activities, the prolbenes lacked these activities supporting a host-mediated stilbene epoxide attenuation mechanism. It is currently unclear whether the prolbenes have other functional roles.
Through an invertible genetic promoter switch, P. luminescens undergoes stochastic phenotypic variation between two major forms, the primary form (P-form) and a minority variant form (M-form or small colony variant form) (39). The P-form is associated with producing the known Photorhabdus secondary metabolites, including the stilbenes, and promotes nematode development (3). The stilbenes are regulated by a LysR family transcriptional repressor HexA, and derepression (or deletion) of this regulator leads to stilbene up-regulation (22, 35). The M-form is biased toward dormancy and produces mad fimbriae, which have been associated with colonizing specific cells in the nematode host. This biological feature of the M-form is thought to contribute to mutualistic colonization of the host nematode (39). Stilbenes, which are produced primarily by the P-form, are required for nematode IJ recovery (3). Much like the Caenorhabditis elegans dauer larvae, Heterorhabditis IJs are in a developmentally arrested stage (state of diapause) that can survive harsh conditions for extended periods of time, allowing them to effectively hunt for insect larvae in the soil. Heterorhabditis nematodes consume Photorhabdus biomass during a successful insect infection, and the bacterial stilbenes contribute to the food signal for recovery. In a stlA deletion mutant that shuts down cinnamic acid biosynthesis, the precursor to stilbenes, IJs are disrupted in their ability to recover. The stlA mutant can be chemically complemented with cinnamic acid or downstream stilbene 1. However, stilbene 1 cannot complement recovery in the absence of bacteria (3, 21). Consequently, stilbene 1 either directly serves as part of the food signal and/or the food signal is downstream of stilbene 1. To test whether downstream stilbene epoxide contributes to recovery, we examined the stilbene epoxidase mutant (Δplu2236) in an IJ recovery assay. Although the epoxidase mutant does not produce detectable levels of the epoxide in liquid cultures or in the insect infection model, the epoxidase gene is dispensable for nematode IJ recovery. Given that we were also not able to observe stilbene epoxide production in the closely related dual insect-human pathogen P. asymbiotica, which also shares a mutualistic relationship with Heterorhabditis nematodes (34), epoxide formation is likely more functionally relevant during insect pathogenesis and bacterial host detoxification.
In Photorhabdus, the stilbenes along with isocyanide metabolites are known to harbor insect immunosuppressive activities (phenoloxidase inhibition) (11, 18). The stilbenes are also known to participate in antibiosis, a phenotype that likely provides Photorhabdus with a competitive advantage in insect colonization. To examine the in vivo role of stilbene epoxidation in virulence, we examined the epoxidase mutant (Δplu2236) relative to wild type P. luminescens in the G. mellonella infection model. Whereas the stilbene epoxide maintained in vitro antimicrobial activities, the stilbene epoxidase gene was dispensable in the in vivo G. mellonella infection model relative to wild type P. luminescens, suggesting that stilbene epoxide 2 only in part contributes to virulence-associated activities (i.e. antibiosis). Importantly, the epoxidation route leads to less toxic molecules and likely provides a strategy for P. luminescens to detoxify accumulating levels of the major stilbene.
Intriguingly, the epoxide functional group represents a crucial structural feature of insect “juvenile hormones.” Formation and degradation of this structural feature widely regulate various aspects across insect developmental biology. Indeed, even juvenile hormone-inspired compounds have been developed into insecticidal agents (40). Our study indicates that P. luminescens has similarly adopted an enzymatic epoxidation strategy as part of its armory against insects. Although the epoxide exhibits reduced bacterial host cytotoxicity, it maintains virulence-associated activities, such as the described antimicrobial activities against other bacteria and yeast (and moderate tyrosinase inhibitory activity). Additionally, the insects use l-proline as a spontaneous chemical defense against the epoxide insults. In sum, our study expands our view on the chemical arms race between pathogen and host, specifically associated with stilbene chemical defenses. With the highly effective insect killing by Photorhabdus, the bacteria have clearly won the war.
Experimental procedures
General experimental procedures
ECD spectra were measured on a J-810 polarimeter (Jasco Inc., Easton, MD). 1H and gradient-selected two-dimensional COSY, HSQC, and HMBC NMR spectra were obtained on a 600 MHz NMR spectrometer with a cold probe (Agilent, Santa Clara, CA). Chemical shifts were recorded using the solvent residual peak signals of pyridine-d5 (δH 8.73 and δC 149.9) or methanol-d4 (δH 3.30) as references. Flash column chromatography was carried out on Sep-Pak® Vac 35cc (10 g) C18 cartridge (Waters). HPLC analysis was performed on an Agilent 1260 Infinity system with a Luna C18(2) (100 Å) 5 μm (4.6 × 150 mm) column (Phenomenex, Torrance, CA) and a PDA detector. An Agilent Prepstar HPLC system with an Agilent Polaris C18-A 5-μm (21.2 × 250 mm) column, a Phenomenex Luna C18(2) or C8(2) (100 Å) 10-μm (10.0 × 250 mm) column, and an Agilent phenyl-hexyl 5-μm (9.4 × 250 mm) column were used for the fractionation, separation, and purification of metabolites. Low-resolution electrospray ionization-mass spectrometry (ESI-MS) data were collected on an Agilent 6120 Quadrupole LC/MS system equipped with a Phenomenex Kinetex C18 (100 Å) 5-μm (4.6 × 250 mm) column. High-resolution (HR) ESI-MS data were obtained using an Agilent iFunnel 6550 QTOF MS instrument fitted with an electrospray ionization (ESI) source coupled to an Agilent 1290 Infinity HPLC system.
