

Abstract
With some advances in modern medicine (such as cancer chemotherapy, broad exposure to antibiotics, and immunosuppression), the incidence of opportunistic fungal pathogens such as Candida albicans has increased. Cases of drug resistance among these pathogens have become more frequent, requiring the development of new drugs and a better understanding of the targeted enzymes. Sterol 14α-demethylase (CYP51) is a cytochrome P450 enzyme required for biosynthesis of sterols in eukaryotic cells and is the major target of clinical drugs for managing fungal pathogens, but some of the CYP51 key features important for rational drug design have remained obscure. We report the catalytic properties, ligand-binding profiles, and inhibition of enzymatic activity of C. albicans CYP51 by clinical antifungal drugs that are used systemically (fluconazole, voriconazole, ketoconazole, itraconazole, and posaconazole) and topically (miconazole and clotrimazole) and by a tetrazole-based drug candidate, VT-1161 (oteseconazole: (R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol). Among the compounds tested, the first-line drug fluconazole was the weakest inhibitor, whereas posaconazole and VT-1161 were the strongest CYP51 inhibitors. We determined the X-ray structures of C. albicans CYP51 complexes with posaconazole and VT-1161, providing a molecular mechanism for the potencies of these drugs, including the activity of VT-1161 against Candida krusei and Candida glabrata, pathogens that are intrinsically resistant to fluconazole. Our comparative structural analysis outlines phylum-specific CYP51 features that could direct future rational development of more efficient broad-spectrum antifungals.
Keywords: Candida albicans, cytochrome P450, enzyme inhibitor, enzyme kinetics, X-ray crystallography, antifungal drugs, sterol 14 alpha-demethylase (CYP51), sterol biosynthesis
Introduction
Candida albicans is the most prevalent fungal human pathogen (1–3). The entire life cycle of this polymorphic fungus occurs in mammalian hosts and involves switches between distinct single-celled yeast and multicellular mycelial forms (1). As a yeast form, C. albicans exists as a ubiquitous commensal of human microbiome, colonizing the skin, mouth, gastrointestinal tract, and female reproductive tract of healthy adults (4, 5). In weakened hosts, however, particularly in immunocompromised or critically ill patients or those who developed microbial dysbiosis, the C. albicans life style changes from commensal to pathogenic. The change involves a cell-type transition between benign yeast and invasive, e.g. hyphal morphology (1, 6), causing diseases varying from relatively easily treatable topical (e.g. athlete's foot and oral thrush) to life-threatening systemic infections (such as disseminated (deep-seated) candidiasis, particularly candidemia), which result in up to 400,000 human deaths annually (2, 4, 7). With the availability of modern medical treatments, including anticancer chemotherapy, organ transplantation, use of different types of implantable medical devises, and administration of broad-spectrum antibiotics and immunosuppressive agents, the number of vulnerable individuals has risen over the past several decades and so has the incidence of candidiasis (8, 9). Candidemia is one of the most common healthcare-associated bloodstream infections in hospitals, especially in intensive care units, in the United States and worldwide, with a mortality of 30–55% (2, 7, 10, 11).
The clinical treatment includes three types of drugs (7), azoles (1,3-imidazole and 1,2,4-triazole derivatives), amphotericin B, and echinocandins. Echinocandins inhibit (1→3)β-d-glucan synthase and thus damage the fungal cell wall. Amphotericin B removes ergosterol from fungal plasma membranes, disrupting their structure. Azoles (the largest class of antimycotic drugs in clinical use (7, 12)) block biosynthesis of ergosterol de novo, not only depleting the source of ergosterol for the membranes but also preventing the formation of physiologically important (hormonal) intracellular sterols, which are required for the cell cycle regulation, cell development and multiplication (13), and apparently for cell transformation as well (5, 12, 14).
During the past 30 years (15), fluconazole has become and remains the first-line agent for treatment and prophylaxis of all types of invasive candidiasis (2, 7, 16). Alternative options for systemic infections include voriconazole and itraconazole. Miconazole and clotrimazole are used in addition to fluconazole as topical agents. Because of its erratic bioavailability and unpredictable trough plasma concentration, the use of posaconazole has been limited mainly for oropharyngeal or esophageal candidiasis and for prophylaxis in high-risk patients (7), but now, when the intravenous formulation of posaconazole has become available, its clinical use has the potential to be extended (17).
Azoles block sterol biosynthesis by inhibiting fungal sterol 14α-demethylase (18), the membrane-bound enzyme in the endoplasmic reticulum that removes the 14α-methyl group from the first cyclized sterol precursor (lanosterol in C. albicans (Fig. 1A)) and thus initiates the advancement of the pathway toward its final products (ergosterol in fungi versus cholesterol in their mammalian hosts) (Fig. 1B). Although these two compounds are structurally similar, host cholesterol cannot replace ergosterol in fungal cells.
Figure 1.
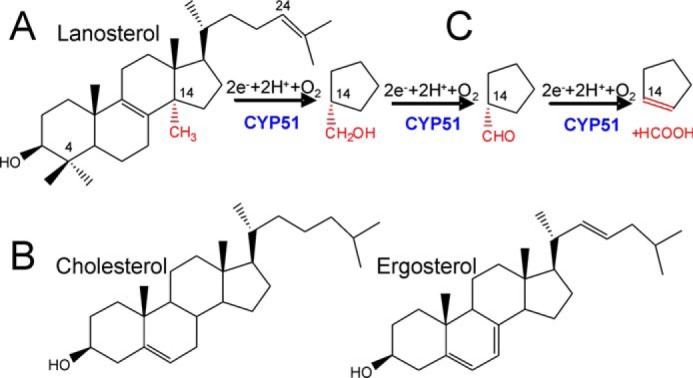
CYP51 in sterol biosynthesis. A, C. albicans CYP51 substrate lanosterol. B, products of the pathway in animals (cholesterol) and in fungi (ergosterol). C, three-step reaction of sterol 14α-demethylation. Each step involves one cycle of monooxygenation. 14α-Demethylation of lanosterol produces 4,4′-dimethylcholesta-8,14,24-triene-3β-ol (14α-desmethyllanosterol in Fig. 5).
Sterol 14α-demethylase (CYP51,2 EC 1.14.13.70) is the most functionally conserved cytochrome P450 (CYP) monooxygenase (19, 20). CYP51 orthologs are found in all biological kingdoms and joined into one (CYP51) family because (despite very low amino acid sequence identity across phylogeny (19)) they all perform one stereo- and regiospecific reaction that involves three consecutive cytochrome P450 catalytic cycles (Fig. 1C). Each cycle requires two electrons, provided by NADPH and delivered to the P450 heme iron via the FAD and FMN prosthetic groups of the redox partner protein NADPH-cytochrome P450 reductase (CPR) and two protons. When the iron is reduced to the ferrous Fe2+ form, it binds a molecule of oxygen, activates it, and then cleaves it into two atoms, inserting one atom into the substrate and reducing the other to a water molecule. During the three monooxygenation cycles, the molecule of the sterol substrate is bound in the CYP51-active site in such a way that its 14α-methyl carbon atom (C30 according to IUPAC nomenclature) is positioned in close proximity to the catalytic heme iron (∼5 Å above the heme plane (21)), a feature that requires high rigidity of the CYP51 substrate-binding cavity (20).
Azole drugs inhibit CYP51 activity by 1) forming an axial coordination bond with the prosthetic heme iron, thus affecting the iron potential to be reduced, and 2) competing with the sterol substrate for the space within the enzyme-active site. The strength of the inhibition profoundly depends on the structural composition of the non-coordinated portion of the azole molecule (22). Interestingly, we found that many antifungal azoles are strong inhibitors of protozoan sterol 14α-demethylases (23, 24) but that none of them inhibit human CYP51 (13). In contrast, many azole-based drugs are known to inhibit (or serve as the substrates for) the human liver drug-metabolizing P450s (families CYP3, CYP2, and CYP1), which can cause drug/drug interaction problems especially if patients are on multiple medications (Intensive Care Unit, AIDS, anticancer chemotherapy) (25). As a result, the systemic use of ketoconazole, one of the major clinical antifungals for more than 40 years, is now limited, mainly due to its high potential for liver injury (www.fda.gov).
New, safer, and more efficient CYP51 inhibitors are highly needed, and the issue is becoming even more urgent as the number of drug-resistant clinical isolates of C. albicans and other pathogenic species of Candida continues to grow (26). In the absence of biochemical and structural information on the target enzyme, however, all of the current clinical antifungal azoles have been discovered empirically by monitoring the effects of a large number of compounds on fungal cell growth. This process has had low efficiency, resulting in slow progress, and eventually led to the loss of interest of many major pharmaceutical companies in the development of new antifungals (27).
Here we describe enzymatic properties of C. albicans CYP51, including substrate binding and catalytic parameters, compare inhibitory potencies of clinical antifungal azoles, and report the first X-ray structures of this enzyme in complexes with posaconazole and a new tetrazole-based drug candidate VT-1161 (oteseconazole: (R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol) (28), which has successfully completed phase 2b clinical trials (29).
Results
General issues regarding C. albicans CYP51
Similar to humans and opposite to some filamentous fungi (e.g. genus Aspergillus (30)), C. albicans has only one CYP51 gene (also known as ERG11). It is located on chromosome 5 and, as do the vast majority of the CYP51 genes, encodes a “B-type” enzyme (with an invariant proline (Pro-375 in C. albicans) instead of a serine that occupies this position in the A-type sterol 14α-demethylases (31)). The C. albicans CYP51 consists of 528 amino acids (62 kDa) (Fig. 2), including the 48-amino acid-long N-terminal membrane anchor sequence. The sequence identity with the orthologs from Aspergillus fumigatus is 45 and 43% (B- and A-type, respectively), 32% with human, 25% with Trypanosoma cruzi/Trypanosoma brucei, and 24% with Leishmania infantum. The family Trypanosomatidae is proposed to have derived ∼1.5 billion years ago (32), and the evolutionary distance between the Candida and Aspergillus genera has been estimated at ∼1 billion years (33).
