

Abstract
RNA cleavage by RNA polymerase (RNAP) is the central step in co-transcriptional RNA proofreading. Bacterial RNAPs were proposed to rely on the same mobile element of the active site, the trigger loop (TL), for both nucleotide addition and RNA cleavage. RNA cleavage can also be stimulated by universal Gre factors, which should replace the TL to get access to the RNAP active site. The contributions of the TL and Gre factors to RNA cleavage reportedly vary between RNAPs from different bacterial species and, probably, different types of transcription complexes. Here, by comparing RNAPs from Escherichia coli, Deinococcus radiodurans, and Thermus aquaticus, we show that the functions of the TL and Gre factors in RNA cleavage are conserved in various species, with important variations that may be related to extremophilic adaptation. Deletions of the TL strongly impair intrinsic RNA cleavage by all three RNAPs and eliminate the interspecies differences in the reaction rates. GreA factors activate RNA cleavage by wild-type RNAPs to similar levels. The rates of GreA-dependent cleavage are lower for ΔTL RNAP variants, suggesting that the TL contributes to the Gre function. Finally, neither the TL nor GreA can efficiently activate RNA cleavage in certain types of backtracked transcription complexes, suggesting that these complexes adopt a catalytically inactive conformation probably important for transcription regulation.
Keywords: bacterial transcription, gene expression, RNA metabolism, RNA polymerase, transcription elongation factor, GreA, RNA proofreading, transcription elongation complex
Introduction
The RNA cleavage reaction is thought to play a crucial role in correction of transcriptional errors during RNA synthesis by RNAP2 (1–4). Because this reaction is performed by the same active site of RNAP as nucleotide addition, RNAP must “backtrack” along the DNA template after nucleotide misincorporation to allow cleavage of internal phosphodiester bonds in RNA. Long backtracking events can result in permanent stalling of RNAP in the backtracked state, and RNA cleavage also serves for reactivation of such “arrested” transcription elongation complexes (TECs) (5–7). After backtracking, the RNA 3′-end is accommodated within the secondary RNAP channel, which normally serves for NTP entry (Fig. 1). Both nucleotide addition and RNA cleavage require two divalent metal ions (usually Mg2+) bound in the RNAP active site, which coordinate the reaction substrates (3, 8). In addition to metal ions, several other factors were shown to contribute to RNA cleavage. In particular, the reaction is greatly stimulated by RNA nucleotide at position +2 relative to the active site through its direct interactions with the second metal ion and the attacking water molecule (1). The +2 nucleotide was also proposed to stimulate the second metal ion binding by changing the geometry of the active site residues involved in metal chelation (“active site tuning”) (3). Recent crystallographic analysis of a bacterial backtracked TEC revealed that the +2 RNA nucleotide is accommodated in an evolutionary conserved RNAP cavity (“proofreading site”), which facilitates catalysis (Fig. 1, left) (9). Similarly, interactions of the +2 nucleotide in a backtracked TEC of yeast RNAP II with a “gating” tyrosine residue in the secondary RNAP channel likely stabilize the backtracked TEC conformation (10). As a result, RNA is preferentially cleaved after one-nucleotide backtracking, resulting in the release of a 2-nt 3′-RNA fragment (illustrated for TEC-2 in Fig. 2), a phenomenon that has been observed in bacterial, eukaryotic, and archaeal RNAPs (1, 4, 11–15).
Figure 1.

Structures of backtracked TEC (PDB code 4WQS, left), elemental paused TEC (PDB code 4GZY, center), and GreA-bound TthRNAP (PDB code 4WQT, right) (9, 28). The TL, BH, and GreA are dark green, violet, and dark red, respectively. The His-936/His-1242 residue in the TL is light green, The Cα atoms at the borders of the TL deletions analyzed in our work (Δ931–1137→3Gly, Δ1254–1272→3Gly, and Δ1237–1255→3Ala in E. coli, D. radiodurans, and T. aquaticus RNAPs, respectively) are shown with black spheres. The position of the SI3 insertion in EcoRNAP is shown as a gray sphere. RNA is red, the 3′-nucleotide in the backtracked TEC is black, the sites of RNA cleavage are indicated with red arrows, and the attacked phosphorus atom is yellow. Mg2+ ions in the RNAP active site are shown with carmine spheres (note that no Me2+ atoms were resolved in the backtracked TEC, and only the first ion is visible in the paused and Gre-bound RNAPs). Acidic residues at the tip of the GreA NTD (Asp-41, Glu-44) are pink. In the paused TEC structure, the central part of the TL is disordered, and the ultimate RNA phosphodiester bond is shifted upstream of the active center (28).
Figure 2.

TECs analyzed in this study. A, schematics of nucleic acid scaffolds used for TEC assembly. RNA oligonucleotides are red. The scaffolds are aligned by the positions of RNA cleavage (indicated with red arrows). The position of the hisP pause site in TEC-3p is indicated. B, representative kinetics of RNA cleavage in the three TECs by wild-type E. coli, D. radiodurans, and T. aquaticus RNAPs at 37 °C. The non-reactive fraction of RNA corresponds to oligonucleotides that were not bound by RNAP during in vitro TEC assembly.
Another RNAP element that has been implicated in RNA cleavage is a mobile trigger loop (TL), which can adopt several functionally important conformations. During nucleotide addition, the TL folds into two α helixes and together with the bridge helix (BH) encloses the NTP substrate within the active site, with residue His-936/His-1242 (Escherichia coli (Eco)/Thermus aquaticus (Taq) numbering; Fig. 1) directly involved in NTP interactions and catalysis (16–18). In this conformation the secondary channel is blocked for both the next NTP entry and RNA backtracking. Thus, during RNA cleavage the TL must accept a partially open conformation, with the same residue, His-936/His-1242, likely participating in catalysis (in the available three-dimensional structure of backtracked TEC, the TL is seen in an inactive conformation as residue His-936/His-1242 is located too far away from the target RNA bond; shown in yellow in Fig. 1, left) (9). Substitutions or deletions in this region of the TL were shown to dramatically impair RNA cleavage in mismatched complexes by TaqRNAP and, to a lesser extent, by EcoRNAP (17, 19). At the same time, deletion of the TL was shown to have little or no effect on RNA cleavage by EcoRNAP in another type of backtracked TECs (TEC-3p and its variants; Fig. 2A) (18, 20), thus raising uncertainty about the actual role of the TL in RNA cleavage. In particular, recent models of RNA cleavage by EcoRNAP did not consider at all the TL as a participant of the cleavage reaction (3) or suggested that its contribution to cleavage is limited to TECs containing certain +2 RNA nucleotides (20).
