

Abstract
Background
Hypertrophic cardiomyopathy (HCM) patients with early repolarization (ER) pattern are at higher risk of ventricular arrhythmia, yet the genetic background of this situation has not been well investigated. Here we report novel trigenic mutations detected in a Chinese family of obstructive HCM with ER and short QT syndrome (SQTS).
Methods
Proband and family members underwent detailed medical assessments. DNAs were extracted from peripheral blood leukocytes for genetic screening with next generation method. The functional characterization of the mutation was conducted in TSA201 cells with patch-clamp experiment.
Results
The proband was a 52-year-old male who had a ER pattern ECG in inferioral-lateral leads with atrioventricular block and QTc of 356 ms. He also suffered from severe left ventricular hypertrophy and dysfunction. Targeted sequencing revealed trigenic mutations: c.700G>A/p.E234K in DES, c.2966G>A/p.R989H in MYPN, and c.5918G>C/p.R1973P in CACNA1C. All mutations were also detected in his daughter with ER and mild myocardium hypertrophy. The CACNA1C-R1973P mutation caused significant reduction (68.4%) of ICa compared to CACNA1C-WT (n = 14 and 14, P < 0.05). The computer modeling showed that all 3 mutations were highly disease-causing. The proband received the CRT-D (cardiac resynchronizing therapy) implantation, which lowered the left ventricular outflow tract gradient (LVOTG, 124 mmHg pre vs. 27 mmHg post) and restored the LV function (LVEF 40% pre vs. 63% post).
Conclusions
The study reveals a novel CACNA1C mutation underlying the unique ER pattern ECGs with SQTS. It also shows the rare trigenic mutations are the pathogenic substrates for the complicated clinical manifestation in HCM patients.
Electronic supplementary material
The online version of this article (doi:10.1186/s12967-017-1180-1) contains supplementary material, which is available to authorized users.
Keywords: Hypertrophic cardiomyopathy (HCM), Early repolarization (ER), Calcium channels, Genetics, Cardiac resynchronization therapy (CRT)
Background
Hypertrophic cardiomyopathy (HCM) is the most common cause of sudden cardiac death (SCD) in the young as well as in the trained athletes, and is also a critical substrate for heart failure disability at any age. HCM is a clinical heterogeneous but relatively common form of genetic heart defect transmitted as autosomal dominant trait (affecting 1 in 500 people). It is characterized by unexplained cardiac hypertrophy, myocyte disarray, and fibrosis [1]. The yield of genetic testing among HCM cases is as high as ~70%, and comprehensive or targeted (MYH7, MYBPC3, TNNI3, TNNT2, TPM1) HCM genetic testing is recommended for patients with HCM and first-degree relatives based on clinical history, family history, and electrocardiographic/echocardiographic phenotype [2, 3]. HCM patients with J point and ST segment elevation are related to higher risk of ventricular arrhythmias and SCD [4]. However, the genetic background of this situation has not been deeply investigated before.
Early repolarization (ER) pattern is defined as J wave (>0.1 mV) with or without ST segment elevation in at least two continuous leads in standard 12-lead ECGs [5]. It has been related with malignant ventricular tachycardia and SCD [6]. ER pattern also has a tendency of heritability [7]. Several ion channel mutations have been linked to ER syndrome, such as genes in IK,ATP and INa [8–10]. It is worth mentioning that, cardiac calcium channel mutations are related to ER with shortened QTc interval because of its influence on the action potential duration (APD). These patients had higher tendencies for cardiac event and SCD [8].
Here we report a Chinese family of HCM with early repolarization pattern, and borderline short QTc interval. The clinical phenotypes included severe cardiac hypertrophy and left ventricular outflow tract obstruction, left ventricular dysfunction, atrial fibrillation, and sustained ventricular tachycardia. Novel trigenic mutations in DES/MYPN/CACNA1C are detected in the proband and his family. The clinical characteristics and genetic background of the family members are investigated in detail.
Methods
For details, please see Additional file 1.
Clinical history
The study was approved by the ethics committee of 3rd People’s Hospital of Wuxi (Wuxi, China) and conducted according to Declaration of Helsinki principles. The informed consents were obtained from all the participants, who belonged to Asian. The clinical assessments included medical history, detailed physical examination, blood tests, electrocardiogram (ECG), ultrasonic cardiogram (UCG), and coronary artery angiography. Patients were clinically diagnosed according to the 2011 ACC/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy [2].