Figure 2.

Amino acid sequence alignment of six structurally characterized eukaryotic sterol 14α-demethylases. The secondary structural elements of C. albicans (3tz1) and T. brucei (3g1q) enzymes are indicated above and below the sequences, respectively. The 34 residues invariant across the whole CYP51 family are marked with blue triangles. CYP51 family signature motifs (signature 1 and signature 2) are underscored (blue line). Black rectangles mark two phyla-specific segments: αG (protozoa) and β5 hairpin (fungi). Sequence alignment was generated in ClustalW, and secondary structural information was added in ESPript.
Optical properties
Like all other heterologously expressed CYP51 orthologs (13, 30, 34–37), the C. albicans enzyme was purified in the oxidized (Fe3+) low-spin hexacoordinate water-bound state, with a Soret band maximum at 417 nm. The reduced difference CO-binding spectrum had a λmax at 446 nm, with no observable trace of the denatured cytochrome P420 form (Fig. 3).
Figure 3.
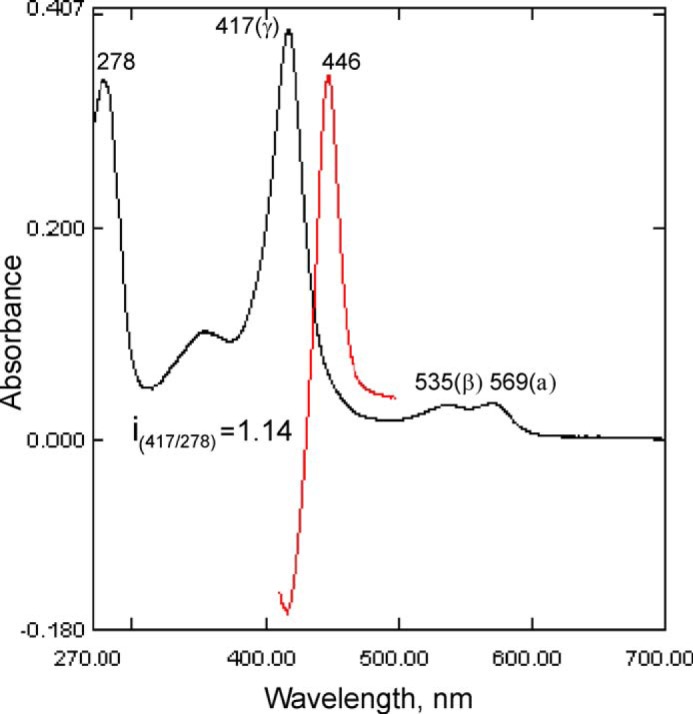
Absorbance spectra of C. albicans CYP51. Absolute (black) and difference CO-binding (red) spectra, P450 concentration 3.3 μm, optical path length 1 cm.
Binding of substrates
The binding of the substrates produced a typical “type 1” spectral response, reflecting the effects of the sterols on the heme iron spin-state equilibrium (Fig. 4A). The greatest low- to high-spin transition (31%) was induced by the natural C. albicans CYP51 substrate lanosterol, followed by eburicol (the preferred substrate of filamentous fungi (30) and a protozoan parasite, T. cruzi (36)) at 27%. The C4-monomethylated sterols obtusifoliol (the CYP51 substrate in plants) and C4-norlanosterol (T. brucei and Leishmania (37)) produced only 13 and 11% of the high-spin (substrate-bound) form. The amplitudes of the response correlated well with the apparent binding affinities (Fig. 4C), so that the differences in the apparent binding efficiencies (high-spin content/Kd) were ∼2-, 5-, and 7-fold, respectively. In our experience, this is the first example of a CYP51 enzyme displaying a clear preference toward lanosterol for binding. When the titration experiments were performed in the presence of a detergent (0.1% Triton X-100 (w/v), ∼7 × cmc) versus the “standard” substrate titration buffer (50 mm potassium phosphate (pH 7.4), containing 200 mm NaCl and 0.1 mm EDTA), the portion of the substrate-bound CYP51 (low- to high-spin transition in the heme iron) was substantially larger for all tested sterols (Fig. 4B), although lanosterol still produced the highest response (44% of water replacement). Moreover, in the presence of the detergent an apparent binding cooperativity was observed in all four cases (Fig. 4D). Because the same effect was observed during the titration of C. albicans CYP51 in the presence of n-tridecyl-β-d-maltoside, the detergent used for gel filtration and co-crystallization (data not shown), we associate this altered response with the monomeric state of the protein, although any physiological relevance is unknown.
Figure 4.

Spectral response of C. albicans CYP51 to the addition of the sterol substrates. Type 1 shift (red line) in the absorbance spectra of C. albicans CYP51 (1.9 μm) upon titration with the four CYP51 sterol substrates is shown. Absolute (top) and difference (bottom) absorbance spectra are shown. A, titration under standard substrate titration conditions (50 mm potassium phosphate buffer (pH 7.4), containing 200 mm NaCl and 0.1 mm EDTA). B, in the presence of 0.1% Triton X-100 (v/v). The titration range was 0.5–7 μm; the titration step was 0.5 μm, and the optical path length was 1 cm. C, titration curves of A; D, titration curves from B. The high-spin form content (the portion of the substrate-bound CYP51 molecules) and the apparent Kd values were calculated as described under “Experimental procedures.”
Catalytic activity and steady-state kinetic parameters
Because lanosterol produced the strongest spectral response in C. albicans CYP51 and serves as the substrate for this enzyme in vivo, it was used in subsequent experiments. The experimentally observed maximal catalytic turnover number was 28 nmol/nmol/min (at 25–35 μm lanosterol) (Fig. 5). The time course and Michaelis-Menten curves indicate that C. albicans CYP51 is stable under the reaction conditions. The steady-state kinetic parameters were determined as follows: kcat 33 min−1, Km 6.3 μm, and kcat/Km 5.2 min−1 μm−1.
Figure 5.

Enzymatic activity of C. albicans CYP51. A, time course of lanosterol conversion (37 °C, 0.5 μm P450, 1 μm CPR, 25 μm lanosterol). Inset, HPLC profile of sterols extracted after a 1.5-min reaction. B, steady-state kinetics (1-min reaction). The experiments were performed in triplicate, and the results are presented as means ± S.E.
Inhibition with clinical antifungal azoles
To compare the potencies of clinical antifungal azoles for inhibition of C. albicans CYP51 activity, we used 60-min reactions because this longer reaction time affords higher sensitivity in these assays (13, 23, 30). The molar ratio of enzyme/inhibitor/lanosterol was 1:2:50. As shown in Fig. 5A, under these conditions C. albicans CYP51 converted all the substrate into the product in the absence of an inhibitor. Among the tested compounds, the weakest inhibitory effect (54% inhibition of the substrate conversion) was observed with fluconazole, which serves as the first-line drug for systemic anticandidiasis (Fig. 6). Two topical antifungals, clotrimazole and miconazole, were stronger and similar to each other in potency (78 and 79% inhibition). The inhibitory effects of voriconazole, ketoconazole, and itraconazole were 84, 85, and 91%, respectively. Posaconazole and the new clinical drug candidate, VT-1161, showed the highest inhibitory potencies (98% inhibition) and therefore were selected for co-crystallization. The spectral responses of C. albicans CYP51 to the binding of posaconazole and VT-1161 (Kd 81 and 21 nm, respectively) are shown in Fig. 7. In general (except for VT-1161), the strength of the inhibition correlated with the length of the inhibitor molecules.
Figure 6.

Comparative inhibitory effects of clinical antifungal drugs on the activity of C. albicans CYP51. The molar ratio of enzyme/inhibitor/substrate was 1:2:50, with the P450 concentration at 0.5 μm (37 °C, 60-min reaction). The experiments were performed in triplicate, and the results are presented as means ± S.D.
Figure 7.

Spectral responses of C. albicans CYP51 to the addition of the triazole posaconazole and tetrazole VT-1161. Type 2 shifts are shown in the Soret band maximum in the absolute (top, red line) and difference (bottom) absorption spectra. The P450 concentration was ∼0.5 μm; the titration range was 0.1–0.8 μm; the titration step was 0.1 μm; and the optical path length was 5 cm. The titration was conducted in 50 mm potassium phosphate buffer (pH 7.4) containing 200 mm NaCl, 0.1 mm EDTA, and 0.1% Triton X-100 (v/v). Apparent Kd values were calculated as described under “Experimental procedures.” A, posaconazole. B, VT-1161. Insets, titration curves.
Structural analysis
C. albicans CYP51 complexes with posaconazole and VT-1161 were crystallized in the same monoclinic C121 space group, and the structures were refined to 2.86 and 2.00 Å (Table 1). In both cases, the asymmetric unit consists of two monomers related via a non-crystallographic 180° rotation axis, with the membrane-bound segments of the P450 molecules facing each other, and helices A and A′ being involved in crystal packing interactions (Fig. 8A). The root-mean-square (r.m.s.) deviations for the Cα atom positions between the two monomers (within the same asymmetric unit, “molecular breathing”) are 0.55 and 0.51 Å in the posaconazole and VT-1161 complexes, respectively. The overall structure exhibits the characteristic P450 fold, and the prosthetic heme group forms contacts with the six protein residues, Cys-470 serving as the fifth (proximal) axial ligand to the iron and the other five forming six hydrogen bonds with the protoporphyrin IX propionates (Tyr-132, Lys-143, and His-468 (ring A) and Tyr-118 and Arg-381 (ring D), shown in Fig. 12A), as discussed below under “Discussion”).