Gre factors (GreA in most bacteria, GreA and GreB in E. coli) stimulate RNA cleavage by binding within the secondary channel of RNAP, which allows them to reach the active site and coordinate the catalytic magnesium ions (21, 22). Transcription factors TFIIS and TFS play similar functions in eukaryotic RNAP II and archaeal RNAP, respectively (11–13). The stimulation of RNA cleavage requires the TL to be open (Fig. 1, right); accordingly, the TL was proposed to have little or no contribution to GreA-dependent RNA cleavage by TaqRNAP (23). In contrast, deletion of the SI3 domain inserted within the TL in EcoRNAP (Fig. 1) was shown to make RNAP completely resistant to the action of the GreB protein, suggesting that this part of the EcoTL is essential for Gre binding and/or function (18). Together, these observations suggested that the details of Gre-dependent stimulation of RNA cleavage may significantly differ between RNAPs.
To resolve the existing controversies on the mechanisms of the cleavage reaction, we analyzed the requirements for the TL and Gre factors during RNA cleavage by three RNAPs: mesophilic E. coli, mesophilic Deinococcus radiodurans (Dra), and thermophilic T. aquaticus. E. coli and T. aquaticus RNAPs were previously used as main models in studies of RNA proofreading by bacterial polymerases (3, 18, 19), but their direct comparison was complicated because of different temperature optima of activity and different reaction conditions used in the assays. Recently, we demonstrated that DraRNAP, which is more closely related to TaqRNAP but operates at the same temperatures as EcoRNAP, displays much higher RNA cleavage rates than the latter (24). In this work we directly compared RNA cleavage by these three RNAPs at various temperatures and in TECs of different structures. We revealed interspecific variations in RNA cleavage but concluded that the general reaction pathway is identical for different RNAPs, with the TL and Gre proteins serving as alternative activators of catalysis (23, 25). We also demonstrated that certain backtracked TECs are resistant to the action of either TL or Gre factors, thus explaining why previous studies arrived to different conclusions on the role of the TL in RNA cleavage.
Results
TECs used for analysis of RNA cleavage
We analyzed RNA cleavage in TECs of distinct architectures that were used in previous studies, (see the Introduction). In particular, the complexes differed in the structures of the RNA 3′-ends, resulting in different translocation registers of the RNAP active site relative to the RNA transcript, and their sensitivity to RNA cleavage (Fig. 2A).
The first complex, TEC-2, contained a single noncomplementary adenine nucleotide at the RNA 3′-end (+2A) and corresponded to mismatched TECs that are formed during transcription after nucleotide misincorporation. This complex and its variants are highly prone to intrinsic RNA cleavage, which releases a 2-nt 3′-RNA fragment (Fig. 2) (1, 19, 24). We also analyzed two variants of TEC-2 with changes in the RNA 3′-end: TEC-2U containing a single noncomplementary uridine nucleotide at the RNA 3′-end and TEC-2+1 containing an additional unpaired 3′ residue and giving rise to 3-nt cleavage products (see below and Fig. 7).
Figure 7.

Comparison of RNA cleavage rates in TEC-2, TEC-2+1, and TEC-2U. The reactions were performed at 37 °C with E. coli and D. radiodurans RNAPs and their ΔTL variants in transcription buffers containing 40 mm Tris-HCl (pH 7.9 at 25 °C), 40 mm KCl, and 10 mm MgCl2. A, TEC structures. B, representative plots of RNA cleavage kinetics in TEC-2+1 and TEC-2U. The data were normalized to the maximal RNA cleavage observed in each reaction. C, comparison of the kobs values for RNA cleavage by EcoRNAP in TEC-2 and its variants. The numbers in bold indicate -fold differences in the reaction rates between WT and ΔTL RNAP variants for each TEC.
The second complex, TEC-3p (for “paused”) contained two unpaired uridine nucleotides at the RNA 3′-end, resulting in the cleavage of a 3-nt RNA fragment (Fig. 2) (18). The reported rates of RNA cleavage by EcoRNAP in this complex (18) were much lower than the rates measured for other mismatched TECs (19). It was also demonstrated that RNA cleavage in TEC-3p was not affected by the TL deletion and was only moderately stimulated by the GreB factor (18). Other variants of this complex containing a single unpaired 3′-nucleotide were also characterized by low RNA cleavage rates and only weak dependence of the reaction on the TL (20). We note that the nucleic acid sequence and the position of RNA cleavage in this complex (Fig. 2) precisely correspond to the elemental pause site found in the hisP pause signal (Fig. 1, center) (26–29), a feature not discussed in previous studies (18, 20).
The third complex, TEC-9, contained eight noncomplementary nucleotides at the RNA 3′-end, resulting in a 9-nt RNA cleavage product (Fig. 2). This complex was previously used to characterize structural transitions of Thermus thermophilus (Tth) RNAP during backtracking using Cys-pair cross-linking (9). The long RNA segment in this backtracked complex was proposed to induce transition of RNAP into a “ratcheted” state with the open main channel, kinked BH, and open TL, a conformation that may be similar to the paused complex (9, 28). This complex was shown to be insensitive to GreA-dependent stimulation of RNA cleavage by TthRNAP, but the role of the TL in the reaction was not tested (9).
Interspecific variations in RNA cleavage
We first focused on reactions in TEC-2, which mimics natural misincorporated complexes that are preferred substrates for RNA cleavage by RNAP (see the Introduction). For this TEC, we measured the RNA cleavage rates for wild-type (WT) and ΔTL E. coli, D. radiodurans, and T. aquaticus RNAPs in the temperature range between 10 and 40 °C both in the absence and in the presence of corresponding GreA factors.