Genetic studies
DNAs were extracted from peripheral blood leukocytes for genetic screenings with next generation method. The identification of known pathogenic variants was based on mutations previously reported to cause cardiovascular disease in the literature. Novel variants considered to be pathogenic were either: (1) stop/frameshift variants; (2) missense mutations positioned in the amino acid conservative region across species; (3) splice-site variations fulfilling the GT-AT rules; or (4) predicted to be possibly damaging or disease-causing by the bioinformatic programs of PolyPhen-2, PROVEAN and MutationTaster2.
The structures of DES-E234K and MYPN-R989H proteins were modeled with protein structure homology modelling, through the online workspace of SWISS-MODEL (http://swissmodel.expasy.org). The mutated sites were highlighted and visualized using VMD1.9.2 (University of Illinois at Urbana-Champaign).
Expanded validation
DNA samples of all the participants were taken for the expanded validations. Coding regions of the mutations identified as described above were amplified by polymerase chain reaction (PCR) for conventional direct sequencing. Purified PCR products were cycle-sequenced on an ABI 3500 Genetic Analyzer (Applied Biosystems, CA). The sequencing results were analyzed by Mutation Surveyor (Softgenetics, PA) and reconfirmed by the same procedure.
Functional characteristics of calcium mutation
Functional characterization of the CACNA1C-R989H mutation was conducted by co-expression of CACNB2B and CACNA2D1 in TSA201 cells, which is a human embryonal kidney with SV40 transformed. Whole cell currents were recorded at room temperature using patch clamp techniques as previously described. Standard whole-cell patch clamp technique was used to measure CACNA1C wild type and mutant calcium currents at room temperature (22–24 °C) with the use of an Axopatch 200 B amplifier, Digidata 1440 A and pclamp version 10.4 software (Axon Instruments, Sunnyvale, CA). Microelectrodes were pulled on a P-97 puller (Sutter Instruments, Novato, CA) and fire polished to a final resistance of 1.5–3 MΏ. Series resistance was compensated by 80–85%. Currents were filtered at 1 kHz and digitized at 5 kHz with an eight-pole Bessel filter. Data were analyzed using Clampfit (Axon Instruments, Sunnyvale, CA), Excel (Microsoft, Redmond, WA), and fitted with Origin 8 (OriginLab Corporation, Northampton, MA) software. The steady-state inactivation curve was fitted with a Boltzmann function: I Ca /I Ca max = {1 + exp [(V − V 1/2 )/k]} −1, where V 1/2 and k are the half-maximal voltage of inactivation and the slope factor respectively.
Statistical analysis
All data points are shown as the mean value and bars represent the standard error of the mean. The Student’s unpaired t test was performed to determine statistical significance between two groups. P < 0.05 was considered to be statistically significant.
Results
Clinical history
The proband was a 52-year-old male presented for chest discomfort and stress-induced dyspnea in 2008. The ECG showed sinus bradycardia (46 bpm), I° atrioventricular block (PR interval = 240 ms) and complete left bundle branch block (CLBBB). ER pattern presented in inferior-lateral leads (II, aVF and V4–V6). Convex ST-segment elevations could be seen in lateral leads V4–V6. Other possible causes of abnormal ST-segment elevations were ruled out, including acute myocardial infarction and ventricular aneurysm. The QTc was 356 ms. The UCG showed LV hypertrophy (LVPW 17 mm, IVS 21 mm) with the basal LVOTG of 48 mmHg. The patient was diagnosed with obstructive hypertrophic cardiomyopathy and received drug therapy of beta-blockers (metoprolol tartrate, 47.5 mg qd) hereafter. Because bradycardia and atrioventricular block has gotten worse since then, metoprolol was withdrawn. In 2009, the patient was admitted for aggravated dyspnea and chest discomfort. UCG showed severe LV hypertrophy (LVPW 18 mm, IVS 25 mm) with the LVOTG of 143 mmHg at rest. He received the percutaneous transluminal alcohol septal ablation. The symptoms were relieved and the LVOTG was decreased to 45 mmHg thereafter.