Table 1.
Data collection and refinement statistics
| Complex | C. albicans CYP51-posaconazole | C. albicans CYP51·VT-1161 |
|---|---|---|
| Data collection | ||
| Wavelength, Å | 0.97856 | 0.97849 |
| Space group | C121 | C121 |
| Cell dimensions | ||
| a, b, c, Å | 179.59 72.91 79.71 | 177.64 71.44 79.19 |
| α, β, γ, ° | 90.00 96.13 90.00 | 90.00 96.63 90.00 |
| Molecules/asymmetric unit | 2 | 2 |
| No. of reflections | 23,729 | 65,813 |
| Resolution (outer shell), Å | 50.39–2.86 (2.93–2.86) | 49.61–2.00 (2.05–2.00) |
| Rmerge (outer shell) | 0.105 (0.787) | 0.075 (0.691) |
| I/σ (outer shell) | 9.7 (2.18) | 12.8 (2.0) |
| Completeness (outer shell), % | 99.4 (99.7) | 98.8 (96.4) |
| Redundancy (outer shell) | 4.8 (4.8) | 5.0 (4.6) |
| Refinement | ||
| Phasing method | Molecular replacement (PDB code 4UYM model) | Molecular replacement (PDB code 5FSA model) |
| R-work | 0.236 | 0.218 |
| R-free | 0.249 | 0.225 |
| r.m.s. deviations from ideal geometry | ||
| Bond lengths, Å | 0.001 | 0.002 |
| Bond angles, ° | 1.29 | 0.99 |
| Ramachandran plot | ||
| Residues in favorable/allowed regions, % | 97.0/99.5 | 97.6/100 |
| Outliers, % | 0.5 | 0 |
| Number of atoms (mean B-factor, Å2) | 8161 (71.4) | 8337 (42.1) |
| No. of residues per molecule | A/B | A/B |
| Protein (mean B-factor, Å2) | 484 (71.9)/483 (73.9) | 484 (41.1)/484 (42.7) |
| Heme (mean B-factor, Å2) | 1 (46.6)/1 (51.0) | 1 (26.2)/1 (25.4) |
| Ligand (mean B-factor, Å2) | 1 (76.6)/1 (88.9) | 1 (37.0)/1 (40.5) |
| Water (mean B-factor, Å2) | 61(53.3)/66 (51.6)/ | 143 (44.5)/173 (45.4) |
| Wilson B-factor, Å2 | 64.32 | 27.12 |
| PDB code | 5FSA | 5TZ1 |
Figure 8.
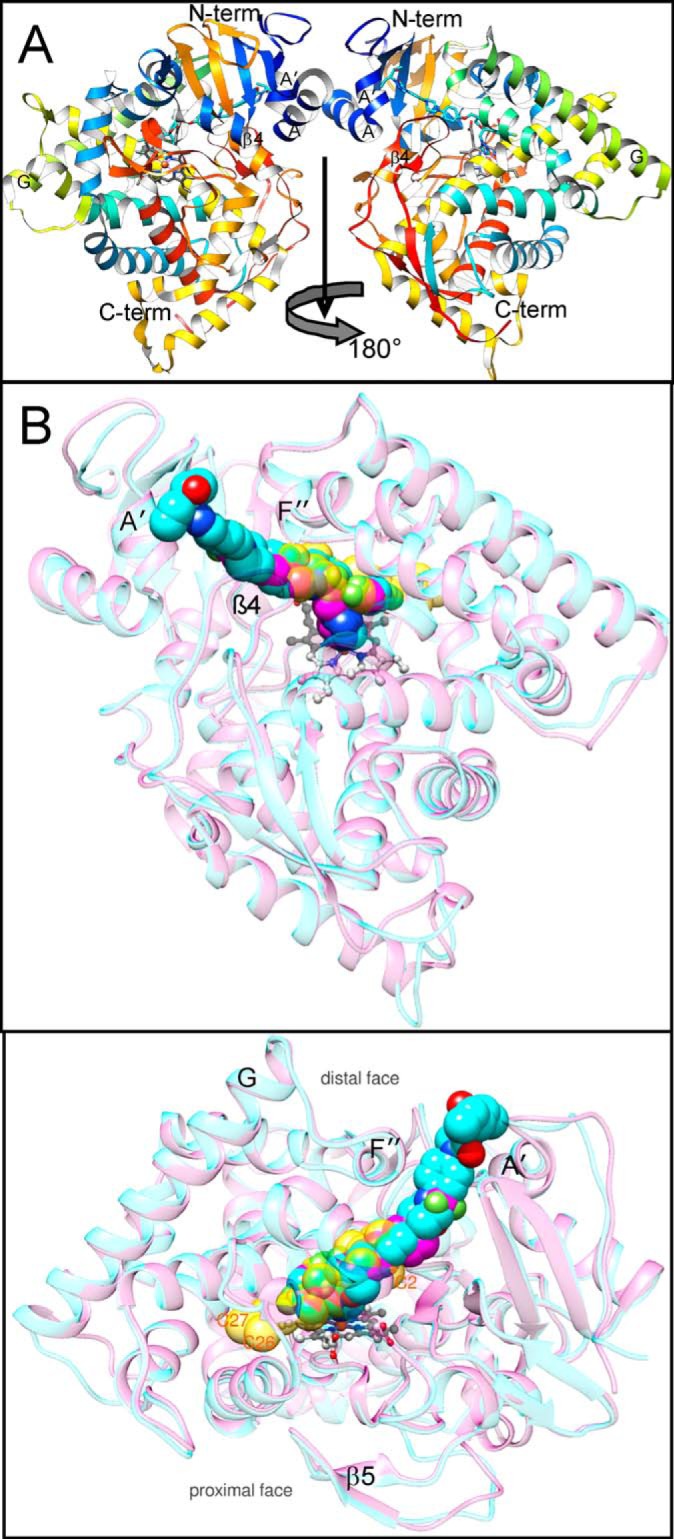
Complexes of C. albicans CYP51 with posaconazole and VT-1161, overall view of the structures. A, asymmetric unit, a view along the rotation axis that runs from top to bottom and relates two monomers. The protein main chain is shown in ribbon representation colored by secondary structure succession from the N (blue) to C (red) termini. Helices A, A′, G, the β4 hairpin, the N terminus, and the C terminus are marked. B, superimposed complexes with posaconazole (cyan) and VT-1161 (magenta). The protein main chains are shown as semitransparent ribbons of the corresponding color. The heme (gray) is depicted in a ball-and-stick representation. The inhibitors and the molecule of the sterol substrate (semitransparent gold, modeled from the costructure with T. brucei CYP51 (PDB code 3P99) are presented as spheres. One atom of the nucleus (C2) and two side chain atoms (C26 and C27) of the sterol molecule are marked as references. Distal (top) and 180°-rotated upper (bottom) view.
Figure 12.

Heme support and proton delivery route in CYP51. Six protein residues located within 3 Å of the heme are noted. A, C. albicans; B, T. cruzi CYP51. The H-bonds with the heme propionates are displayed as red dashes, and the iron-coordinated cysteine is seen at the bottom. C, fragment of CYP51 sequence alignment showing the porphyrin ring D supporting lysine in fungi/animals (Lys-143 in C. albicans) versus located one turn upstream of the C-helix arginine (Arg-124) in protozoa. D, surface-exposed Asp-225 in C. albicans CYP51, via the CYP51 signature His-310 and “conserved P450 threonine” (Thr-311), supplies protons to the oxygenated heme iron. E, heme support in human P450scc (CYP11A1 (PDB code 3N9Y)) is provided for comparison. F, proton delivery route in P450cam (CYP101A1 (PDB code 1DZ4)) (56) presented as a comparison. Overall, the heme support in CYP11A1 is probably stronger, because, opposite to CYP51, all the H-bonds here are formed between the N and O atoms (salt bridges). The charges in the conserved salt bridge pair involved into the proton delivery route in CYP101 (as well as in most other CYP families) are reversed.
No major ligand accommodating rearrangements are seen in the C. albicans CYP51 structure upon binding of posaconazole versus VT-1161, and the r.m.s. deviation for all Cα atoms between the two complexes is only 0.69 Å. Moreover, when presented as spheres (van der Waals radii of atoms), the posaconazole and VT-1161 molecules superimpose very well, both acquiring the conformation that is adjusted to the shape of the C. albicans CYP51 active site and occupying an area to accommodate the nucleus and the proximal half (1 isoprenoid unit) of the arm of the sterol substrate (Fig. 8B) (21).
The fluorinated β-phenyl ring of each inhibitor (see also Fig. 9) is buried deep in the substrate-binding cavity, and the basic azole ring nitrogen forms the sixth (distal) axial coordination bond with the heme iron. As expected, the long arm resides in the substrate access channel, reaching the channel entrance (VT-1161) or even surpassing it and being exposed above the protein surface (posaconazole). The channel entrance is gated by the A′- and F″-helices and the β4-hairpin (Fig. 8B).
Figure 9.

Posaconazole and VT-1161 bound in the C. albicans CYP51 active site. The 2Fo − Fc omit electron density maps (gray mesh) of posaconazole (cyan) and VT-1161 (magenta) are contoured at 1.2 and 1.5 σ, respectively. A, posaconazole; B, VT-1161. The inhibitor contacting residues (within 4.5 Å, stick representation) and the corresponding secondary structural elements (ribbon representation) of C. albicans CYP51 are depicted in green and marked (see also Table 2). The heme is depicted as gray spheres. H-bonds are shown as red dashes. The heme-coordinating nitrogen atoms in the structural formulas of the inhibitors are circled.