We observed huge variations in the rates of RNA cleavage in different reactions (kobs from ∼0.000002 s−1 for ΔTL DraRNAP at 20 °C to 50 s−1 for WT TaqRNAP+GreA at 40 °C, a 25-million time difference) (Table 1 and supplemental Table S1). In accordance with the Arrhenius equation, we revealed linear dependences of the logarithms of cleavage rates on reverse temperature for reactions performed with WT RNAPs both in the absence and in the presence of GreA (Fig. 3). The slopes of these plots were used to calculate activation energies (Ea) of the reactions, which illustrate the heat dependences of RNA cleavage for each RNAP (Fig. 3, supplemental Table S2).
Table 1.
Rates of RNA cleavage by EcoRNAP and DraRNAP in various TECs
Reactions were performed at 37 °C in transcription buffers containing 40 mm Tris-HCl (pH 7.9 at 25 °C), 40 mm KCl, and 10 mm MgCl2 for TEC-2 and TEC-9 or 40 mm Tris-HCl (pH 8.5 at 25 °C), 40 mm KCl, and 20 mm MgCl2 for TEC-3p. ND, not determined. Means and S.D. from 3–5 independent experiments are shown. The numbers in bold indicate -fold differences in the reaction rates relative to WT RNAPs in the same TEC.
| RNAP |
kobs |
|||||
|---|---|---|---|---|---|---|
| TEC-2 |
TEC-3p |
TEC-9 |
||||
| E. coli | D. radiodurans | E. coli | D. radiodurans | E. coli | D. radiodurans | |
| s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | |
| WT | 0.017 ± 0.001 | 0.27 ± 0.10 | 0.00034 ± 0.00007 | 0.00048 ± 0.00008 | 0.00057 ± 0.00018 | 0.00097 ± 0.00013 |
| 1 | 1 | 1 | 1 | 1 | 1 | |
| ΔTL | 0.00016 ± 0.00001 | 0.000085 ± 0.000017 | 0.00031 ± 0.00008 | 0.00014 ± 0.00001 | 0.00065 ± 0.00005 | 0.00012 ± 0.00003 |
| 0.01 | 0.0003 | 0.9 | 0.3 | 1.1 | 0.12 | |
| WT + GreA | 21.3 ± 3.6 | 30.4 ± 5.5 | 0.0014 ± 0.0003 | 0.0023 ± 0.0007 | 0.00038 ± 0.00008 | ND |
| 1280 | 110 | 4.2 | 4.9 | 0.7 | ||
| ΔTL + GreA | 0.67 ± 0.17 | 0.75 ± 0.01 | 0.00049 ± 0.00001 | 0.0016 ± 0.0009 | 0.00018 ± 0.00007 | ND |
| 40 | 2.8 | 1.5 | 3.3 | 0.3 | ||
Figure 3.

Arrhenius plots of the rates of RNA cleavage in the TEC-2 for WT (left) and ΔTL (right) E. coli, D. radiodurans, and T. aquaticus RNAPs. The reactions were performed at 4 different temperatures (10, 20, 30, and 37 °C for E. coli and D. radiodurans RNAPs or 40 °C for TaqRNAP) in transcription buffer containing 40 mm Tris-HCl (pH 7.9 at 25 °C), 40 mm KCl, and 10 mm MgCl2. The plots show the dependence of the logarithm of reaction rates on reverse absolute temperature (log(kobs) versus T−1) (see supplemental Table S1 for kobs values). The Arrhenius equation is shown on the upper right.
Mesophilic DraRNAP was significantly faster in RNA cleavage than mesophilic EcoRNAP at all temperatures (from 43 to 16-fold difference at 10 °C and 37 °C, respectively; Fig. 3 andsupplemental Table S1), which is in agreement with our recent findings (24). Thermophilic TaqRNAP revealed a much stronger temperature dependence of the reaction rates than the mesophilic polymerases. At 10 °C, the rate of RNA cleavage by TaqRNAP was comparable to EcoRNAP, whereas at 40 °C, it was equal to the rate of DraRNAP. The steeper Arrhenius plot for TaqRNAP corresponded to a much higher Ea value of the reaction (190 kJ/mol for TaqRNAP in comparison with 148 and 115 kJ/mol for E. coli and D. radiodurans RNAPs, respectively; supplemental Table S2).
Most experiments in this study were performed in transcription buffers containing Tris-HCl, equilibrated to pH 7.9 at 25 °C, to facilitate comparison with previously published works, all of which also used Tris-based buffers (with reaction temperatures varying between 20 and 40 °C) (3, 18–20, 30). However, temperature-dependent changes in the pH values of Tris buffers (∼0.026 pH decrease per 1 °C temperature increase) could by themselves affect the rates of RNA cleavage in this and other studies. We, therefore, performed control experiments on RNA cleavage by wild-type E. coli and T. aquaticus RNAPs in transcription buffers calibrated for the temperature-dependent change so that the reaction pH was exactly 7.9 at each temperature. We found that this indeed resulted in a certain shift in the cleavage rates (supplemental Fig. S1). In particular, the rates became lower at low temperatures (2.5-fold change at maximum), because the buffer equilibrated to pH 7.9 at 25 °C had higher pH at 10 °C, which stimulated RNA cleavage (15). However, the observed differences in most cases were within the sum of standard deviations of the two values, and the ratio of the cleavage rates of E. coli and T. aquaticus RNAPs at each temperature remained almost the same in both buffers (supplemental Fig. S1). Thus, we conclude that temperature-dependent pH changes do not strongly affect the relative rates of RNA cleavage by various RNAPs.
The differences between E. coli and D. radiodurans RNAPs completely disappeared in the presence of the corresponding GreA factors, which more strongly activated EcoRNAP than DraRNAP with more efficient stimulation at low temperatures for both RNAPs (23,200/1,300- and 580/110-fold increase in the reaction rates at 10 °C and 37 °C, respectively; supplemental Table S1). This corresponded to significantly lower Ea values in comparison with GreA-less reactions (69 and 78 kJ/mol for E. coli and D. radiodurans RNAPs, respectively, supplemental Table S2). Thus, GreA minimizes the temperature-dependent rearrangements of the active site required for RNA cleavage (probably, by alleviating the need for TL restructuring).