X-ray crystallographic characterization of the orphan FMO
A synthetic DNA fragment encoding the P. luminescens FMO (plu2236 in strain TT01) was cloned into pSGC-His (also known as pNIC28-Bas4) (41) vector using 5′-tacttccaatccatgcatgtcggcattattggggcg-3′ and 5′-tatccacctttactgttaacgtcggtgttgatgactttgag-3′ as forward and reverse primers, respectively, via ligation-independent cloning (LIC sites are underscored in the primer sequences). The plasmid expressing the FMO with an N-terminal tobacco etch virus protease-cleavable His6 tag was chemically transformed into E. coli BL21-CodonPlus(DE3)-RIL cells (Agilent, Santa Clara, CA). A 20-ml culture grown overnight at 37 °C in 2× YT medium was used to inoculate 2 liters of ZYP-5052 auto-induction medium (42) containing selenomethionine, 200 μg/ml kanamycin, and 34 μg/ml chloramphenicol in a LEX48 fermenter (Epiphyte3 Inc., Toronto, Ontario, Canada). After 4 h of growth at 37 °C, the temperature was reduced to 22 °C, and the culture was allowed to grow overnight prior to harvesting by centrifugation. Cells were lysed by sonication in a buffer containing 20 mm Tris, pH 8.0, 500 mm sodium chloride, 20 mm imidazole, 10% glycerol (v/v), 0.1% sodium azide (w/v), and 0.1% Tween 20 (v/v), purified over a 1-ml HisTrap column (GE Healthcare) using standard protocols on a AKTA-Xpress FPLC system, followed by further elution on a HighLoad Superdex S200 column pre-equilibrated with 20 mm HEPES, pH 7.5, 150 mm sodium chloride, 5% glycerol (v/v), and 5 mm DTT. FMO containing fractions were pooled and concentrated to 7.63 mg/ml prior to crystallization.
Crystallization screening by sitting-drop vapor diffusion method was performed using a Phoenix (Art Robins Instruments, Sunnyvale, CA) liquid handling system with MCSG1–4 screens (Anatrace Inc., Maumee, OH), and FMO crystals diffracting to high resolution were obtained in 0.1 m BisTris propane-HCl, pH 7.0, containing 1 m ammonium citrate tribasic, pH 7.0. Prior to flash-cooling, crystals were cryoprotected in the mother liquor containing 30% ethylene glycol (v/v). A SeMet-SAD (single-wavelength anomalous dispersion) dataset was collected at X29A beamline of NSLS (National Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY) and processed using HKL2000 (43). The structure of the FMO was determined using SHELX package as implemented within CCP4 (44, 45). An initial model built using Buccaneer was improved with iterative model building and refinement cycles with Coot (46, 47) and Refmac5 (48) programs. The refined structure of the FMO showed excellent stereochemical parameters (Table 1), and residues 90–103 and 390–412 were not modeled due to poor electron density maps.
Genetic deletion of plu2236 in P. luminescens TT01
The coding sequence of plu2236 was cleanly excised by allelic-exchange mutagenesis using previously described procedures (10). P. luminescens TT01 gDNA was used as a PCR template. The direct upstream and downstream regions of plu2236 were amplified by PCR using primer pairs (Plu2236-UpF, gaaaaatgagctcaaatcaagcacatattttcccgttaaataatc/Plu2236-UpR, aatttactcggaataagtatttctcctgttgtagatacgagcgat; Plu2236-DownF, acaacaggagaaatacttattccgagtaaattgtcataccggaac/Plu2236-DownR, gtaaaaatgagctcgaaaaaagaaccgaagattaccacaacc). The amplified products were purified and used as templates in an overlap extension PCR using the primer pair Plu2236-UpF/Plu2236-DownR. The full-length exchange sequence lacking the plu2236 gene was digested with SacI and ligated into the corresponding site in the suicide plasmid pDS132 (pDΔ2236). The plasmid was cloned and propagated in an E. coli DH5α λ-pir strain on lysogeny broth (LB) agar plates (1% tryptone (w/v), 0.5% yeast extract (w/v), 1% sodium chloride (w/v), 1.5% agar (w/v)) supplemented with 25 μg/ml chloramphenicol and in LB liquid medium similarly supplemented with 25 μg/ml chloramphenicol. The plasmid was verified by restriction analysis and sequencing.
The deletion construct (pDΔ2236) was transformed into an E. coli DH5α λ-pir diaminopimelic acid (DAP) auxotroph donor strain by heat-shock transformation. Transformants were selected on LB agar plates supplemented with 25 μg/ml chloramphenicol and DAP. Selected colonies were inoculated into 5 ml of LB liquid media with DAP and chloramphenicol and grown overnight at 37 °C and 250 rpm. Wild type P. luminescens TT01 colonies were also inoculated into LB broth and grown overnight at 30 °C and 250 rpm. Fresh cultures were inoculated and grown to A600 = 0.5–0.6. These mid-exponential E. coli and P. luminescens cultures were mixed (1 ml total) in three different ratios (1:1, 1:4, and 1:9, respectively) and diluted in culture tubes containing 5 ml of fresh LB. Each mixture was passed through a 0.2-μm filter, and the dried filters were placed on LB agar plates supplemented with DAP and incubated overnight at 30 °C. The filters were added to sterile 50-ml tubes containing 3 ml of fresh LB media and then vortexed vigorously to resuspend the cells. The suspension was plated onto LB agar plates supplemented with 25 μg/ml chloramphenicol and lacking DAP. The plates were incubated over 4 days at 30 °C. The resulting colonies were picked, and counter-selection was carried out on LB sucrose (5%, w/v) plates for 2 days at 30 °C. Successful mutant colonies were identified by over-spanning colony PCR using a diagnostic primer pair (Plu2236-Up-Genome, atacatacttgccagccctaacaaacgat/Plu2236-Down-Genome, gcaatggcagaaatacggttattgatagt). The Δplu2236 strains were re-streaked three times on LB sucrose plates and re-verified by over-spanning colony PCR and sequencing.
Metabolite analysis of P. luminescens wild type and Δplu2236 strains
P. luminescens TT01 wild type and Δplu2236 strains were grown on LB agar plates at 30 °C for 2 days. Single colonies from the agar plates were inoculated into 5 ml of LB liquid media and then further cultivated at 30 °C and 250 rpm for 48 h. The 5-ml bacterial cultures were centrifuged at 2000 × g for 20 min at 4 °C, and the supernatants from each sample were collected and subsequently extracted twice with 5 ml of ethyl acetate. The combined ethyl acetate-soluble fraction was dried under reduced pressure on a HT-4X evaporation system (Genevac Inc., Gardiner, NY). The crude materials were subsequently resuspended in 200 μl of methanol, and 10 μl of these samples were analyzed on an Agilent single quadrupole LC/MS system (column, Phenomenex Kinetex C18 (100 Å) 5 μm (4.6 × 250 mm); flow rate, 0.7 ml/min; mobile phase composition, water/acetonitrile (ACN) gradient system containing 0.1% formic acid (v/v): 0–30 min, 10–100% ACN; hold for 5 min, 100% ACN; 2 min, 100–10% ACN; 5 min post-time, 10% ACN).