As the shorter structure, VT-1161 interacts with 22 amino acid residues of C. albicans CYP51 (Table 2 and Fig. 9B). Its potency in inhibiting the enzyme activity, however, is enhanced by the H-bond between the imidazole ring of His-377 and the trifluoroethoxyphenyl oxygen of VT-1161 (1–5 kcal/mol, which is at least 1 order of magnitude stronger than a van der Waals interaction). Posaconazole (Fig. 9A) does not form any H-bonds with the protein but has contacts with a set of 28 residues, which in addition to those interacting with VT-1161 includes four more residues from helix A′ (Phe-58, Ala-61, Ala-62, and Gly-65), one more residue from the β4 hairpin (Ser-506), and one residue from the β1–2 turn (Leu-88) (Table 2). All of these side chains line the surface of the C. albicans CYP51 substrate channel in close proximity to the entry.
Table 2.
Ligand contacting residues (<4.5 Å) in C. albicans and in T. cruzi CYP51 structures
| Secondary structural element | Drug |
|||
|---|---|---|---|---|
| Posaconazole |
VT-1161 |
|||
| T. cruzi, PDB code 3K1O | C. albicans PDB code 5FSA | T. cruzi, PDB code 5AJR | C. albicans PDB code 5TZ1 | |
| Helix A′ | Phe-58 | |||
| Ile-45 | Ala-61 | |||
| Val-46 | Ala-62 | |||
| Phe-48 | Tyr-64 | Phe-48 | Tyr-64 | |
| Gly-49 | Gly-65 | |||
| β1-β2 turn | Leu-88 | |||
| Helix B′ | Tyr-103 | Tyr-118 | Tyr-103 | Tyr-118 |
| Leu-121 | Leu-121 | |||
| Met-106 | Thr-122 | Met-106 | Thr-122 | |
| Phe-110 | Phe-126 | Phe-110 | Phe-126 | |
| B″ helical turn | Ile-131 | Ile-131 | ||
| Tyr-116 | Tyr-132 | Tyr-116 | Tyr-132 | |
| Helix C | Leu-127 | |||
| Helix F″ | Phe-228 | Phe-228 | ||
| Pro-210 | Pro-230 | Pro-210 | Pro-230 | |
| Ala-211 | ||||
| Val-213 | Val-213 | |||
| Phe-214 | Phe-233 | Phe-214 | Phe-233 | |
| Helix I | Ala-287 | Gly-303 | Ala-287 | Gly-303 |
| Ile-304 | Ala-288 | Ile-304 | ||
| Phe-290 | Phe-290 | |||
| Ala-291 | Gly-307 | Ala-291 | Gly-307 | |
| Gly-308 | Gly-292 | Gly-308 | ||
| Thr-295 | Thr-311 | Thr-295 | Thr-311 | |
| K/β1–4 loop | Leu-356 | Leu-376 | Leu-356 | Leu-376 |
| Leu-357 | His-377 | Leu-357 | His-377, H-bond | |
| Met-358 | Ser-378 | Met-358 | Ser-378 | |
| β1–4 strand | Met-360 | Phe-380 | Met-360 | Phe-380 |
| β4 hairpin | Tyr-457 | Tyr-505 | Tyr-505 | |
| His-458 | Ser-506 | |||
| Thr-459 | Ser-507 | Ser-507 | ||
| Met-460 | Met-508 | Met-460 | Met-508 | |
The length of the Fe-N coordination bond is 2.1 Å in the posaconazole complex and 2.2 Å in the VT-1161 complex, reflecting the higher basicity of the N4 atom in the 1,2,4-triazole ring of posaconazole in comparison with the N4 atom in the 1,2,3,4-tetrazole ring in VT-1161 (38) and supporting higher selectivity (39) of VT-1161 toward the target CYP51 enzymes versus human “drug-metabolizing” P450s (28). This result also corresponds to the shorter red shift (39) induced by VT-1161 in the C. albicans CYP51 Soret band maximum (417–421 nm versus 417–423 nm in the case of posaconazole (see Fig. 7)).
Notably, none of the inhibitors disrupted the Tyr-118 or Tyr-132 H-bond with the heme propionates in the C. albicans CYP51 (seen in Fig. 9). These two tyrosine side chain hydroxyl-based hydrogen bonds with the heme appear to be a characteristic feature of the CYP51 family (CYP51 family signature 1 (Fig. 2)), and they have been found broken in many protozoan CYP51·inhibitor complexes (e.g. T. brucei, VNI ((R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imida-zol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide) (40), VFV ((R)-N-(1-(3,4′-difluorobiphenyl-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide), and VNT ((R)-N-(1-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide) (41) (Tyr-103 corresponds to C. albicans Tyr-118); and T. cruzi, VNF ((R)-N-(2-(1H-imidazol-1-yl)-1-phenylethyl)-4′-chlorobiphenyl-4-carboxamide) (24), UDD (N-[4-(trifluoromethyl)phenyl]-N-[1-[5-(trifluoromethyl)-2-pyridyl]-4-piperidyl]pyridin-3-amine) (39), NEE (1-(3-(4-chloro-3,5-dimethylphenoxy)benzyl)-1H-imidazole) (42), LFT ((S)-1-(4-chlorophenyl)-2-(1H-imidazol-1-yl)ethyl-4-isopropylphenylcarbamate), and LFS ((S)-1-(4-fluorophenyl)-2-(1H-imidazol-1-yl)ethyl-4-isopropylphenylcarbamate) (43) (Tyr-116 corresponds to C. albicans Tyr-132), as well as in the complexes of A. fumigatus CYP51B with voriconazole and VNI (30) (Tyr-122 corresponds to Tyr-118 in C. albicans)).
Comparison of the C. albicans CYP51 structures with the posaconazole and VT-1161-bound T. cruzi CYP51
In the fungal and protozoan CYP51 structures (protein sequence identity 25%, r.m.s. deviation for the Cα atoms 1.9 Å), posaconazole and VT-1161 are positioned very similarly (Fig. 10). Small alterations in the inhibitor conformations are defined by the differences in the topologies of the substrate binding cavities of these enzymes and are due to the following: 1) the side chains of the phyla-specific residues that shape the cavity surface, e.g. Tyr-64 versus Phe-48, Leu-121 versus Ile-105, Thr-122 versus Met-106, His-377 versus Leu-357, and Ser-378 versus Met-376 in C. albicans and T. cruzi CYP51, respectively (Fig. 10 and Table 2); and 2) the FG arm that in the protozoan structure is ∼6 Å longer: thus, Phe-233 in C. albicans corresponds to T. cruzi Phe-214 in the sequence alignment (Fig. 2) but in the structure it aligns with T. cruzi Val-213 (Fig. 10A). Interestingly, although no H-bond is formed between VT-1161 and T. cruzi CYP51 (it has a non-polar leucine (Leu-357) instead of basic histidine (His-377) in this position (Fig. 10B)), and the number of inhibitor-contacting residues is also smaller (19 versus 22 (Table 2)), the inhibitory effect of VT-1161 on the T. cruzi CYP51 enzyme is still very potent (94% inhibition under the same conditions (38)). The inhibitory effect of posaconazole on T. cruzi CYP51 activity is 100% (24). We propose that the stronger inhibition is connected with the energetically richer FG-loop (the lid covering the entrance into the substrate access channel) in the protozoan CYP51 structures.
Figure 10.

Superimposition of C. albicans and T. cruzi (gray) CYP51 complexes with posaconazole (PDB code 3K1O) and VT-1161 (PDB code 5AJR). A, posaconazole; B, VT-1161. Some phyla-specific residues that line the enzyme substrate-binding cavity, altering the conformation of the inhibitors, are shown as examples. A complete list of the corresponding ligand-contacting residues aligned in the fungal and protozoan CYP51 structures is shown in Table 2. Insets, C. albicans CYP51 in a surface representation (same view). The C-atoms of posaconazole and VT-1161 are colored in cyan and magenta, respectively.
Comparison with the VNI-bound A. fumigatus CYP51B structure
Although the structures of C. albicans and A. fumigatus CYP51 (protein sequence identity 45% and active site identity 65%) display even higher similarity, the r.m.s. deviation for the Cα atoms being only 1.3 Å, the orientation of VNI in the A. fumigatus complex substantially differs from that of posaconazole (or VT-1161) (Fig. 11) in C. albicans CYP51. It appears that the altered binding modes of the inhibitors reflect the best fit of each ligand molecule to the topology of the enzyme binding cavity, which, again, is being achieved without any considerable rearrangements in the protein backbone, except for some changes in the spatial positions of the FG-loop (primarily the F″ area), although the side chains of the identical residues in the two proteins often adopt different conformations (Fig. 11). Thus, regardless of the high structural similarity of the CYP51 enzymes, determination of CYP51-inhibitor co-structures remains highly desirable, especially when dealing with novel inhibitory scaffolds.
Figure 11.