GreA also strongly activated RNA cleavage by TaqRNAP, but this RNAP retained the stronger temperature dependence of the reaction, with little changes in the Ea value (177 kJ/mol). As a result, the rates of Gre-dependent cleavage at 10 and 20 °C by TaqRNAP were significantly lower than the rates of E. coli and D. radiodurans RNAPs, whereas at higher temperatures all three RNAPs had comparable rates of cleavage in the presence of GreA (Fig. 3, supplemental Table S1). Thus, the TaqRNAP active site requires more extensive heat-dependent rearrangements during both intrinsic and Gre-dependent RNA cleavage reactions.
The observed differences between RNAPs can be further illustrated by comparison of the corresponding values of free energy of activation (ΔG#). According to the activation theory of catalysis, the ΔG# values correspond to the difference in free energies (“energetic barrier”) between the ground state and activated enzyme-substrate complexes (see Fig. 4 and Refs. 31 and 32 for details). In the absence of GreA, these values for TaqRNAP were higher than for DraRNAP but lower than for the EcoRNAP, which corresponded to lower and higher energetic barriers of the reaction for D. radiodurans and E. coli RNAPs, respectively (negative and positive ΔΔG# differences relative to TaqRNAP) (Fig. 4; supplemental Table S2). The addition of GreA significantly lowered the ΔG# values for all three RNAPs, but the values for TaqRNAP remained higher than those for E. coli and D. radiodurans RNAPs at low temperatures (10–20 °C).
Figure 4.

Temperature dependence of the ΔΔG# values for RNA cleavage in TEC-2 by various RNAPs relative to TaqRNAP. Shown are the differences in the free energy of activation for each reaction relative to the reaction performed with TaqRNAP in the absence of GreA at the same temperature (ΔΔG# = ΔG#x − ΔG#Taq). The ΔG#Taq values for WT and ΔTL T. aquaticus RNAPs were used for comparisons with WT and ΔTL RNAP variants, respectively (see supplemental Table S2 for primary data). The upper left image illustrates the meaning of the ΔG# value in terms of “reaction coordinates”: E, enzyme; S, substrate; ES, ground-state enzyme-substrate complex; ES#, activated enzyme-substrate complex; P, product (31). The ΔG# and ΔΔG# values were calculated according to the formulae shown on the upper right as described in Refs. 31 and 32.
Interplay between the TL and GreA factors in RNA cleavage
We then analyzed RNA cleavage by ΔTL RNAPs. Deletions strongly impaired activity of all three RNAPs. Remarkably, the rates of cleavage at each temperature became comparable for all RNAPs (Fig. 3, supplemental Table S1), suggesting that the differences observed for WT RNAPs depend on the presence of the TL in the active site (24).
For the mutant RNAPs, we could not accurately measure RNA cleavage at 10 °C due to extremely low rates of the reaction (the half-times of several days). Although the remaining three temperature points (20, 30, and 37/40 °C) were insufficient for reliable calculation of the Ea values, we used these data for comparison of the corresponding ΔG# values. We found that the ΔG# values were highly similar for all three mutant RNAPs and were much higher than the values for WT RNAPs at all the tested temperatures (Fig. 4, supplemental Table S2). Thus, in WT RNAPs the TL significantly lowers the energetic barrier for the cleavage reaction.
GreA factors activated RNA cleavage by all three ΔTL RNAPs to comparable levels (5,000–20,000-fold activation in various reactions), making them significantly faster than WT RNAPs in GreA-less reactions (Fig. 3, supplemental Table S1). This corresponded to a significant decrease in the ΔG# values and lowering of the energetic barriers of the reaction (Fig. 4, supplemental Table S2). Remarkably, all three GreA factors acted similarly on their cognate ΔTL RNAPs; in contrast, the activity of E. coli GreB was shown to strictly depend on the presence of the SI3 insertion in the TL of EcoRNAP (18) (which was deleted in our ΔTL EcoRNAP).
At the same time, the rates of RNA cleavage by all three ΔTL RNAPs remained significantly lower than the rates for WT RNAPs in Gre-containing reactions (20–400× for various reactions, supplemental Table S1), suggesting that the TL contributes to GreA-dependent RNA cleavage. The TL also likely determines the difference in the cleavage rates in GreA reactions between TaqRNAPs and EcoRNAP/DraRNAPs, which was observed for WT but not for ΔTL RNAP variants (see above). To determine whether the TL deletion could affect GreA affinity to the TEC, we analyzed RNA cleavage by wild-type and ΔTL EcoRNAPs at different GreA concentrations (Fig. 5). GreA titration suggested that it binds both RNAPs with the same affinities (apparent Kd values of 4.6 ± 0.9 and 3.6 ± 0.8 μm for WT and ΔTL RNAPs, respectively).
Figure 5.
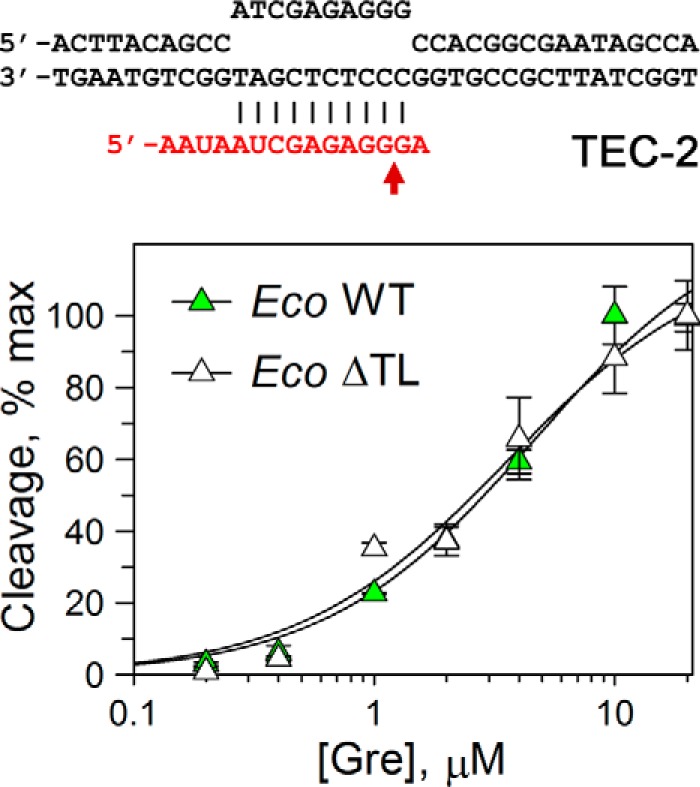
Comparison of apparent GreA affinities to WT and ΔTL Eco RNAPs. The reactions were performed in TEC-2 at 0 °C (to make the measurements possible by manual mixing) for 1 min for WT RNAP and for 10 min for ΔTL RNAP; under these conditions the reactions did not go to completion, and <20–30% of active TECs reacted even at maximal GreA concentration. For each RNAP, the data were normalized relative to the maximal RNA cleavage measured at high GreA concentration. The data were fitted to a hyperbolic equation.