In vitro reconstitution of Plu2236
PlFMO was expressed in E. coli BAP1. A 5-ml overnight starter culture was used to inoculate 1 liter of Terrific Broth (1.2% tryptone (w/v), 2.4% yeast extract (w/v), 0.4% glycerol (v/v), 0.2% KH2PO4 (w/v), 1.3% K2HPO4 (w/v)) mixed with ZYM-5052 auto-induction medium (42), and the culture was grown at 37 °C and 250 rpm until the A600 reached 0.5–0.6. The culture was then slowly cooled and incubated at 22 °C and 250 rpm for 18 h. The cells were centrifuged at 12,000 × g for 30 min at 4 °C, and the cell pellets were lysed by sonication with native lysis buffer (NLB: 50 mm NaH2PO4, pH 8, 300 mm NaCl, 10 mm imidazole) supplemented with lysozyme (1 mg/ml, American Bioanalytical), benzonase nuclease (1 μl/ml), and protease inhibitors (cOmpleteTM, EDTA-free Protease Inhibitor Mixture, Roche Applied Science). The lysate was centrifuged at 30,000 × g for 30 min at 4 °C, and the supernatant was subsequently filtered through a 0.45- and a 0.22-μm syringe filter. The filtrates were mixed with nickel-nitrilotriacetic acid resin and subsequently washed with two wash buffers, WB 20 (50 mm NaH2PO4, pH 8, 300 mm NaCl, 20 mm imidazole) and WB 50 (50 mm NaH2PO4, pH 8, 300 mm NaCl, 50 mm imidazole). The protein was eluted with an imidazole step gradient (100 and 250 mm) in the buffer. Purity was assessed by SDS-PAGE analysis and Coomassie staining. The purified protein was buffer-exchanged (Amicon Ultra Centrifugal Filter Unit 30-kDa molecular mass cutoff; EMD Millipore, Darmstadt, Germany) into a reaction buffer (50 mm NaH2PO4, pH 7.0). In vitro experiments of Plu2236 were prepared on a 50-μl scale and were analyzed in triplicate (50 mm NaH2PO4, pH 7.0, 200 μm stilbene substrate, 100 μm NADH or NADPH, 5 μm Plu2236 enzyme). The reaction mixtures were incubated at 25 °C for 30 min and extracted with ethyl acetate. The ethyl acetate-soluble materials were then dried under N2 gas, and the reaction materials were analyzed on an Agilent iFunnel 6550 ESI-QTOF-MS instrument with a Phenomenex Kinetex C18 (100 Å) 5 μm (4.6 × 250 mm) column.
Stilbene analysis of P. luminescens TT01-infected G. mellonella larvae
G. mellonella waxmoth larvae were purchased from Carolina Biological Supply and stored in the dark at 28 °C. Prior to injection, insects were starved overnight. P. luminescens TT01 was streaked onto an LB agar plate and incubated at 30 °C for 2 days. The bacterial colonies were inoculated into LB liquid media, cultivated overnight at 30 °C and 250 rpm, and pelleted by centrifugation at 2000 × g for 20 min. The bacterial cell pellet was washed twice with an equal volume of sterile PBS (0.8% NaCl (w/v), 0.02% KCl (w/v), 0.14% Na2HPO4 (w/v), 0.02% KH2PO4 (w/v)), and the resuspension was diluted in fresh PBS to A600 = 0.1 followed by further 1:1000 dilution with PBS. Waxmoth larvae were chilled on ice for 10 min and surface-sterilized with 100% ethanol prior to inoculation. 10 μl of the bacterial suspension was injected into the hemocoels of the larvae, and control groups were injected with sterile PBS. The larvae infected by P. luminescens TT01 were split and stored in Petri dishes at room temperature. At each time point, 10 inoculated larvae were randomly collected, snap frozen, and stored at −80 °C (five time points: 6, 12, 24, 36, and 48 h; 50 larvae total/condition). All of the P. luminescens-infected larvae were dead within 48 h as expected, although none of the control larvae died. To avoid cross-contamination, collected larvae, including the control group, were separately pound and ground using a sterilized mortar and pestle, and the crushed materials were extracted with 100% methanol. The methanol slurries were filtered through cheesecloth. The filtrates were dried by rotary evaporation, and the crude materials were resuspended in 10 ml of distilled water. The aqueous suspensions were partitioned with n-hexane, dichloromethane, ethyl acetate, and n-butanol. The organic fractions were individually dried using a HT-4X evaporation system for 2 h. The dried residues were resuspended in methanol and analyzed by LC/MS as described above.
Larger-scale cultivation of P. luminescens TT01 in hemolymph-mimetic medium
To isolate the new stilbene metabolites detected in G. mellonella larvae infected with P. luminescens TT01, we prepared an HMM based on the concentrations of the proteinogenic amino acids present in the hemolymph of G. mellonella (10) and examined the production of the stilbene molecules in this medium. Single colonies of P. luminescens TT01 were inoculated into 5 ml of HMM and incubated at 30 °C and 250 rpm. After 48 h of cultivation, the bacterial cultures were centrifuged, and the supernatants were extracted with ethyl acetate (two times 5 ml) in 14-ml polypropylene culture tubes. The organic layers were evaporated under reduced pressure. Crude materials were dissolved in 200 μl of 100% methanol and then monitored on an Agilent 6120 single quadrupole LC/MS system (column, Phenomenex Kinetex C18 (100 Å) 5-μm (4.6 × 250 mm) column; flow rate, 0.7 ml/min; mobile phase composition, water/ACN gradient solvent system containing 0.1% formic acid (v/v): 0–30 min, 10–100% ACN; hold for 5 min, 100% ACN; 2 min, 100–10% ACN; 5 min post-time, 10% ACN). LC/MS analysis of samples extracted from 5-ml cultures led to the detection of the targeted stilbene metabolites, confirming that HMM suitably mimicked the G. mellonella hemolymph for prolbene production. With this confirmation, we initiated a 16-liter scale P. luminescens culture in HMM to isolate the molecules. P. luminescens TT01 seed cultures (16 × 5 ml) were transferred to 16 × 1 liter of HMM in 4-liter Erlenmeyer flasks. After 48 h, the whole culture broth was extracted with twice with equal proportions of ethyl acetate (32 liters total), and the organic layer was dried using a rotary evaporation system to yield a crude extract (2.3 g).