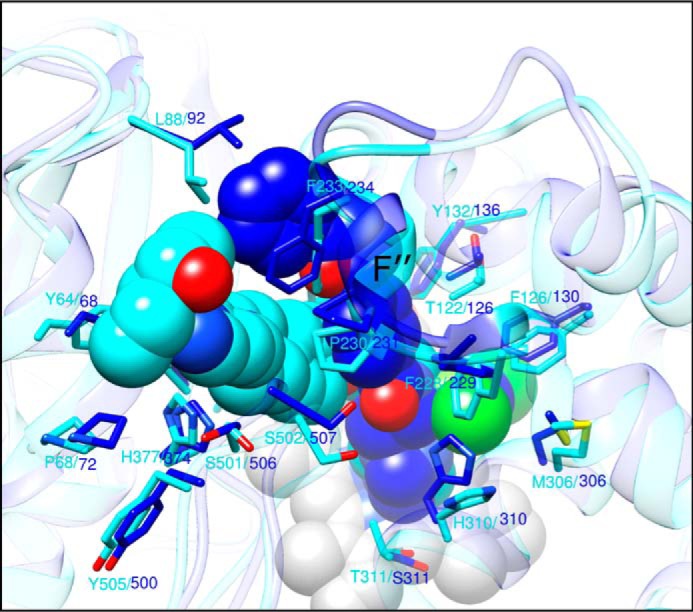
Superimposition of C. albicans CYP51 and A. fumigatus CYP51B (PDB code 4UYL) complexes with posaconazole (cyan) and VNI (blue), respectively. The heme (gray), posaconazole, and VNI are depicted as spheres; the binding cavity-forming residues, conserved in both proteins, are shown in stick representation and labeled.
Discussion
The structures of C. albicans CYP51 support our hypothesis about high rigidity of the substrate-binding cavity as a molecular basis for the exclusive functional conservation of this P450 enzyme across phylogeny (20, 24, 30, 37, 40, 41), the most conserved area being the B′-helix/BC-loop (CYP51 signature 1 (Fig. 2)), which interacts with the β-surface of the sterol substrate (21): the r.m.s. deviation values for the Cα atoms here between the posaconazole and VT-1161-bound C. albicans CYP51 structures are 0.26 ± 0.1 Å, and increase only to 0.53 ± 0.3 and 0.82 ± 0.3 Å between e.g. C. albicans/A. fumigatus and C. albicans/T. cruzi CYP51, respectively. The structures also confirm two CYP51-characteristic features that distinguish sterol 14α-demethylases from other P450s and most likely play a role in their susceptibility to inhibition. First, as mentioned above, there is the heme support from the protein moiety, which involves two comparatively weaker hydrogen bonds between the porphyrin ring propionates and the CYP51-family characteristic tyrosines (Fig. 12, A, B, and E) and might influence the redox potential of the CYP51 heme iron. Second, there is the “reverse” proton delivery pair, His-310 (helix I, CYP51 signature 2)–Asp-226 (helix F) in C. albicans (Fig. 12, D and F), because in other P450 families the surface-exposed residue in helix F is usually positively charged and forms a salt bridge with an I-helix aspartate or glutamate (20). It remains to be determined whether switching the charges in this salt bridge pair would influence the CYP51 enzymatic properties.
Structural comparison of C. albicans CYP51 with the CYP51 orthologs from other biological kingdoms reveals three phylum-distinguishing features, which are likely to be of functional importance: 1) the β5-hairpin (the segment unique for fungal CYP51 and so far not observed in any other P450 folded proteins); 2) the I-helix (“undistorted” in fungal and protozoan versus “broken” in human CYP51); and 3) the FG-loop (shorter and “single-helical” in fungi and human versus “two-helical” in protozoan CYP51) (Fig. 13). The β5-hairpin (Fig. 13A) is formed by the fungi-specific insert between the meander and the heme bulge regions in the P450 sequence (25 amino acid residues in C. albicans CYP51 (see Fig. 2)). In the structure, the β5-hairpin lies above the heme bulge covering the positively charged residues on the proximal surface of the P450 molecule and thus may play a role in the interaction of fungal CYP51 with NADPH-cytochrome P450 reductase (CPR), most likely by moving aside so that the positive charges on the proximal P450 surface become available for the interaction with the negatively charged surface of CPR (30), although it may also provide some additional P450/CPR interactions, perhaps making the redox complex stronger. Helix I is the core structure in the P450 fold. It runs just above the right site of the distal surface of the heme, parallel to the heme plane, carries two proton delivery residues (His-310 and Thr-311 in C. albicans), and forms a wall in the substrate-binding cavity (substrate recognition site (SRS) 4 (44)). It also holds the azole and halogenated β-phenyl rings of the inhibitors. In the CYP51 enzymes, the I-helix is positioned 1–2 Å closer to the heme iron, on the average, than it is in other P450 structures (40). Similar to the protozoan and A. fumigatus orthologs and opposite to human CYP51, the I-helix in the C. albicans CYP51 structure is “whole” (Fig. 13B), supporting the notion that the disordered loop-like region in this portion of the human CYP51 active site (the lack of the main chain α-helical H-bonding in the middle of the I-helix) might be the reason for its drastically weaker susceptibility to inhibition (13). In comparison with the protozoan enzymes, the FG-loop in C. albicans CYP51 is shorter and carries only one α-helical insert (F″ versus F″ and G′ in the protozoan structures) (Fig. 13C). The single-helical loop is energetically weaker and therefore might be more flexible. This explains why the length of the drug molecule would generally correlate with its potency to inhibit C. albicans CYP51 (see Fig. 6), because the longer structures (like posaconazole) can form additional contacts around the substrate access channel and thus stabilize the closed state of the entry. Taken together, these features provide a molecular background for the experimentally observed phylum-dependent changes in CYP51 sensitivity to inhibition, protozoan > fungal ≫ human (mammalian).
Figure 13.

Three phylum-specific segments (magenta) mapped on the structure of C. albicans CYP51 (semitransparent magenta). A, β5 hairpin is unique for fungal CYP51 and forms the proximal surface of the P450 molecule. B, similar to protozoan and opposite to human CYP51, the I-helix in the C. albicans enzyme does not have a loop-like region in the middle portion. C, FG-loop in C. albicans CYP51 is shorter and has only one α-helical region (F″), and the G′-helix is unique for the protozoan enzymes. Superimposition with the structure of T. brucei CYP51 (PDB code 3G1Q) is shown in semi-transparent gold. View is from distal face of the P450 molecule.
Prolonged/frequent use of any drug, including antifungal azoles, often causes drug resistance. Because fluconazole has served as the major drug for treatment of C. albicans infections for >30 years, data on fluconazole-resistant clinical isolates of C. albicans are abundant (26, 45–48). Different mechanisms of acquired azole resistance have been proposed (48, 49), and mutations in the C. albicans enzyme are listed as one of them, although their contribution to the whole phenomenon remains unclear. However, taking into account the high structural similarity of CYP51 across phylogeny and the ability of these enzymes to preserve their conserved biological function at only 34 invariant amino acid residues, the frequency of such mutations is unlikely to be high. Indeed, as has been summarized recently, most of the 140 mutations reported in CYP51 from fluconazole-resistant strains of C. albicans were also found in drug-sensitive strains and therefore are probably irrelevant to resistance mechanisms (46). The C. albicans CYP51 structure shows that of 10 mutations, which so far have not been found upon sequencing of drug-sensitive strains (46), five (Y132H, Y132F, K143R, G307S, and S405F) involve residues that are exposed in the active-site cavity, suggesting that they might potentially influence fluconazole binding affinity (Fig. 14A). Thus, Tyr-132 (ηB″) and Lys-143 (αC) form the H-bonds with the heme ring D propionate. Changes in the H-bond strength (Y132H and K143R) or abolishing an H-bond (Y132F) might possibly alter the redox potential of the heme iron or otherwise affect the ability of the iron to coordinate the basic nitrogen of the azole ring. The G307S mutation (helix I) might slightly decrease the binding cavity volume. It is not clear, however, how this would influence the interaction of the mutant enzyme with the substrate. Ser-405 (β1–3) lies outside the fluconazole-contacting area, yet it cannot be excluded that the bulky phenylalanine inserted here (S405F) might affect fluconazole binding by weakening the β1–3/β1–4 (SRS5) interaction. The other four mutations (Y447H, G448E, G448V, and G450E) are all located on the tip of the β5 hairpin (Fig. 14B), on the proximal surface of the CYP51 molecule. It is conceivable that these mutations might potentially contribute to azole resistance indirectly, by influencing the CYP51 interaction with CPR, and therefore these mutants could be an interesting subject for further studies. Finally, although Asn-136 is also exposed to the proximal surface of the CYP51 molecule, its mutation to tyrosine (Fig. 14B), in our opinion, is unlikely to influence the C. albicans CYP51 drug sensitivity. All protozoan CYP51 enzymes have a tyrosine residue in this position (Fig. 2), and yet their susceptibility to inhibition is on the average higher than that of the fungal orthologs. These are only speculations, and the effects of mutations must be evaluated experimentally before reaching conclusions. If drug resistance of a mutant is confirmed, such mutations/sequence variations could serve as markers of CYP51-associated fluconazole resistance in C. albicans and in other fungal pathogens. We propose that development of novel, alternative, and more potent CYP51 inhibitory scaffolds should be helpful in resolving the problem, particularly because activation of the drug efflux transporters has been experimentally proven to be a prime mechanism of fluconazole resistance in C. albicans (45, 48, 49).
Figure 14.

Amino acid substitutions in CYP51 from fluconazole-resistant clinical isolates of C. albicans that have not been found in fluconazole-sensitive strains, mapped on the C. albicans CYP51 structure. A, mutations within the substrate-binding cavity, distal view. B, mutations on the proximal P450 surface, proximal view. The side chains of the wild-type residues are shown as green lines, and the side chains of the mutant residues are presented as tan sticks. Fluconazole (red sticks) was modeled from the complex with Leishmania infantum CYP51 (PDB code 3L4D).