Variations in RNA cleavage between TECs
We next compared the roles of the TL and GreA factors in RNA cleavage in the mismatched TECs of different structures (Fig. 2). To facilitate direct comparisons of the reaction rates, we performed these experiments with RNAPs from mesophilic E. coli and D. radiodurans, which belong to diverse bacterial phyla but have similar temperature optima (24, 33).
The rates of intrinsic RNA cleavage by WT RNAPs in TEC-3p and TEC-9 were dramatically lower than the rates of cleavage in TEC-2 (∼50/30× and 550/300× in TEC-3p/TEC-9 for E. coli and D. radiodurans RNAPs, respectively). Moreover, these rates were comparable to the rates of the ΔTL RNAPs measured in the same TECs (Fig. 6 and Table 1). This suggested that the TL has no role in intrinsic RNA cleavage in paused or long-backtracked TECs, represented by TEC-3p and TEC-9, respectively.
Figure 6.
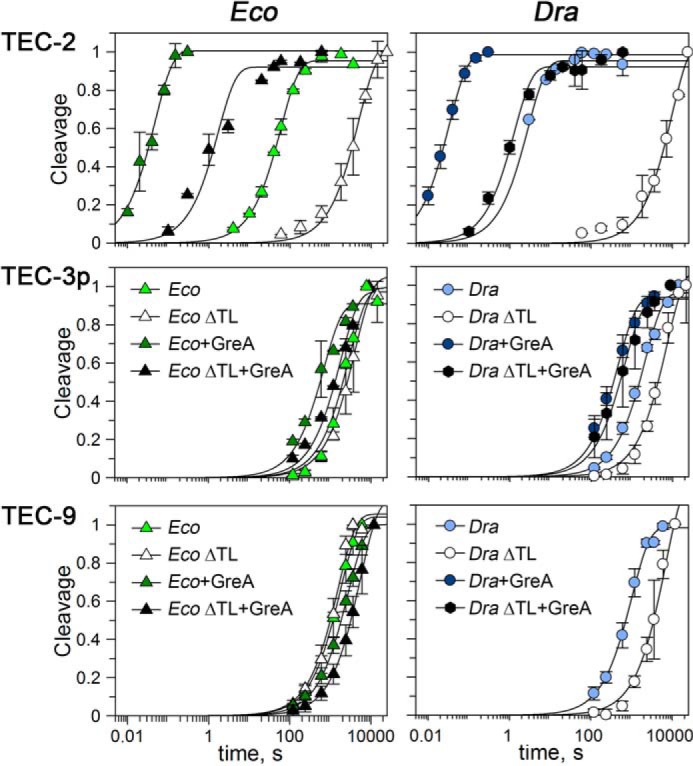
Kinetics of RNA cleavage by WT and ΔTL E. coli and D. radiodurans RNAPs in the absence and in the presence of GreA in various TECs. The reactions were performed at 37 °C, and the data were normalized by the maximal efficiencies of RNA cleavage in each reaction. The data for TEC-2 correspond to Fig. 3 and supplemental Table S1.
It was recently proposed (20) that the TL may be important for RNA cleavage in only a subset of TECs containing adenine nucleotide at the RNA 3′-end (such as TEC-2, Fig. 2A). Indeed, adenine-containing TECs are characterized by the highest rates of RNA cleavage in comparison with other nucleotide variants (1, 3, 20). We, therefore, measured RNA cleavage in TEC-2U containing uridine instead of adenine at the +2 position (Fig. 7A). The rate of RNA cleavage by EcoRNAP in this TEC was ∼7× lower than in TEC-2. However, deletion of the TL still greatly reduced the cleavage rate in this complex (∼17-fold; Fig. 7, B and C). Similarly, TL deletion severely affected RNA cleavage by TaqRNAP in another non-paused TEC-containing uridine residue at the +2 position (19). We further note that the low rate of RNA cleavage in TEC-3p was not due to the presence of two noncomplementary nucleotides at the RNA 3′-end, as similar noncomplementary RNA ends did not compromise RNA cleavage and did not change its dependence on the TL in non-paused mismatched TECs (see Ref. 19 and our data for TEC-2+1 in Fig. 7).
We further measured the rates of Gre-dependent RNA cleavage in TEC-3p and TEC-9. In contrast to TEC-2, GreA could not efficiently activate the reaction in TEC-3p by both E. coli and D. radiodurans RNAPs (5-fold stimulation at maximum) (Fig. 6 and Table 1). Notably, E. coli GreB also had a much weaker stimulatory effect on RNA cleavage in TEC-3p (18) than the effects of Gre factors in non-paused complexes (e.g. Ref. 23 and this work).
TEC-9 was also resistant to GreA-dependent stimulation of RNA cleavage by EcoRNAP (Fig. 6 and Table 1). It was suggested that the presence of a long RNA segment in this complex might prevent GreA binding to RNAP (9). However, because TEC-9 is similarly resistant to the TL- and Gre-dependent activation of RNA cleavage (Fig. 6), we propose that its low reactivity could also result from the specific ratcheted RNAP conformation, which may be similar to the paused state and require significant RNAP rearrangements before catalysis (9).
Discussion
From our experiments we can draw several conclusions on the mechanism of RNA cleavage.