Isolation and purification of stilbene metabolites
The crude organic extract (2.3 g) was subjected to a Waters Sep-Pak® Vac 35cc (10 g) C18 cartridge with a step gradient elution (20, 40, 60, 80, and 100% methanol in water). The 60% methanol fraction was further fractionated using an Agilent Prepstar HPLC system (column, Agilent Polaris C18-A 5 μm (21.2 × 250 mm)) with a linear gradient elution (20–80% aqueous methanol over 60 min; 8 ml/min; 1-min fraction collection interval). Fraction 26 was then separated by reversed-phase HPLC (column, Phenomenex Luna C18 (100 Å) 10 μm (10.0 × 250 mm)) with a linear gradient elution (10–100% acetonitrile in water for 60 min; 4 ml/min) to yield partially purified compound 3. Compound 3 was then purified by reversed phase C8-HPLC (column, Phenomenex Luna C8(2) (100 Å) 10 μm (10.0 × 250 mm)) with a gradient solvent system (10–100% methanol in water for 60 min; 4 ml/min; tR = 20.5 min; 1.5 mg of pure product). For compound 4, the combined preparatory-scale fraction (27 + 28) was separated by reversed-phase HPLC (column, Phenomenex Luna C8(2) (100 Å) 5 μm (4.6 × 150 mm)) using an isocratic solvent system (35% aqueous acetonitrile over 30 min; 4 ml/min) to afford partially pure compound 4 (tR = 7.5 min). Purification of compound 4 (1.8 mg, tR = 18.9 min) was carried out over an Agilent phenyl-hexyl 5-μm (9.4 × 250 mm) column using a gradient separation (10–100% acetonitrile in water over 60 min; 4 ml/min). Known compound, 3,5-dihydroxy-4-isopropyl-trans-stilbene (tR = 25.5 min, 1), was isolated from preparatory fraction 45 via a Phenomenex Luna C8(2) (100 Å) 10-μm (10.0 × 250 mm) column eluting with a gradient system of 60–100% methanol in water for 30 min to yield 5.0 mg of pure product (23). 2-Isopropyl-5-(3-phenyl-oxiranyl)-benzene-1,3-diol (2) from preparatory fraction 43 was isolated and purified over a Phenomenex Luna C8(2) (100 Å) 10-μm (10.0 × 250 mm) column and an Agilent phenyl-hexyl 5 μm (9.4 × 250 mm) column eluting with a gradient system of 50–100% acetonitrile in water for 30 min to yield 3.0 mg of pure metabolite.
3,5-Dihydroxy-4-isopropyl-trans-stilbene (1) consists of the following: amorphous solid; 1H NMR (CDCl3, 600 MHz) δ 7.47 (2H, d, J = 7.5 Hz, H-10 and H-14), 7.35 (2H, t, J = 7.5 Hz, H-11 and H-13), 7.25 (1H, t, J = 7.3 Hz, H-12), 6.99 (1H, d, J = 16.0 Hz, H-8), 6.90 (1H, d, J = 16.0 Hz, H-7), 6.50 (2H, s, H-2 and H-4), 4.75 (2H, s, OH), 3.53–3.38 (1H, m, H-15), 1.38 (6H, d, J = 7.0 Hz, H-16 and H-17); ESI-MS m/z 255 [M + H]+, 277 [M + Na]+.
2-Isopropyl-5-(3-phenyl-oxiranyl)-benzene-1,3-diol (2) consists of the following: amorphous solid; 1H NMR (CD3OD, 600 MHz) δ 7.36–7.29 (5H, m, H-10, H-11, H-12, H-13, H-14), 6.24 (2H, s, H-2 and H-4), 3.78 (1H, d, J = 1.8 Hz, H-8), 3.63 (1H, d, J = 1.8 Hz, H-7), 3.48 (1H, dq, J = 14.0, 7.0 Hz, H-15), 1.28 (6H, d, J = 7.0 Hz, H-16 and H-17); ESI-MS m/z 271 [M + H]+, 293 [M + Na]+.
Prolbene A (3) consists of the following: amorphous solid; 1H and 13C NMR spectra, see Table 2; HR-ESI-QTOF-MS [M + H]+ m/z 386.1964 (calculated for C22H28NO5, 386.1967).
Prolbene B (4) consists of the following: amorphous solid; 1H and 13C NMR spectra, see Table 2; HR-ESI-QTOF-MS [M + H]+ m/z 386.1969 (calculated for C22H28NO5, 386.1967).
Marfey's analysis of prolbenes
Standard d- and l-proline were purchased from Sigma. Compounds 3 and 4 (each 0.5 mg) were hydrolyzed in 500 μl of 6 n HCl at 110 °C for 1 h, and the reaction mixture was dried in vacuo overnight. The hydrolysate was dissolved in distilled water and completely dried for 24 h in a Genevac HT-4X evaporation system to remove excess residual acid. The hydrolyzed materials and standard amino acids were treated with 50 μl of a solution of Nα-(2,4-dinitro-5-fluorophenyl)-l-alaninamide (FDAA) (10 mg/ml in acetone) followed by the addition of 100 μl of 1 n NaHCO3 (49). The reaction mixture was heated at 80 °C for 3 min and quenched with 50 μl of 2 n HCl. The derivatized materials were then diluted to 300 μl with 50% aqueous acetonitrile for LC/MS analysis. 10-μl samples were analyzed using the single quadrupole LC/MS system equipped with a Phenomenex Kinetex C18 (100 Å) 5-μm (4.6 × 250 mm) column using a flow rate (0.7 ml/min) and a solvent system of water and acetonitrile containing 0.1% formic acid (v/v). The retention times of derivatized amino acids were as follows: gradient, 0–40 min, 20–60% acetonitrile, l-proline 16.5 min, and d-proline 17.8 min.