To summarize, C. albicans CYP51 prefers its natural substrate lanosterol, has relatively high catalytic efficiency, and in general is more strongly inhibited by those clinical azoles that have a longer side chain arm. VT-1161 represents a successful exception because its interaction with the enzyme is strengthened by the H-bond with His-377. The fact that this His residue is conserved in all CYP51 enzymes from the Candida genera explains the high potency of VT-1161 against Candida krusei and Candida glabrata (50), two pathogens that are intrinsically resistant to fluconazole. Thus, the X-ray structures of C. albicans CYP51 open new opportunities for more efficient, structure-based design of novel, more potent inhibitors. The consideration of common fungus-distinguishing structural characteristics should afford easier development of the antifungal drugs with the broad-spectrum activity, which is very important as thorough diagnostic analysis can take valuable time, and the outcome of systemic fungal infections strongly depends on how quickly the treatment has been initiated (7).
Experimental procedures
Reagents
Voriconazole, ketoconazole, itraconazole, and posaconazole were purchased from Santa Cruz Biotechnology, and fluconazole, clotrimazole, and miconazole were from ICN Biomedicals. VT-1161 was provided by Viamet Pharmaceuticals (Durham, NC). Hydroxypropyl-β-cyclodextrin (HPCD) was from CTD (Alachua, FL). DEAE- and CM-Sepharose were from GE Healthcare; Ni2+-nitrilotriacetate (NTA)-agarose was from Qiagen, and the Superdex 200 10/300GL column was from GE Healthcare. n-Tridecyl-β-d-maltoside and other chemicals were purchased from Sigma.
Proteins
Full-length C. albicans CYP51 and rat CPR were expressed in Escherichia coli and purified as described previously (35). Full-length C. albicans CYP51 was used for all functional studies (spectral titration, enzymatic activity, and inhibition). For crystallization purposes, the C. albicans protein was truncated as follows: the 48-amino acid membrane anchor sequence at the N terminus (up to the conserved CYP51 proline, Pro-49, in C. albicans CYP51) was replaced with the 6-amino acid sequence fragment MAKKTP-, using the upstream primer5′-GCGCATATGGCTAAGAAAACCCCGCCATTAGTGTTTTATTGGATTCC-3′ and the downstream primer 5′-CGCAAGCTTCTAGTGATGGTGATGAAACATACAAG-3′. The PCR was performed as described for A. fumigatus CYP51B (30) except that FailSafe PCR Premix D was used. The correctness of the insert was confirmed by DNA sequencing. For protein expression, the truncated CYP51 gene was subcloned into the pCW expression vector at NdeI (5′-) and HindIII (3′-) sites, and the plasmid was transformed into E. coli HMS174 (DE3) (Novagen)-competent cells. The expression conditions were the same as for the full-length protein.
Purification of C. albicans CYP51 for crystallization
The truncated C. albicans CYP51 (490 amino acid residues, 56 kDa) was solubilized from E. coli cells and purified in two steps, including affinity chromatography on Ni2+-NTA-agarose and gel filtration chromatography on Superdex 200 10/300GL as follows. All purification steps were done at 4 °C, and all buffers contained 0.1 mm phenylmethylsulfonyl fluoride and 0.1 mm dithiothreitol, which were added fresh daily. The pellet was homogenized in 200 mm potassium phosphate buffer (pH 7.4) containing 100 mm NaCl, 0.1 mm EDTA, 10% glycerol (v/v), and 0.1% Triton X-100 (v/v). The suspension was sonicated on ice (Sonic Dismembrator model 500, Thermo Fisher Scientific); Triton X-100 was added to 0.4% (v/v), and the mixture was stirred at 4 °C for 2 h. The solubilized protein was separated from the insoluble material by centrifugation at 150,000 × g for 40 min (Optima l-80 Ultracentrifuge, Beckman). The supernatant was frozen in liquid nitrogen and stored at −80 °C until use. Then it was thawed, diluted 2-fold with 20 mm potassium phosphate buffer (pH 7.4) containing 500 mm NaCl, 10% glycerol (v/v), and 2 m imidazole, and applied to an Ni2+-NTA-agarose column equilibrated with 100 mm potassium phosphate buffer (pH 7.4) containing 300 mm NaCl, 10% glycerol (v/v), 0.2% Triton X-100 (v/v), and 1 mm imidazole. The column was washed with 10 bed volumes of equilibration buffer and then with 20 bed volumes of 50 mm potassium phosphate buffer (pH 7.4) containing 500 mm NaCl, 10% glycerol (v/v), 10 mm imidazole, and 0.1% Triton X-100 (v/v), and then with 40 bed volumes of 50 mm potassium phosphate (pH 7.4) containing 500 mm NaCl, 10% glycerol, 10 mm imidazole, and 0.06 mm (2.5 × cmc) n-tridecyl-β-d-maltoside, and then with 10 bed volumes of the same buffer containing 20 mm imidazole. The P450 was eluted in the same buffer with a linear gradient of imidazole (20–180 mm); the fractions with a spectrophotometric index (A422/A280) of ≥0.7 were pooled and concentrated using an Amicon Ultra 50 K (Millipore) concentration device to ∼0.25 mm. Tris carboxyethylphosphine (TCEP) was added to a final concentration of 5.2 mm, and the solution was incubated on ice for 30 min and centrifuged at 16,000 × g for 5 min (TL-100 ultracentrifuge, Beckman Instruments). The supernatant was applied to the Superdex 200 10/300GL column, using an ÄKTA purifier system equipped with UNICORN 5.11 software (GE Healthcare). The column was equilibrated with 50 mm potassium phosphate buffer (pH 7.4) containing 150 mm NaCl, 10% glycerol (v/v), 0.06 mm n-tridecyl-β-d-maltoside, and 0.1 mm EDTA. The flow rate was 0.2 ml/min. The major peak was eluted in a 2-ml volume between 12.5 and 14.5 ml, and the fractions with spectrophotometric indices (A417/A280) of >1 were pooled and concentrated to about 400 μm P450. The absorption spectrum is shown in Fig. 3. TCEP was added (to 5.2 mm), and the protein was aliquoted, frozen in liquid nitrogen, and stored at −80 °C until use. The purity was verified by SDS-PAGE.
Spectral characterization
UV-visible absorption spectra were recorded at 20 °C using a dual-beam Shimadzu UV-240IPC spectrophotometer. The C. albicans CYP51 concentration was determined from the Soret band absorbance in the absolute spectrum, using an absolute molar extinction coefficient ϵ417 = 117 mm−1 cm−1 for the low-spin oxidized form of the protein or a difference molar extinction coefficient Δϵ(446–490) = 91 mm −1 cm−1 for the reduced carbon monoxide (CO) complex in the difference spectra (37). The spin state of the P450 samples was estimated from the absolute spectra as the ratio (ΔA393–470/ΔA418–470), with values of 0.4 and 2.0 corresponding to 100% low- and 100% high-spin iron, respectively (51).
Ligand binding affinities were measured by spectral titration. Of the five known CYP51 substrates (19), we tested four in this work (Fig. 4). Sterols induced a shift in the heme iron spin-state equilibrium toward the high-spin form (the shift in the Soret band maximum from 417 to 393 nm), reflecting the displacement of the heme-coordinated water molecule from the iron coordination sphere (termed a type 1 spectral response). For these experiments, sterols were added from 0.5 mm stock solutions in 45% HPCD (w/v) (35) to the sample cuvette (1-cm optical path length), and the same volume of HPCD was added to the reference cuvette. The P450 concentration was ∼2 μm, and the titration range was 0.5–7 μm. The apparent dissociation constants of the enzyme·substrate complex (Kd) were calculated in GraphPad Prism 6 software (La Jolla, CA) by fitting the data for the substrate-induced absorbance changes in the difference spectra Δ(A390 − A424) versus substrate concentration to a one site-total binding equation (binding − saturation). When the titration experiments were conducted in the presence of 0.1% Triton X-100 (v/v), the data were fit to a one site-specific binding model with a Hill slope equation (binding − saturation).
Binding of azole inhibitors caused a red shift in the Soret band maximum, from 417 to 421–423 nm (termed a type 2 spectral response), reflecting replacement of the water molecule in the (low-spin) iron coordination sphere with the stronger ligand (39). Posaconazole and VT-1161 were added from 0.2 mm stock solutions in (CH3)2SO to the sample cuvette (5-cm optical path length), and the same volume of (CH3)2SO was added to the reference cuvette. The P450 concentration was ∼0.5 μm, and the titration range was 0.1–0.8 μm. The apparent dissociation constants of the enzyme·ligand complex (Kd) were calculated in GraphPad Prism 6 software by fitting the data for the ligand-induced absorbance changes in the difference spectra Δ(Amax − Amin) versus ligand concentration to the quadratic Equation 1 (tight-binding ligands) (39),
| (Eq. 1) |
where [L] and [E] are the total concentrations of ligand and enzyme used for the titration, respectively. Although, as we reported before (13, 23, 24, 39, 43), the apparent binding affinities of many ligands do not necessarily exactly reflect their potencies as CYP51 inhibitors, in these experiments both posaconazole and VT-1161 produced Kd values in the low nanomolar range.