During the follow-up in 2013, the patient complained of occasional palpitation, dizziness and dyspnea. The ECG showed atrial fibrillation with slow ventricular response of 40–50 bpm. UCG showed the recurrence of LV hypertrophy and increased LVOTG. Transaortic septal myectomy and pacemaker implantation were advised, but the patient refused. In Dec 2013 the patient was presented to the emergency department for continuous palpitation and dyspnea, with compromised blood pressure. The ECG showed persistent ventricular tachycardia (VT) with the heart rate of 150 bpm. The UCG showed LV dysfunction (LVEF, 40%), LV hypertrophy and severe LVOT obstruction (LVOTG, 124 mmHg at rest). After the VT was terminated with intravenous application of lidocaine, he presented slow heart rate (50 bpm) with frequent ventricular premature contractions (PVCs). Prominent J wave presented in leads I, II, aVL, aVF, and V4–V6, with the amplitudes ranging from 0.2 to 0.4 mV. Covex ST-segment elevation could be seen in lateral leads V4–V6. The QTc averaged 356 ms in V5 and V6 leads. Furthermore, VT tended to recur easily. The heart rhythm was stabilized with temporary pacing (lifting the HR to 80 bpm) combined with amiodarone. In January 2014, the patient received the implantation of a cardiac resynchronizing therapy (CRT-D). The symptoms and the heart function were greatly increased thereafter. Interestingly, the LVOTG was dramatically decreased to 27 mmHg. The effectiveness of the CRT-D persisted during the follow-up, and the LVOTG was further lowered to 17 mmHg at 12 months after the implantation. Figure 1a–d showed the ECGs and the UCGs of the index patient.
Fig. 1.
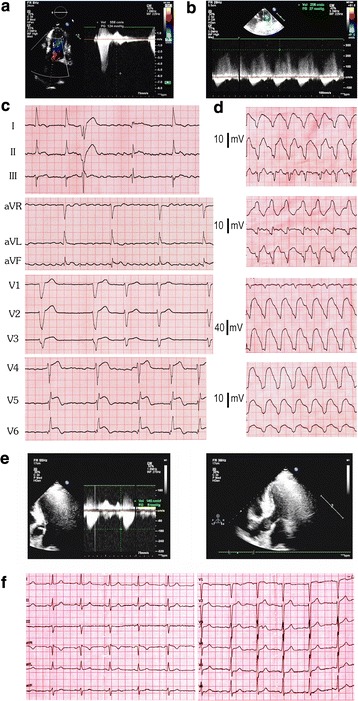
The UCG and ECG of the proband and his daughter. a The UCG before the CRT-D implantation. Severe myocardial hypertrophy and left ventricle outflow tract obstruction with the gradient of 124 mmHg. b The LVOT obstruction was greatly relieved and the gradient dropped to 27 mmHg after 1 week of the CRT-D implantation. c 12 Lead ECG in 2013 showed atrial fibrillation with slow ventricular rates. J waves could be seen in leads I, II, aVL, aVF, and V4–V6, with the amplitudes ranging from 0.2 to 0.4 mV. Covex-shaped ST segment elevation could be seen in lateral leads V4–V6. The QTc averaged 356 ms in V5 and V6 leads. d The ECG of the sustained ventricular tachycardia. e The UCG of the daughter showed left ventricle hypertrophy and mild LVOT obstructions. f The ECG of the daughter showed abnormal Q waves in leads V1–V3, but we didn’t find any ER waves
The patient’s 30-year-old daughter was asymptomatic. The UCG showed left ventricle hypertrophy with LVOTG of 8 mmHg. Her ECG showed abnormal Q waves in leads V1–V3 without ER pattern in any leads (Fig. 1e, f). The patient’s elder sister had no symptoms, and her UCG and ECG were normal. The mother of the proband died suddenly at the age of 70 for unknown origin. Table 1 displayed the UCG parameters of the patient and his family members.
Table 1.