Electronic circular dichroism (ECD) and computational NMR chemical shift calculations (DP4 analysis)
Conformational searches for ECD and DP4 analyses were performed using the MacroModel (Version 9.9, Schrödinger LLC) program with a mixed torsional/low-mode sampling method in the MMFF force field. The searches were examined in the gas phase with a 50 kJ/mol energy window limit and 10,000 maximum number of steps to thoroughly probe low-energy conformers. Minimization processes were achieved utilizing Polak-Ribiere conjugate gradient (PRCG) method, 10,000 maximum iterations, and a 0.001 kJ (mol Å)−1 convergence threshold on the root mean square gradient. The geometry of conformers within 10 kJ/mol was further optimized at the B3LYP/6–31+G(d) level with tight convergence, and ECD calculations were performed at the identical theory level and basis sets. The generated excitation energies and rotational strengths were Boltzmann-averaged based on the calculated Gibbs free energy of each conformer and proceeded for the ECD visualization utilizing SpecDis (50). Advanced searches for DP4 analysis were carried out in the MMFF force field, in the gas phase with a 50 kJ/mol energy window and 10,000 maximum iterations based on the original authors' recommendations (33). NMR chemical shift calculations of all conformers within 10 kJ/mol of the relative energy were performed using the Gaussian 09 package (Gaussian Inc.) in B3LYP/6–31G(d,p) theory level. Chemical shifts values were calculated using GIAO magnetic shielding tensor values. The DP4 probability analysis was conducted using an applet available on line.
MIC antimicrobial assays
The growth inhibitory activities of 1-4 against three microbial strains, including one Gram-positive bacterium B. subtilis, one Gram-negative bacterium E. coli, and one unicellular fungus S. cerevisiae, were evaluated using the in vitro MIC methods. Bacterial strains B. subtilis and E. coli were inoculated onto LB agar and grown at 30 and 37 °C overnight, respectively. Similarly, S. cerevisiae yeast was grown on potato dextrose agar (PDA: 0.4% potato starch (w/v), 2% dextrose (w/v), 1.5% agar (w/v)) at 30 °C. Single colonies were appropriately inoculated in either LB or YM (0.5% yeast extract (w/v), 3% malt extract (w/v)). Each microbial growth was then diluted with fresh media to A600 = 0.1. Compounds were dissolved in DMSO at a concentration of 10 mg/ml. 50-μl aliquots of fresh media were added to each well along a row of a 96-well plate, not including the first well to which 96 μl was added. 4 μl of the samples were added to the first well to yield a final concentration of 400 μg/ml, and this well was then serially diluted throughout the row. 50 μl of the cell culture broth was then added to each well with a maximum tester concentration of 200 μg/ml compound. DMSO was used as a negative control, and samples were prepared in triplicate. Plates were then sealed and incubated at the 30 °C for 24 h. Growth was assessed by measurement of A600. The MIC values were determined as the minimal concentration exhibiting complete inhibition of cell growth.
Mushroom tyrosinase inhibitory assays
Mushroom tyrosinase, 3,4-dihydroxy-l-phenylalanine (l-DOPA), and kojic acid were purchased from Sigma. The enzyme inhibitory assay was performed in a 96-well plate and completed in triplicate for each compound. Briefly, compounds 1-4 were dissolved in 10% DMSO (v/v) in 0.1 m phosphate buffer, pH 6.8, and 50 μl of the compounds was dispensed into the wells. 50 μl of 2 mm l-DOPA solution dissolved in 0.1 m phosphate buffer, pH 6.8, was added to the compound solutions followed by incubation for 10 min at room temperature. The 50 μl of mushroom tyrosinase solution in phosphate buffer (135 units/ml) was finally dispensed into each well, and the plate was measured through the absorbance at 475 nm. Kojic acid was used as positive control.
Nematode cultivation
Nematodes used were the H. bacteriophora M31e strain. To generate axenic H. bacteriophora IJs, symbiont IJs (IJs containing their bacterial endosymbiont) were grown to the adult stage on plates (51) containing 5 μg/ml cholesterol + 0.1% sodium pyruvate (w/v) seeded with Photorhabdus temperata NC1 TRN16 (RET16) bacteria (52). Gravid adults were washed off the plates and incubated in a bleach solution consisting of 4.05 ml of ddH2O, 0.85 ml of 5 m NaOH, and 0.25 ml of commercial bleach (Clorox, concentrated, 8.25% NaOCl (w/v)) until worms were dissolved and only eggs remained. The eggs were rinsed twice in 10 ml of sterile ddH2O and plated on RET16 bacteria grown on 1× lipid agar plates containing 5 μg/ml cholesterol, 0.1% sodium pyruvate (w/v), 100 μg/ml carbenicillin, and 0.75 μg/ml gentamycin. IJs collected from the plates were surface-sterilized by incubating in 0.75% commercial bleach (Clorox, concentrated, 8.25% NaOCl (w/v)) for 5 min. IJs were then rinsed three times in sterile ddH2O and stored in sterile ddH2O at 15 °C until use.
Infective juvenile recovery assay
P. luminescens TT01 wild type and mutant strain Δplu2236 (two independently established mutants: Δplu2236-1 and Δplu2236-2) were maintained in separate boxes to avoid cross-contamination. For IJ recovery assays, WT and Δplu2236 were grown in PP3 broth at 25 °C for ∼24 h. A 2-μl suspension was then spread on 0.5× nutrient agar plates containing 5 μg/ml cholesterol and 0.1% sodium pyruvate (w/v) with yeast extract omitted and grown at 25 °C for 2–3 days until the bacteria produced pigments indicative of supporting nematode growth. Axenic IJs generated as described above were surface-sterilized and 10–12 healthy IJs were placed on the bacterial lawn and allowed to grow at 25 °C for 4 days. In parallel, the IJ population was confirmed to be axenic by spreading a 100-μl aliquot containing ∼500–1000 IJs on a 1× lipid agar + cholesterol + pyruvate plate and confirming the absence of bacterial growth after incubation at 25 °C for 4 days. Recovery was assayed as an increase in the body size of the worm. We note that some of the IJs placed on the plates could not be located at the end of the assay; these IJs were assumed to have not recovered.