Catalytic activity and inhibition assays
The standard reaction mixture (35) contained 0.5 μm P450 and 1.0 μm CPR, 100 μm l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine, 0.4 mg/ml isocitrate dehydrogenase, and 25 mm sodium isocitrate in 20 mm potassium MOPS buffer (pH 7.4) containing 50 mm KCl, 5 mm MgCl2, and 10% glycerol (v/v). After the addition of the radiolabeled ([3-3H]) lanosterol (∼4,000 dpm/nmol, final concentration 50 μm), the mixture was preincubated for 2 min at 37 °C in a shaking water bath. The reaction was initiated by addition of 100 μm NADPH and stopped by extraction of the sterols with ethyl acetate. The extracted sterols were analyzed by a reversed-phase HPLC system (Waters) equipped with a β-RAM detector (INUS Systems) using a Nova Pak octadecylsilane (C18) column as described (30). For steady-state kinetic analysis, the reactions were run for 60 s at 37 °C, and the lanosterol concentration range was 6–50 μm. Michaelis-Menten parameters were calculated using GraphPad Prism 6, with the reaction rates (nmol of product formed/nmol of P450/min) being plotted versus total substrate concentration and using non-linear regression. The inhibitory potencies of azoles on C. albicans CYP51 activity were compared as percentage of inhibition of lanosterol 14α-demethylation in 60-min reactions at a molar ratio of enzyme/substrate/inhibitor = 1:50:2 (23, 30, 40, 43). Under these reaction conditions, in the absence of an inhibitor, C. albicans CYP51 converted all lanosterol into the product.
Crystallization
To obtain the crystals of C. albicans CYP51·posaconazole complex, 10 mm posaconazole (dissolved in (CH3)2SO) was added in a 1:3 (P450/inhibitor) molar ratio to the C. albicans CYP51 protein diluted to 200 μm with 10 mm potassium phosphate buffer (pH 7.4) containing 0.06 mm n-tridecyl-β-d-maltoside. The mixture was incubated for 60 min on ice, centrifuged at 16,000 × g for 5 min (Galaxy 16D, VWR Scientific), and used for crystallization. The crystals were grown using a hanging drop vapor diffusion technique by mixing equal volumes of complex solution and mother liquor (0.2 m calcium acetate hydrate (pH 7.3) and 20% (w/v) polyethylene glycol 4000), equilibrating with the reservoir solution at 18 °C. To obtain the crystals of the C. albicans CYP51·VT-1161 complex, 10 mm VT-1161 (dissolved in (CH3)2SO) was added in a 1:1.3 (P450/inhibitor) molar ratio to the 400 μm C. albicans CYP51 protein. The mixture was incubated for 60 min on ice, centrifuged at 16,000 × g for 5 min (Galaxy 16D, VWR Scientific), and used for crystallization. The crystals were grown using hanging drop vapor diffusion technique by mixing equal volumes of complex solution and mother liquor (0.1 m HEPES (pH 7.4) containing 0.2 m NaCl, and 10% (w/v) polyethylene glycol 6000) and equilibrating with the reservoir solution at 22 °C. In both cases crystals appeared after 3–5 days. The crystals were mounted in nylon loops, cryoprotected by swiping them through a droplet of 50% (v/v) propylene glycol in mother liquor, and flash-cooled in liquid nitrogen.
X-ray data collection, structure determination, and refinement
Diffraction data were collected at 100 K using synchrotron radiation on the insertion device of the Life Sciences Collaborative Access Team, Sector 21 of the Advanced Photon Source, Argonne National Laboratory (Argonne, IL), beam line/detector 21-ID-F/Rayonix MX-225 and 21-ID-D/Dectris Eiger 9 M for C. albicans CYP51 complexes with posaconazole and VT-1161, respectively. The data were processed with Xia2 and scaled with Aimless software (CCP4 Program Suite (52) 7.0.021). Data collection and refinement statistics are summarized in Table 1. The structure of the C. albicans CYP51·posaconazole complex was determined by molecular replacement with the program MOLREP (CCP4 Program Suite) using the coordinates of A. fumigatus CYP51B (Protein Data Bank code 4UYM (30)) as a template, and the resolution range of data was 50.39–2.86 Å; the correlation coefficient was 0.782; and the Rfactor of the correct solution was 0.3043. The structure of C. albicans CYP51·VT-1161 complex was solved in PhaserMR (CCP4 suite), with the coordinates of C. albicans CYP51-posaconazole as a template, and the resolution range of data was 49.61–2.00 Å; the correlation coefficient was 0.944; one solution was found; and the R-factor of the solution was 0.263. The refinement was performed with Phenix (CCP4 suite), and the model building was carried out with Coot (53). Structure superpositions for r.m.s. deviation calculations were done in LSQkab (CCP4 suite) using the SSM algorithm (54), and all structural figures were prepared with PyMOLTM (Schrödinger LLC), and UCSF Chimera (55).
Author contributions
G. I. L., W. J. H., and R. J. S. conceived the study. G. I. L. coordinated the study. T. Y. H., L. F., Z. W., A. Q., and G. I. L. performed the experiments. G. I. L., W. J. H., J. D. Y., R. J. S., and F. P. G. analyzed the data; G. I. L. and F. P. G. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at Sector 21 of the Advanced Photon Source (Argonne, IL). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02–06CH11357.
This work was supported by National Institutes of Health Grant R01 GM067871 (to G. I. L.) and in part by Viamet Pharmaceuticals (Durham, NC). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5FSA and 5TZ1) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CYP51
- sterol 14α-demethylase
- CYP or P450
- cytochrome P450
- CPR
- NADPH-cytochrome P450 reductase
- r.m.s.
- root-mean-square
- SRS
- substrate recognition site
- TCEP
- tris carboxyethylphosphine
- VT-1161
- (R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol)
- Ni-NTA
- Ni2+-nitrilotriacetate
- PDB
- Protein Data Bank
- cmc
- critical micelle concentration
- HPCD
- hydroxypropyl-β-cyclodextrin.
References
- 1. Noble S. M., Gianetti B. A., and Witchley J. N. (2017) Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat. Rev. Microbiol. 15, 96–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kullberg B. J., and Arendrup M. C. (2015) Invasive candidiasis. N. Engl. J. Med. 373, 1445–1456 [DOI] [PubMed] [Google Scholar]
- 3. Jampilek J. (2016) How can we bolster the antifungal drug discovery pipeline? Future Med. Chem. 8, 1393–1397 [DOI] [PubMed] [Google Scholar]
- 4. da Silva Dantas A., Lee K. K., Raziunaite I., Schaefer K., Wagener J., Yadav B., and Gow N. A. (2016) Cell biology of Candida albicans–host interactions. Curr. Opin. Microbiol. 34, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ene I. V., Adya A. K., Wehmeier S., Brand A. C., MacCallum D. M., Gow N. A., and Brown A. J. (2012) Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell. Microbiol. 14, 1319–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Desai J. V., and Mitchell A. P. (2015) Candida albicans biofilm development and its genetic control. Microbiol. Spectr. 3, 10.1128/microbiolspec.MB-0005-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pappas P. G., Kauffman C. A., Andes D. R., Clancy C. J., Marr K. A., Ostrosky-Zeichner L., Reboli A. C., Schuster M. G., Vazquez J. A., Walsh T. J., Zaoutis T. E., and Sobel J. D. (2016) Clinical practice guideline for the management of candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 62, e1-e50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yapar N. (2014) Epidemiology and risk factors for invasive candidiasis. Ther. Clin. Risk Manag. 10, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson D. T., Dimondi V. P., Johnson S. W., Jones T. M., and Drew R. H. (2016) Role of isavuconazole in the treatment of invasive fungal infections. Ther. Clin. Risk Manag. 12, 1197–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Antinori S., Milazzo L., Sollima S., Galli M., and Corbellino M. (2016) Candidemia and invasive candidiasis in adults: a narrative review. Eur. J. Intern. Med. 34, 21–28 [DOI] [PubMed] [Google Scholar]
- 11. Brown G. D., Denning D. W., Gow N. A., Levitz S. M., Netea M. G., and White T. C. (2012) Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165rv13 10.1126/scitranslmed.3004404 [DOI] [PubMed] [Google Scholar]
- 12. Lass-Flörl C. (2011) Triazole antifungal agents in invasive fungal infections. Drugs 71, 2405–2419 [DOI] [PubMed] [Google Scholar]
- 13. Hargrove T. Y., Friggeri L., Wawrzak Z., Sivakumaran S., Yazlovitskaya E. M., Hiebert S. W., Guengerich F. P., Waterman M. R., and Lepesheva G. I. (2016) Human sterol 14α-demethylase as a target for anticancer chemotherapy: towards structure-aided drug design. J. Lipid Res. 57, 1552–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnson E. M., Richardson M. D., and Warnock D. W. (1983) Effect of imidazole antifungals on the development of germ tubes by strains of Candida albicans. J. Antimicrob. Chemother. 12, 303–316 [DOI] [PubMed] [Google Scholar]
- 15. Saag M. S., and Dismukes W. E. (1988) Azole antifungal agents: emphasis on new triazoles. Antimicrob. Agents Chemother. 32, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pappas P. G., Kauffman C. A., Andes D., Benjamin D. K. Jr., Calandra T. F., Edwards J. E. Jr., Filler S. G., Fisher J. F., Kullberg B.-J., Ostrosky-Zeichner L., Reboli A. C., Rex J. H., Walsh T. J., Sobel J. D., and Infectious Diseases Society of America (2009) Clinical practice guidelines for the management candidiasis: 2009 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 503–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeong W., Haywood P., Shanmuganathan N., Lindsay J., Urbancic K., Ananda-Rajah M. R., Chen S. C., Bajel A., Ritchie D., Grigg A., Seymour J. F., Peleg A. Y., Kong D. C., and Slavin M. A. (2016) Safety, clinical effectiveness and trough plasma concentrations of intravenous posaconazole in patients with haematological malignancies and/or undergoing allogeneic haematopoietic stem cell transplantation: off-trial experience. J. Antimicrob. Chemother. 71, 3540–3547 [DOI] [PubMed] [Google Scholar]