First, bacterial RNAPs may significantly differ in the rates of intrinsic RNA cleavage but share the same reaction pathway. Among RNAPs that were analyzed in this study, RNAP from the highly stress-resistant D. radiodurans has the highest rate of intrinsic RNA cleavage in mismatched TECs at all temperatures. As was proposed previously, this might help it to cope with high RNA error rates and increased levels of TEC backtracking resulting from increased DNA damage under stress conditions (24). In its turn, thermophilic TaqRNAP has the strongest temperature dependence of the RNA cleavage activity, which parallels the strong temperature dependence of RNA synthesis by this RNAP (32). This likely results from a decrease in the conformational mobility of the active site, which thus requires more heat energy for restructuring during catalysis. This property is likely related to thermal adaptation and is necessary for stabilization of the protein structure at high temperatures (31, 32, 34).
Second, most of the observed interspecific differences in both intrinsic and Gre-dependent RNA cleavage between E. coli, D. radiodurans, and T. aquaticus RNAPs depend on the TL and disappear when the TL is deleted (see also Ref 24). The drastic decrease in the reaction rates in the ΔTL RNAPs confirms that the TL plays an essential role in the cleavage reaction in mismatched TECs in all RNAPs. Previously, temperature-dependent differences in both RNA synthesis and RNA cleavage between thermophilic T. aquaticus and mesophilic D. radiodurans RNAPs were proposed to depend on the F-loop, a β′ subunit region near the active site (32). During nucleotide addition, the F-loop directly contacts the folded TL, thus explaining its influence on RNA synthesis (35). Recent structure of the backtracked complex revealed that the F-loop is re-oriented and accommodated between the open TL and the RNA 3′-end (9), which explains how it could also affect RNA cleavage through the TL refolding.
Interestingly, the TL seems to have little role in both intrinsic and factor-dependent RNA cleavage by archaeal RNAP and eukaryotic RNAP II, and consistently, the reported rates of intrinsic RNA cleavage by these RNAPs are much lower than the rates measured for bacterial RNAPs (30, 36–38) At the same time, the TL can be “recruited” to activate the reaction in RNAP II by certain amino acid substitutions (30), suggesting that the underlying architecture of the active site is conserved in all RNAPs.
Third, in all three analyzed RNAPs, both the TL and GreA can efficiently stimulate RNA cleavage in non-paused mismatched TECs, and GreA-dependent cleavage is partially dependent on the physical presence of the TL. Based on available data we propose that the role of the TL in Gre action is independent of its role in intrinsic RNA cleavage. Indeed, the TL must be unfolded to allow GreA binding (Fig. 1) (9), and substitutions of the catalytic residue His-1242 in TaqRNAP were shown to have little effect on GreA-dependent cleavage (23). Furthermore, GreA reorients the RNA 3′-end from the proofreading site to another cavity, which is only formed after GreA binding and is inaccessible for the TL (9). Therefore, the TL might indirectly participate in Gre-dependent RNA cleavage in mismatched TECs by interacting with GreA and positioning it for catalysis.
Fourth, previously reported interspecific variations in RNA cleavage (1, 3, 18–20) mainly resulted from the use of different types of TECs, which differed in their sensitivity to TL- and Gre-mediated cleavage. In contrast to short-backtracked mismatched TECs, neither the TL nor GreA can activate RNA cleavage in specific types of transcription complexes, i.e. paused-backtracked and long-backtracked TECs. Previous biochemical and structural analyses of hisP-paused complexes revealed a specific RNAP conformation with a partially open main channel, distorted BH, and unfolded TL (Fig. 1, center) (26–29). Furthermore, depending on the secondary RNA structure, the RNA 3′-end in the paused complex (corresponding to the third nucleotide from the RNA 3′-end in TEC-3p) was shown to fluctuate between distorted post-translocated and frayed pre-translocated conformations, both of which are unfavorable for RNA cleavage (26, 28, 39). We, therefore, propose that the paused RNAP conformation is preserved in TEC-3p, which is based on the hisP sequence, thus making the target phosphodiester bond inaccessible for the TL action. In support of this notion, other variants of this TEC with substitutions in the RNA 3′-end are also characterized by significantly lower RNA cleavage rates in comparison with our measurements for TEC-2 and its variants (kobs of 0.0025/0.0005 s−1 versus 0.017/0.0024 s−1 for +2A/+2U complexes; Ref. 20 and Fig. 7). The resistance of TEC-3p to Gre-induced cleavage suggests that RNAP conformation in this complex is also incompatible with the Gre action. In particular, the kinked BH may prevent correct positioning of the RNA transcript and GreA in the active site (Fig. 1, center).
Recent studies demonstrated that long backtracked RNA induces similar changes in TEC-9 (RNAP ratcheting) (9), probably explaining the low reactivity of this complex. Thus, both paused and long-backtracked complexes likely adopt an inactive conformation with distorted active site in which the TL or GreA cannot activate RNA cleavage. Interestingly, the rates of TL/Gre-independent RNA cleavage are comparable for different RNAPs and different types of transcription complexes (Figs. 6 and 7 and Table 1), suggesting that they correspond to an “unactivated” state of the active site. In addition, the presence of a long RNA segment in the secondary RNAP channel may directly prevent GreA binding (9). The GreB factor has likely evolved to specifically activate RNA cleavage in such extensively backtracked TECs, but its existence is limited to E. coli and related bacteria (40, 41). Thus, RNAP stalling at genomic pause sites or long backtracking events, which make it resistant to both intrinsic and Gre-dependent RNA cleavage, might have important consequences for transcription regulation, resolution of the transcription-replication conflicts, and coordination of transcription with translation (42–44).
Experimental procedures
DNA and proteins
DNA and RNA oligonucleotides used for site-directed mutagenesis and for TEC assembly were purchased from Syntol and DNA Synthesis (Moscow). Mutant variants of rpoC genes with deletions of the TL-encoding segments (Δ931–1137→3Gly, Δ1254–1272→3Gly, and Δ1237–1255→3Ala in E. coli, D. radiodurans, and T. aquaticus RNAPs, respectively) were obtained by site-directed mutagenesis. Wild-type and mutant E. coli, D. radiodurans, and T. aquaticus core RNAPs and corresponding GreA factors were purified as previously described (24, 32, 45, 46).