UCG characteristics of the proband and family members
| Proband | Daughter | Elder sister | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 2008 | 2009 | 2010 | 2013 | 2014 (1 w post CRT-D) | 2014 (6 m post CRT-D) | 2015 (12 m post CRT-D) | |||
| IVS (mm) | 21 | 25 | 15 | 23 | 23 | 22 | 21 | 13 | 10 |
| LVPW (mm) | 17 | 18 | 18 | 18 | 23 | 21 | 20 | 13 | 9 |
| LVOTG (mmHg) | 48 | 143 | 45 | 124 | 27 | 16 | 17 | 8 | – |
| LVEDd (mm) | 50 | 52 | 50 | 56 | 54 | 54 | 52 | 55 | 55 |
| LVESd (mm) | 31 | 32 | 31 | 40 | 30 | 31 | 30 | 31 | 34 |
| LVEF % | 60 | 55 | 60 | 40 | 63 | 60 | 60 | 64 | 60 |
IVS intraventricular septum, LVPW left ventricular post wall, LVOTG left ventricle outflow tract gradient, LVEDd left ventricle end-diastolic diameter, LVESd left ventricle end-systolic diameter, LVEF left ventricular ejection fraction
Genetic screening identified candidate mutations
DNA samples of the proband were applied for targeted exome sequencing of 120 genes implicated in inherited cardiovascular diseases (Additional file 1: Table S1). The average sequencing depths on the targeted regions exceeded 100.0. The sample covered more than 98.0% of the targeted regions. We identified 3 novel missense mutations in the proband: c.700G>A/p.E234K in the exon3 of desmin (DES), c.2966G>A/p.R989H in the exon24 of myopalladin (MYPN), and c.5918G>C/p.R1973P in exon46 of CACNA1C (Fig. 2; Table 2). All mutations were indicated as pathogenic mutation by MutationTaster2, Polyphen2, and PROVEAN. Meanwhile, no pathogenic variant was discovered in typical HCM candidate genes, such as β-myosin heavy chain (MYH7), cardiac myosin binding protein-C (MYBPC3), α-tropomyosin (TPM1), cardiac troponin T (TNNT2), myosin regulatory light chain (MYL2), myosin essential light chain (MYL3), cardiac troponin I (TNNI3), and cardiac a-actin (ACTC1), Burke et al. [11].
Fig. 2.
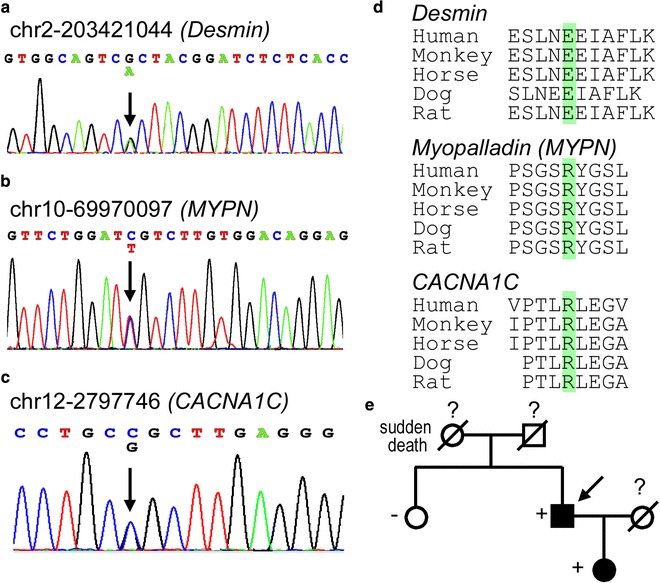
The gene mutations detected in the family members. a The mutated site of the desmin (DES), c.700G>A/p.E234K. b The mutated site of the Myopalladin (MYPN), c.2966G>A/p.R989H. c The mutated site of the gene CACNA1C, c.5918G>C/p.R1973P. d The conservation of the mutated site. All 3 sites were highly conserved among different species. e The pedigree chart of the family. The symptom-free daughter showed mild myocardial hypertrophy and left ventricle outflow tract obstruction, but her ECG did not show abnormal ST segments elevations. The DES, MYPN and CACNA1C mutations were detected in the daughter but not in the older sister, whose UCG did not show any abnormalities
Table 2.
Genetic mutations carried by the proband
| Position | Sequence | Protein | DNA changes | AA changes | MutationTaster2 | Polyphen2 | PROVEAN | |
|---|---|---|---|---|---|---|---|---|
| CACNA1C | chr12-2797746 | NM_000719 | Q13936 | c.5918G>C | p.R1973P | Disease causing (prob: 0.999) | Possibly damaging (score: 0.887) | Deleterious (score: −3.153) |
| DES | chr2-220285033 | NM_001927 | P17661 | c.700G>A | p.E234K | Disease causing (prob: 0.999) | Probably damaging (score 0.999) | Deleterious (score: −3.292) |
| MYPN | chr10-69970097 | NM_001256268 | NP_001243197.1 | c.2966G>A | p.R989H | Disease causing (prob: 0.999) | Probably damaging (score 1.000) | Deleterious (score: −2.693) |
In the expanded validation test, the elder sister of the proband did not carry any of the mutations detected above. However, the 3 mutations were also detected in his daughter. All the mutated amino acid residues were conserved among different species, indicating the sites were conserved through the evolution.