- 18. van den Bossche H. (1988) in Sterol Biosynthesis Inhibitors (Berg D., and Plempel M., eds) pp. 79–119, Ellis Horwood, Chichester, UK [Google Scholar]
- 19. Lepesheva G. I., and Waterman M. R. (2007) Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 1770, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lepesheva G. I., and Waterman M. R. (2011) Structural basis for conservation in the CYP51 family. Biochim. Biophys. Acta 1814, 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hargrove T. Y., Wawrzak Z., Liu J., Waterman M. R., Nes W. D., and Lepesheva G. I. (2012) Structural complex of sterol 14α-demethylase (CYP51) with 14α-methylenecyclopropyl-Δ7–24,25-dihydrolanosterol. J. Lipid Res. 53, 311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Correia M. A., and Ortiz de Montellano P. R. (2005) in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano P. R., ed) pp. 246–322, Kluwer Academic-Plenum Publishing, New York [Google Scholar]
- 23. Lepesheva G. I., Ott R. D., Hargrove T. Y., Kleshchenko Y. Y., Schuster I., Nes W. D., Hill G. C., Villalta F., and Waterman M. R. (2007) Sterol 14α-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem. Biol. 14, 1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lepesheva G. I., Hargrove T. Y., Anderson S., Kleshchenko Y., Furtak V., Wawrzak Z., Villalta F., and Waterman M. R. (2010) Structural insights into inhibition of sterol 14 α-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 285, 25582–25590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang W., Ramamoorthy Y., Kilicarslan T., Nolte H., Tyndale R. F., and Sellers E. M. (2002) Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Dispos. 30, 314–318 [DOI] [PubMed] [Google Scholar]
- 26. White T. C., Marr K. A., and Bowden R. A. (1998) Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin. Microbiol. Rev. 11, 382–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Denning D. W., and Bromley M. J. (2015) How to bolster the antifungal pipeline. Science 347, 1414–1416 [DOI] [PubMed] [Google Scholar]
- 28. Hoekstra W. J., Garvey E. P., Moore W. R., Rafferty S. W., Yates C. M., and Schotzinger R. J. (2014) Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 24, 3455–3458 [DOI] [PubMed] [Google Scholar]
- 29. Garvey E. P., Hoekstra W. J., Schotzinger R. J., Sobel J. D., Lilly E. A., and Fidel P. L. (2015) Efficacy of the clinical agent VT-1161 against fluconazole-sensitive and -resistant Candida albicans in a murine model of vaginal candidiasis. Antimicrob. Agents Chemother. 59, 5567–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hargrove T. Y., Wawrzak Z., Lamb D. C., Guengerich F. P., and Lepesheva G. I. (2015) Structure-functional characterization of cytochrome P450 sterol 14α-demethylase (CYP51B) from Aspergillus fumigatus and molecular basis for the development of antifungal drugs. J. Biol. Chem. 290, 23916–23934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cherkesova T. S., Hargrove T. Y., Vanrell M. C., Ges I., Usanov S. A., Romano P. S., and Lepesheva G. I. (2014) Sequence variation in CYP51A from the Y strain of Trypanosoma cruzi alters its sensitivity to inhibition. FEBS Lett. 588, 3878–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Knoll A. (1992) The early evolution of eukaryotes: a geological perspective. Science 256, 622–627 [DOI] [PubMed] [Google Scholar]
- 33. Heckman D. S., Geiser D. M., Eidell B. R., Stauffer R. L., Kardos N. L., and Hedges S. B. (2001) Molecular evidence for the early colonization of land by fungi and plants. Science 293, 1129–1133 [DOI] [PubMed] [Google Scholar]
- 34. Lepesheva G. I., Podust L. M., Bellamine A., and Waterman M. R. (2001) Folding requirements are different between sterol 14α-demethylase (CYP51) from Mycobacterium tuberculosis and human or fungal orthologs. J. Biol. Chem. 276, 28413–28420 [DOI] [PubMed] [Google Scholar]
- 35. Lepesheva G. I., Nes W. D., Zhou W., Hill G. C., and Waterman M. R. (2004) CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry 43, 10789–10799 [DOI] [PubMed] [Google Scholar]
- 36. Lepesheva G. I., Zaitseva N. G., Nes W. D., Zhou W., Arase M., Liu J., Hill G. C., and Waterman M. R. (2006) CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B′ helix defines substrate preferences of sterol 14α-demethylase. J. Biol. Chem. 281, 3577–3585 [DOI] [PubMed] [Google Scholar]
- 37. Hargrove T. Y., Wawrzak Z., Liu J., Nes W. D., Waterman M. R., and Lepesheva G. I. (2011) Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14α-demethylase (CYP51) from Leishmania infantum. J. Biol. Chem. 286, 26838–26848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoekstra W. J., Hargrove T. Y., Wawrzak Z., da Gama Jaen Batista D., da Silva C. F., Nefertiti A. S., Rachakonda G., Schotzinger R. J., Villalta F., Soeiro Mde N., and Lepesheva G. I. (2016) Clinical candidate VT-1161's antiparasitic effect in vitro, activity in a murine model of Chagas disease, and structural characterization in complex with the target enzyme CYP51 from Trypanosoma cruzi. Antimicrob. Agents Chemother. 60, 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hargrove T. Y., Wawrzak Z., Alexander P. W., Chaplin J. H., Keenan M., Charman S. A., Perez C. J., Waterman M. R., Chatelain E., and Lepesheva G. I. (2013) Complexes of Trypanosoma cruzi sterol 14α-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: Structural basis for pathogen selectivity. J. Biol. Chem. 288, 31602–31615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lepesheva G. I., Park H. W., Hargrove T. Y., Vanhollebeke B., Wawrzak Z., Harp J. M., Sundaramoorthy M., Nes W. D., Pays E., Chaudhuri M., Villalta F., and Waterman M. R. (2010) Crystal structures of Trypanosoma brucei sterol 14α-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 285, 1773–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lepesheva G. I., Hargrove T. Y., Rachakonda G., Wawrzak Z., Pomel S., Cojean S., Nde P. N., Nes W. D., Locuson C. W., Calcutt M. W., Waterman M. R., Daniels J. S., Loiseau P. M., and Villalta F. (2015) VFV as a new effective CYP51 structure-derived drug candidate for Chagas disease and visceral leishmaniasis. J. Infect. Dis. 212, 1439–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Andriani G., Amata E., Beatty J., Clements Z., Coffey B. J., Courtemanche G., Devine W., Erath J., Juda C. E., Wawrzak Z., Wood J. T., Lepesheva G. I., Rodriguez A., and Pollastri M. P. (2013) Antitrypanosomal lead discovery: identification of a ligand-efficient inhibitor of Trypanosoma cruzi CYP51 and parasite growth. J. Med. Chem. 56, 2556–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Friggeri L., Hargrove T. Y., Rachakonda G., Williams A. D., Wawrzak Z., Di Santo R., De Vita D., Waterman M. R., Tortorella S., Villalta F., and Lepesheva G. I. (2014) Structural basis for rational design of inhibitors targeting Trypanosoma cruzi sterol 14α-demethylase: two regions of the enzyme molecule potentiate its inhibition. J. Med. Chem. 57, 6704–6717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gotoh O. (1992) Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 267, 83–90 [PubMed] [Google Scholar]
- 45. Mane A., Vidhate P., Kusro C., Waman V., Saxena V., Kulkarni-Kale U., and Risbud A. (2016) Molecular mechanisms associated with fluconazole resistance in clinical Candida albicans isolates from India. Mycoses 59, 93–100 [DOI] [PubMed] [Google Scholar]
- 46. Morio F., Loge C., Besse B., Hennequin C., and Le Pape P. (2010) Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagn. Microbiol. Infect. Dis. 66, 373–384 [DOI] [PubMed] [Google Scholar]
- 47. Marichal P., Koymans L., Willemsens S., Bellens D., Verhasselt P., Luyten W., Borgers M., Ramaekers F. C., Odds F. C., and Bossche H. V. (1999) Contribution of mutations in the cytochrome P450 14α-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans. Microbiology 145, 2701–2713 [DOI] [PubMed] [Google Scholar]
- 48. Morschhäuser J. (2016) The development of fluconazole resistance in Candida albicans–an example of microevolution of a fungal pathogen. J. Microbiol. 54, 192–201 [DOI] [PubMed] [Google Scholar]
- 49. Lupetti A., Danesi R., Campa M., Del Tacca M., and Kelly S. (2002) Molecular basis of resistance to azole antifungals. Trends Mol. Med. 8, 76–81 [DOI] [PubMed] [Google Scholar]
- 50. Schell W. A., Jones A. M., Garvey E. P., Hoekstra W. J., Schotzinger R. J., and Alexander B. D. (2017) Fungal CYP51 inhibitors VT-1161 and VT-1129 exhibit strong in vitro activity against Candida glabrata and C. krusei isolates clinically resistant to azole and echinocandin antifungal compounds. Antimicrob. Agents Chemother. 61, e01817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lepesheva G. I., Strushkevich N. V., and Usanov S. A. (1999) Conformational dynamics and molecular interaction reactions of recombinant cytochrome P450scc (CYP11A1) detected by fluorescence energy transfer. Biochim. Biophys. Acta 1434, 31–43 [DOI] [PubMed] [Google Scholar]
- 52. Potterton E., Briggs P., Turkenburg M., and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 53. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 55. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 56. Schlichting I., Berendzen J., Chu K., Stock A. M., Maves S. A., Benson D. E., Sweet R. M., Ringe D., Petsko G. A., and Sligar S. G. (2000) The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287, 1615–1622 [DOI] [PubMed] [Google Scholar]