P. luminescens virulence assays in a G. mellonella infection model
Larvae of the Greater Waxmoth were reared as described previously (“Stilbene analysis of P. luminescens TT01-infected G. mellonella larvae”). Wild type P. luminescens TT01 along with the Δplu2236 strain were streaked onto LB agar and grown for 2 days at 30 °C. Single well defined colonies from each strain were then inoculated into 5 ml of LB for overnight growth at 30 °C and 250 rpm. The overnight growths were subcultured at 1:1000 into 5 ml of LB medium and grown at 30 °C and 250 rpm to approximately A600 = 0.1. The cells were collected via centrifugation at 2000 × g and subsequently washed twice with sterile PBS. The cells were resuspended in sterile PBS to an A600 = 0.1 and further diluted 1000-fold. Immediately prior to injection, the larvae were maintained on ice to slow movement and surface-sterilized around the hindmost right proleg. 10 μl of each cell suspension was injected into the hemocoels of 10 larvae. Sterile PBS was used as a vehicle control. The infected larvae were maintained at 30 °C and monitored for death. There were no observable differences in killing between the two P. luminescens strains. All infected larvae were dead within 48 h post-infection, whereas all larvae receiving the PBS vehicle control survived.
Insecticidal metabolite assays in G. mellonella
The insecticidal activity of stilbenes 1 and 2 was assessed against G. mellonella. The stilbenes were dissolved in DMSO to 0.1 mg/μl. Insects were reared as described above. 10 larvae were selected per condition (30 larvae total) having an average mass of 250 mg. Insects were maintained on ice prior to injection. The area surrounding the hindmost right proleg was surface-sterilized with 100% ethanol immediately prior to injection. 2.5 μl of 1, 2, or DMSO was injected into the hemocoel of the larvae, and the larvae were maintained at 30 °C. The larvae were periodically monitored for death over the course of 5 days. Within this time frame, no death was observed under any condition.
Growth inhibitory assays against producing strain P. luminescens TT01
Stilbenes 1 and 2 were examined for the growth inhibitory activity against their producing strain P. luminescens TT01 by using the minimal inhibitory concentration method. P. luminescens TT01 wild type and a mutant, ΔhexA, a knock-out strain of a LysR-type transcriptional repressor (35), was tested for comparison. The P. luminescens hexA mutant was generously provided by Prof. David J. Clarke (University College Cork, Cork, Ireland). We also tested the epoxidase plu2236 mutant. Strains P. luminescens TT01, ΔhexA, and Δplu2236 were streaked onto LB agar plates and grown at 30 °C for 2 days. Single colonies of each strain were subsequently inoculated into 5 ml of LB liquid medium and incubated at 30 °C and 250 rpm until A600 = 0.1. The bacterial cultures were then dispensed into fresh LB medium at a 1:100 dilution. Stilbenes 1 and 2 were subsequently prepared in the fresh LB medium containing DMSO in the 1.5-ml sterilized Eppendorf tubes, and 50 μl of sample was mixed with 50 μl of cell broth in the 96-well plate. The 10% DMSO was required to dissolve stilbenes in the medium. The assays were completed in triplicate, and the absorbances of the cells were measured at 600 nm over three time points, 12, 24, and 48 h after inoculation.
Author contributions
H. B. P. and J. M. C. designed the study. H. B. P., P. S., S. C. A., and J. M. C. wrote the paper. H. B. P. carried out bacterial cultivations, LC/MS analyses, metabolite isolations, NMR-based structural characterizations, computational chemistry, in vitro enzyme assays, and antimicrobial assays. P. S., J. B. B., and S. C. A. oversaw and carried out the X-ray-based protein structure analyses. C. E. P. carried out the invertebrate animal infection model experiments. J. T. constructed bacterial mutant strains. J. H. L. and E. A. H. oversaw and carried out the nematode cultivation and infective juvenile recovery assays. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Prof. David J. Clarke for providing P. luminescens hexA mutant strain. We thank the Yale Center for Research Computing for guidance and use of the research computing infrastructure. We also thank Dr. Joonseok Oh at Crawford laboratory for advice and comments regarding NMR computational chemistry. X-ray data presented in this publication was collected at the X29A beamline. We acknowledge the high-throughput X-ray crystallography team: M. Ahmed, N. Banu, R. Bhosle, S. Chamala, S. Chowdhury, A. Fiser, A. Gizzi, A. S. Glenn, J. Hammonds, B. Hillerich, K. Khafizov, J. Lafleur, J. D. Love, M. Stead, R. Seidel, and R. Toro. Use of the NSLS is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-98CH10886. The New York Structural Genomics Research Consortium was supported by U54 GM094662 to SCA.
This work on the characterization of metabolites from bacterial symbionts was supported by the Searle Scholars Program Grant 13-SSP-210 (to J. M. C.), Damon Runyon Cancer Research Foundation Grant DRR-39-16 (to J. M. C.), and National Institutes of Health Grant 1DP2-CA186575 (to J. M. C.). Our structural genomics work was supported by the National Institutes of Health Grant U54-GM094662 (to S. C. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1–S8 and Figs. S1–S21.