Analysis of RNA cleavage
Analysis of RNA cleavage was performed in TECs assembled from RNAP core enzymes and synthetic oligonucleotides as described (24). In all cases, RNA oligonucleotides were 5′-labeled with γ-[32P]ATP and T4 polynucleotide kinase. Most measurements were performed in transcription buffer containing 40 mm Tris-HCl (pH 7.9 at 25 °C) and 40 mm KCl. The samples were incubated at different temperatures (10, 20, 30, and 37 °C for E. coli and D. radiodurans RNAPs or 40 °C for TaqRNAP), and the reactions were started by the addition of 10 mm MgCl2. For experiments presented in supplemental Fig. S1, the buffers were equilibrated at 25 °C using correction coefficients so that their pH values at 10, 20, 30, and 37/40 °C were 7.9. For TEC-3p, the reactions were performed in buffer containing 40 mm Tris-HCl (pH 8.5 at 25 °C) and 40 mm KCl in the presence of 20 mm MgCl2 (18). GreA factors were added to 3 μm 3–5 min before the addition of MgCl2, when indicated. The reactions were terminated after increasing time intervals by the addition of stop solution containing 8 m urea, 1× Tris borate-EDTA and 0.5 m EDTA. In the case of very fast kinetics, the measurements were performed in a rapid quench-flow instrument (QFM-400, BioLogic). RNA products were separated by 23% (acrylamide:bisacrylamide = 20:3) denaturing PAGE. The data were fitted to the single-exponential equation, and the observed reaction rates (kobs) were calculated as previously described (24, 32). All experiments were independently repeated 3–5 times, and mean values and standard deviations were determined. The Ea, ΔG#, and ΔΔG# values were derived from the kobs values using the formulae shown in Figs. 3 and 4 as described (31, 32). The apparent Kd values for GreA were determined from the GreA titration curves using the hyperbolic equation,
| (Eq. 1) |
where P is the amount of cleaved RNA product at a given GreA concentration, and Pmax is the maximal cleavage at saturation.
Author contributions
N. M., D. E., and A. K. designed the study and analyzed the data. N. M. and D. E. performed the experiments. A. K. wrote the manuscript with input from the other authors.
Supplementary Material
This work was supported by the Russian Science Foundation (14-14-01074; to A. K.), a grant from the President of Russian Federation for Young Scientists (MK 8983.2016.4; to N. M.), Russian Foundation for Basic Research (16-34-60237; to D. E.), and the Russian Academy of Sciences Presidium Programme on Molecular and Cellular Biology. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1 and S2 and Fig. S1.
- RNAP
- RNA polymerase
- TEC
- transcription elongation complex
- TL
- trigger loop
- Eco
- E. coli
- Dra
- D. radiodurans
- Taq
- Thermus aquaticus
- BH
- bridge helix
- ΔG#
- free energy of activation.
References
- 1. Zenkin N., Yuzenkova Y., and Severinov K. (2006) Transcript-assisted transcriptional proofreading. Science 313, 518–520 [DOI] [PubMed] [Google Scholar]
- 2. Erie D. A., Hajiseyedjavadi O., Young M. C., and von Hippel P. H. (1993) Multiple RNA polymerase conformations and GreA: control of the fidelity of transcription. Science 262, 867–873 [DOI] [PubMed] [Google Scholar]
- 3. Sosunova E., Sosunov V., Epshtein V., Nikiforov V., and Mustaev A. (2013) Control of transcriptional fidelity by active center tuning as derived from RNA polymerase endonuclease reaction. J. Biol. Chem. 288, 6688–6703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sydow J. F., Brueckner F., Cheung A. C., Damsma G. E., Dengl S., Lehmann E., Vassylyev D., and Cramer P. (2009) Structural basis of transcription: mismatch-specific fidelity mechanisms and paused RNA polymerase II with frayed RNA. Mol. Cell 34, 710–721 [DOI] [PubMed] [Google Scholar]
- 5. Komissarova N., and Kashlev M. (1997) RNA polymerase switches between inactivated and activated states By translocating back and forth along the DNA and the RNA. J. Biol. Chem. 272, 15329–15338 [DOI] [PubMed] [Google Scholar]
- 6. Komissarova N., and Kashlev M. (1997) Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3′ end of the RNA intact and extruded. Proc. Natl. Acad. Sci. U.S.A. 94, 1755–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nudler E., Mustaev A., Lukhtanov E., and Goldfarb A. (1997) The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell 89, 33–41 [DOI] [PubMed] [Google Scholar]
- 8. Sosunov V., Sosunova E., Mustaev A., Bass I., Nikiforov V., and Goldfarb A. (2003) Unified two-metal mechanism of RNA synthesis and degradation by RNA polymerase. EMBO J. 22, 2234–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sekine S., Murayama Y., Svetlov V., Nudler E., and Yokoyama S. (2015) The ratcheted and ratchetable structural states of RNA polymerase underlie multiple transcriptional functions. Mol. Cell 57, 408–421 [DOI] [PubMed] [Google Scholar]
- 10. Cheung A. C., and Cramer P. (2011) Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature 471, 249–253 [DOI] [PubMed] [Google Scholar]
- 11. Guo H., and Price D. H. (1993) Mechanism of DmS-II-mediated pause suppression by Drosophila RNA polymerase II. J. Biol. Chem. 268, 18762–18770 [PubMed] [Google Scholar]
- 12. Izban M. G., and Luse D. S. (1993) SII-facilitated transcript cleavage in RNA polymerase II complexes stalled early after initiation occurs in primarily dinucleotide increments. J. Biol. Chem. 268, 12864–12873 [PubMed] [Google Scholar]
- 13. Lange U., and Hausner W. (2004) Transcriptional fidelity and proofreading in Archaea and implications for the mechanism of TFS-induced RNA cleavage. Mol. Microbiol. 52, 1133–1143 [DOI] [PubMed] [Google Scholar]
- 14. Whitehall S. K., Bardeleben C., and Kassavetis G. A. (1994) Hydrolytic cleavage of nascent RNA in RNA polymerase III ternary transcription complexes. J. Biol. Chem. 269, 2299–2306 [PubMed] [Google Scholar]
- 15. Orlova M., Newlands J., Das A., Goldfarb A., and Borukhov S. (1995) Intrinsic transcript cleavage activity of RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 92, 4596–4600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vassylyev D. G., Vassylyeva M. N., Zhang J., Palangat M., Artsimovitch I., and Landick R. (2007) Structural basis for substrate loading in bacterial RNA polymerase. Nature 448, 163–168 [DOI] [PubMed] [Google Scholar]
- 17. Yuzenkova Y., Bochkareva A., Tadigotla V. R., Roghanian M., Zorov S., Severinov K., and Zenkin N. (2010) Stepwise mechanism for transcription fidelity. BMC Biol. 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang J., Palangat M., and Landick R. (2010) Role of the RNA polymerase trigger loop in catalysis and pausing. Nat. Struct. Mol. Biol. 17, 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuzenkova Y., and Zenkin N. (2010) Central role of the RNA polymerase trigger loop in intrinsic RNA hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 107, 10878–10883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bae B., Nayak D., Ray A., Mustaev A., Landick R., and Darst S. A. (2015) CBR antimicrobials inhibit RNA polymerase via at least two bridge-helix cap-mediated effects on nucleotide addition. Proc. Natl. Acad. Sci. U.S.A. 112, E4178–E4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laptenko O., Lee J., Lomakin I., and Borukhov S. (2003) Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 22, 6322–6334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sosunova E., Sosunov V., Kozlov M., Nikiforov V., Goldfarb A., and Mustaev A. (2003) Donation of catalytic residues to RNA polymerase active center by transcription factor Gre. Proc. Natl. Acad. Sci. U.S.A. 100, 15469–15474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roghanian M., Yuzenkova Y., and Zenkin N. (2011) Controlled interplay between trigger loop and Gre factor in the RNA polymerase active centre. Nucleic Acids Res. 39, 4352–4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Esyunina D., Turtola M., Pupov D., Bass I., Klimašauskas S., Belogurov G., and Kulbachinskiy A. (2016) Lineage-specific variations in the trigger loop modulate RNA proofreading by bacterial RNA polymerases. Nucleic Acids Res. 44, 1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zenkin N. (2014) Multiple personalities of the RNA polymerase active centre. Microbiology 160, 1316–1320 [DOI] [PubMed] [Google Scholar]
- 26. Hein P. P., Kolb K. E., Windgassen T., Bellecourt M. J., Darst S. A., Mooney R. A., and Landick R. (2014) RNA polymerase pausing and nascent-RNA structure formation are linked through clamp-domain movement. Nat. Struct. Mol. Biol. 21, 794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Toulokhonov I., Zhang J., Palangat M., and Landick R. (2007) A central role of the RNA polymerase trigger loop in active-site rearrangement during transcriptional pausing. Mol. Cell 27, 406–419 [DOI] [PubMed] [Google Scholar]
- 28. Weixlbaumer A., Leon K., Landick R., and Darst S. A. (2013) Structural basis of transcriptional pausing in bacteria. Cell 152, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nayak D., Voss M., Windgassen T., Mooney R. A., and Landick R. (2013) Cys-pair reporters detect a constrained trigger loop in a paused RNA polymerase. Mol. Cell 50, 882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Čabart P., Jin H., Li L., and Kaplan C. D. (2014) Activation and reactivation of the RNA polymerase II trigger loop for intrinsic RNA cleavage and catalysis. Transcription 5, e28869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lonhienne T., Gerday C., and Feller G. (2000) Psychrophilic enzymes: revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim. Biophys. Acta 1543, 1–10 [DOI] [PubMed] [Google Scholar]
- 32. Miropolskaya N., Esyunina D., Klimasauskas S., Nikiforov V., Artsimovitch I., and Kulbachinskiy A. (2014) Interplay between the trigger loop and the F loop during RNA polymerase catalysis. Nucleic Acids Res. 42, 544–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kulbachinskiy A., Bass I., Bogdanova E., Goldfarb A., and Nikiforov V. (2004) Cold sensitivity of thermophilic and mesophilic RNA polymerases. J. Bacteriol. 186, 7818–7820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fields P. A. (2001) Review: Protein function at thermal extremes: balancing stability and flexibility. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 129, 417–431 [DOI] [PubMed] [Google Scholar]
- 35. Miropolskaya N., Artsimovitch I., Klimasauskas S., Nikiforov V., and Kulbachinskiy A. (2009) Allosteric control of catalysis by the F loop of RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 106, 18942–18947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fouqueau T., Zeller M. E., Cheung A. C., Cramer P., and Thomm M. (2013) The RNA polymerase trigger loop functions in all three phases of the transcription cycle. Nucleic Acids Res. 41, 7048–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaplan C. D., Larsson K. M., and Kornberg R. D. (2008) The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by α-amanitin. Mol. Cell 30, 547–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nielsen S., and Zenkin N. (2013) Transcript assisted phosphodiester bond hydrolysis by eukaryotic RNA polymerase II. Transcription 4, 209–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Petushkov I., Pupov D., Bass I., and Kulbachinskiy A. (2015) Mutations in the CRE pocket of bacterial RNA polymerase affect multiple steps of transcription. Nucleic Acids Res. 43, 5798–5809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koulich D., Orlova M., Malhotra A., Sali A., Darst S. A., and Borukhov S. (1997) Domain organization of Escherichia coli transcript cleavage factors GreA and GreB. J. Biol. Chem. 272, 7201–7210 [DOI] [PubMed] [Google Scholar]
- 41. Borukhov S., Sagitov V., and Goldfarb A. (1993) Transcript cleavage factors from E. coli. Cell 72, 459–466 [DOI] [PubMed] [Google Scholar]
- 42. Dutta D., Shatalin K., Epshtein V., Gottesman M. E., and Nudler E. (2011) Linking RNA polymerase backtracking to genome instability in E. coli. Cell 146, 533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Larson M. H., Mooney R. A., Peters J. M., Windgassen T., Nayak D., Gross C. A., Block S. M., Greenleaf W. J., Landick R., and Weissman J. S. (2014) A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science 344, 1042–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang J., and Landick R. (2016) A two-way street: regulatory interplay between RNA polymerase and nascent RNA structure. Trends Biochem. Sci. 41, 293–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Esyunina D. M., and Kulbachinskiy A. V. (2015) Purification and characterization of recombinant Deinococcus radiodurans RNA polymerase. Biochemistry 80, 1271–1278 [DOI] [PubMed] [Google Scholar]
- 46. Svetlov V., and Artsimovitch I. (2015) Purification of bacterial RNA polymerase: tools and protocols. Methods Mol. Biol. 1276, 13–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.