The atomic coordinates and structure factors (code 4HB9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- IJ
- infective juvenile
- FMO
- flavin adenosine dinucleotide-dependent monooxygenase
- HR-ESI-QTOF-MS
- high-resolution electrospray ionization quadrupole-time-of-flight mass spectrometry
- HMM
- hemolymph mimetic medium
- COSY
- correlation spectroscopy
- HSQC
- heteronuclear single-quantum correlation
- HMBC
- heteronuclear multiple-bond correlation
- ECD
- electronic circular dichroism
- MMFF
- Merck molecular force field
- GIAO
- gauge-including atomic orbital
- MIC
- minimal inhibitory concentration
- PKS
- polyketide synthase
- PDA
- photo diode array
- LB
- lysogeny broth
- DAP
- diaminopimelic acid
- ACN
- acetonitrile
- BisTris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- PDB
- Protein Data Bank
- l-DOPA
- 3,4-dihydroxy-l-phenylalanine
- HMM
- hemolymph-mimetic medium
- FDAA
- Nα-(2,4-dinitro-5-fluorophenyl)-l-alaninamide
- ddH2O
- double distilled H2O.
References
- 1. Clarke D. J. (2008) Photorhabdus: a model for the analysis of pathogenicity and mutualism. Cell. Microbiol. 10, 2159–2167 [DOI] [PubMed] [Google Scholar]
- 2. Clarke D. J. (2014) The genetic basis of the symbiosis between Photorhabdus and its invertebrate hosts. Adv. Appl. Microbiol. 88, 1–29 [DOI] [PubMed] [Google Scholar]
- 3. Waterfield N. R., Ciche T., and Clarke D. (2009) Photorhabdus and a host of hosts. Annu. Rev. Microbiol. 63, 557–574 [DOI] [PubMed] [Google Scholar]
- 4. Gerrard J., Waterfield N., Vohra R., and ffrench-Constant R. (2004) Human infection with Photorhabdus asymbiotica: an emerging bacterial pathogen. Microbes Infect. 6, 229–237 [DOI] [PubMed] [Google Scholar]
- 5. Bode H. B. (2009) Entomopathogenic bacteria as a source of secondary metabolites. Curr. Opin. Chem. Biol. 13, 224–230 [DOI] [PubMed] [Google Scholar]
- 6. Vizcaino M. I., Guo X., and Crawford J. M. (2014) Merging chemical ecology with bacterial genome mining for secondary metabolite discovery. J. Ind. Microbiol. Biotechnol. 41, 285–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brachmann A. O., Joyce S. A., Jenke-Kodama H., Schwär G., Clarke D. J., and Bode H. B. (2007) A type II polyketide synthase is responsible for anthraquinone biosynthesis in Photorhabdus luminescens. Chembiochem 8, 1721–1728 [DOI] [PubMed] [Google Scholar]
- 8. Richardson W. H., Schmidt T. M., and Nealson K. H. (1988) Identification of an anthraquinone pigment and a hydroxystilbene antibiotic from Xenorhabdus luminescens. Appl. Environ. Microbiol. 54, 1602–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brachmann A. O., Kirchner F., Kegler C., Kinski S. C., Schmitt I., and Bode H. B. (2012) Triggering the production of the cryptic blue pigment indigoidine from Photorhabdus luminescens. J. Biotechnol. 157, 96–99 [DOI] [PubMed] [Google Scholar]
- 10. Crawford J. M., Kontnik R., and Clardy J. (2010) Regulating alternative lifestyles in entomopathogenic bacteria. Curr. Biol. 20, 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crawford J. M., Portmann C., Zhang X., Roeffaers M. B., and Clardy J. (2012) Small molecule perimeter defense in entomopathogenic bacteria. Proc. Natl. Acad. Sci. U.S.A. 109, 10821–10826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brachmann A. O., Brameyer S., Kresovic D., Hitkova I., Kopp Y., Manske C., Schubert K., Bode H. B., and Heermann R. (2013) Pyrones as bacterial signaling molecules. Nat. Chem. Biol. 9, 573–578 [DOI] [PubMed] [Google Scholar]
- 13. Nollmann F. I., Heinrich A. K., Brachmann A. O., Morisseau C., Mukherjee K., Casanova-Torres Á. M., Strobl F., Kleinhans D., Kinski S., Schultz K., Beeton M. L., Kaiser M., Chu Y. Y., Phan Ke L., et al. (2015) A Photorhabdus natural product inhibits insect juvenile hormone epoxide hydrolase. ChemBioChem 16, 766–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bode H. B., Brachmann A. O., Jadhav K. B., Seyfarth L., Dauth C., Fuchs S. W., Kaiser M., Waterfield N. R., Sack H., Heinemann S. H., and Arndt H. D. (2015) Structure elucidation and activity of kolossin A, the d-/l-pentadecapeptide product of a giant nonribosomal peptide synthetase. Angew. Chem. Int. Ed. Engl. 54, 10352–10355 [DOI] [PubMed] [Google Scholar]
- 15. Bian X., Plaza A., Zhang Y., and Müller R. (2012) Luminmycins A–C, cryptic natural products from Photorhabdus luminescens identified by heterologous expression in Escherichia coli. J. Nat. Prod. 75, 1652–1655 [DOI] [PubMed] [Google Scholar]
- 16. Dudnik A., Bigler L., and Dudler R. (2013) Heterologous expression of a Photorhabdus luminescens syrbactin-like gene cluster results in production of the potent proteasome inhibitor glidobactin A. Microbiol. Res. 168, 73–76 [DOI] [PubMed] [Google Scholar]
- 17. Theodore C. M., King J. B., You J., and Cichewicz R. H. (2012) Production of cytotoxic glidobactins/luminmycins by Photorhabdus asymbiotica in liquid media and live crickets. J. Nat. Prod. 75, 2007–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eleftherianos I., Boundy S., Joyce S. A., Aslam S., Marshall J. W., Cox R. J., Simpson T. J., Clarke D. J., ffrench-Constant R. H., and Reynolds S. E. (2007) An antibiotic produced by an insect-pathogenic bacterium suppresses host defenses through phenoloxidase inhibition. Proc. Natl. Acad. Sci. U.S.A. 104, 2419–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu K., Li J., Li B., Webster J. M., and Chen G. (2006) A novel antimicrobial epoxide isolated from larval Galleria mellonella infected by the nematode symbiont, Photorhabdus luminescens (Enterobacteriaceae). Bioorg. Med. Chem. 14, 4677–4681 [DOI] [PubMed] [Google Scholar]
- 20. Hu K., and Webster J. M. (2000) Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus-Heterorhabditis infected Galleria mellonella larvae. FEMS Microbiol. Lett. 189, 219–223 [DOI] [PubMed] [Google Scholar]
- 21. Joyce S. A., Brachmann A. O., Glazer I., Lango L., Schwär G., Clarke D. J., and Bode H. B. (2008) Bacterial biosynthesis of a multipotent stilbene. Angew. Chem. Int. Ed. Engl. 47, 1942–1945 [DOI] [PubMed] [Google Scholar]
- 22. Kontnik R., Crawford J. M., and Clardy J. (2010) Exploiting a global regulator for small molecule discovery in Photorhabdus luminescens. ACS Chem. Biol. 5, 659–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Park H. B., and Crawford J. M. (2015) Lumiquinone A, an α-aminomalonate-derived aminobenzoquinone from Photorhabdus luminescens. J. Nat. Prod. 78, 1437–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paul V. J., Frautschy S., Fenical W., and Nealson K. H. (1981) Antibiotics in microbial ecology. J. Chem. Ecol. 7, 589–597 [DOI] [PubMed] [Google Scholar]
- 25. Akhurst R. J. (1982) Antibiotic activity of Xenorhabdus spp., bacteria symbiotically associated with insect pathogenic nematodes of the families Heterorhabditidae and Steinernematidae. J. Gen. Microbiol. 128, 3061–3065 [DOI] [PubMed] [Google Scholar]
- 26. Hu K., Li J., and Webster J. M. (1997) Quantitative analysis of a bacteria-derived antibiotic in nematode-infected insects using HPLC-UV and TLC-UV methods. J. Chromatogr. B Biomed. Sci. Appl. 703, 177–183 [DOI] [PubMed] [Google Scholar]
- 27. Williams J. S., Thomas M., and Clarke D. J. (2005) The gene stlA encodes a phenylalanine ammonia-lyase that is involved in the production of a stilbene antibiotic in Photorhabdus luminescens TT01. Microbiology 151, 2543–2550 [DOI] [PubMed] [Google Scholar]
- 28. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 29. Eswaramoorthy S., Bonanno J. B., Burley S. K., and Swaminathan S. (2006) Mechanism of action of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 9832–9837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Volkers G., Palm G. J., Weiss M. S., Wright G. D., and Hinrichs W. (2011) Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. FEBS Lett. 585, 1061–1066 [DOI] [PubMed] [Google Scholar]
- 31. Park H. B., Perez C. E., Perry E. K., and Crawford J. M. (2016) Activating and attenuating the amicoumacin antibiotics. Molecules 21, E824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park H. B., and Crawford J. M. (2016) Pyrazinone protease inhibitor metabolites from Photorhabdus luminescens. J. Antibiot. 69, 616–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith S. G., and Goodman J. M. (2010) Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: the DP4 probability. J. Am. Chem. Soc. 132, 12946–12959 [DOI] [PubMed] [Google Scholar]
- 34. Gerrard J. G., Joyce S. A., Clarke D. J., ffrench-Constant R. H., Nimmo G. R., Looke D. F., Feil E. J., Pearce L., and Waterfield N. R. (2006) Nematode symbiont for Photorhabdus asymbiotica. Emerg. Infect. Dis. 12, 1562–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Joyce S. A., and Clarke D. J. (2003) A hexA homologue from Photorhabdus regulates pathogenicity, symbiosis and phenotypic variation. Mol. Microbiol. 47, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 36. Hertweck C. (2009) The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. Engl. 48, 4688–4716 [DOI] [PubMed] [Google Scholar]
- 37. Fuchs S. W., Bozhüyük K. A., Kresovic D., Grundmann F., Dill V., Brachmann A. O., Waterfield N. R., and Bode H. B. (2013) Formation of 1,3-cyclohexanediones and resorcinols catalyzed by a widely occurring ketosynthase. Angew. Chem. Int. Ed. Engl. 52, 4108–4112 [DOI] [PubMed] [Google Scholar]
- 38. Brameyer S., Kresovic D., Bode H. B., and Heermann R. (2015) Dialkylresorcinols as bacterial signaling molecules. Proc. Natl. Acad. Sci. U.S.A. 112, 572–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Somvanshi V. S., Sloup R. E., Crawford J. M., Martin A. R., Heidt A. J., Kim K.-S., Clardy J., and Ciche T. A. (2012) A single promoter inversion switches Photorhabdus between pathogenic and mutualistic states. Science 337, 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilson T. G. (2004) The molecular site of action of juvenile hormone and juvenile hormone insecticides during metamorphosis: how these compounds kill insects. J. Insect Physiol. 50, 111–121 [DOI] [PubMed] [Google Scholar]
- 41. Savitsky P., Bray J., Cooper C. D., Marsden B. D., Mahajan P., Burgess-Brown N. A., and Gileadi O. (2010) High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol. 172, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 43. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 44. Sheldrick G. M. (2015) Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 71, 3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cowtan K. (2008) Fitting molecular fragments into electron density. Acta Crystallogr. D Biol. Crystallogr. 64, 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Skubák P., Murshudov G. N., and Pannu N. S. (2004) Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D Biol. Crystallogr. 60, 2196–2201 [DOI] [PubMed] [Google Scholar]
- 49. Marfey P. (1984) Determination of d-amino acids. II. Use of a bifunctional reagent, 1, 5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 49, 591–596 [Google Scholar]
- 50. Bruhn T., Schaumlöffel A., Hemberger Y., and Bringmann G. (2013) SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 25, 243–249 [DOI] [PubMed] [Google Scholar]
- 51. Vivas E. I., and Goodrich-Blair H. (2001) Xenorhabdus nematophilus as a model for host-bacterium interactions: rpoS is necessary for mutualism with nematodes. J. Bacteriol. 183, 4687–4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ciche T. A., Kim K. S., Kaufmann-Daszczuk B., Nguyen K. C., and Hall D. H. (2008) Cell invasion and matricide during Photorhabdus luminescens transmission by Heterorhabditis bacteriophora nematodes. Appl. Environ. Microbiol. 74, 2275–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. de Beer T. A., Berka K., Thornton J. M., and Laskowski R. A. (2014) PDBsum additions. Nucleic Acids Res. 42, D292–D296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.