Computational analysis of the DES and MYPN mutations
For DES and MYPN, the structures of the mutated domains were modeled with SWISS-MODEL by protein structure homology modeling. The model of DES was built from the segment of 149-253 amino-acid sequence (Fig. 3a). The c.700G>A mutation in DES predicted the 234th acidic glutamic acid to be replaced by an alkaline residue, lysine, which influence the dimerization of the gene. The rod structure of normal Desmin was predicted to be lost. The model of MYPN was built from the segment of amino-acid sequence between 944 and 1044 (Fig. 3b). The 989th aliphatic arginine was replaced by a heterocyclic residue, histidine, in mutated myopalladin, which would lead to change the protein conformation and influence the myopalladin interaction with ACTN. The function of this region was predicted to be totally lost.
Fig. 3.
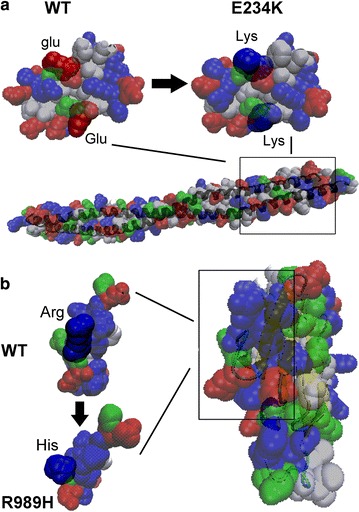
Structure modeling of the mutated domains of Desmin and Myopalladin. a The model of DES built from the segment of 149th–253th amino-acids sequence. The mutation caused the 234th acidic residue glutamic acid to be replaced by an alkaline residue lysine, which influence the dimerization of protein. b The model of MYPN built from the segment of 944th–1044th amino-acids sequence. The mutation caused the 989th liphatic amino-acid arginine to be replaced by a heterocyclic residue histidine in mutated myopallin, which changed the protein conformation and was predicted to influence the myopalladin interaction with ACTN
Functional characterization of R1937P mutant in CACNA1C
CACNA1C encodes for the α-subunit of cardiac L-type calcium channels. The mutation of c.5918G>C was predicted to cause the 1973th basic arginine to be replaced by the nonpolar/hydrophobic proline in the C-terminal tail of the ion channel (Fig. 4a).
Fig. 4.
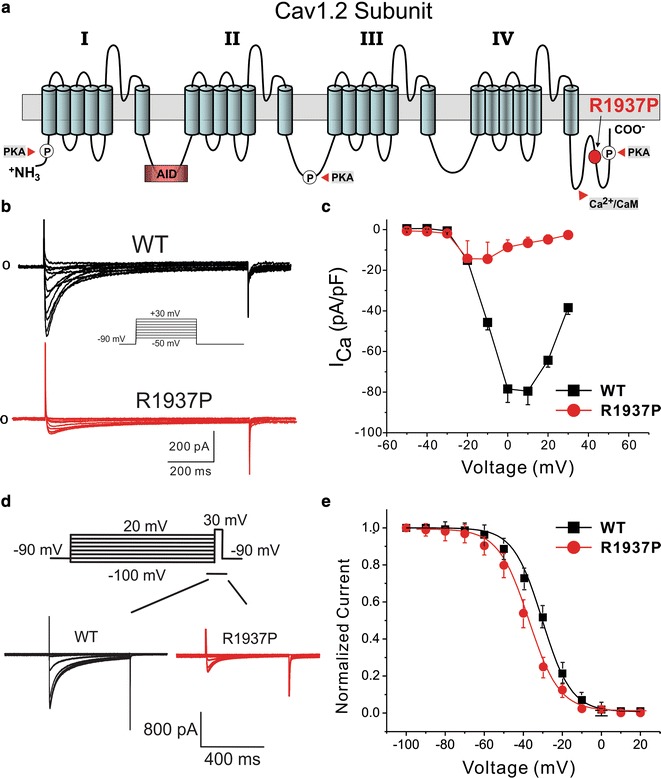
Functional expression studies of the R1973P mutant in CACNA1C. a 2D Topology of the mutated site. b Representative whole-cell calcium current traces (ICa) recorded in TSA201 cells transfected with WT-CACNA1Cand R1973-CACNA1C co-expressed with CACNB2b and CACNA2D1 subunit genes. ICa traces recorded are in response to the voltage clamp protocol shown at the top inset. c Current–voltage relationship (I–V curve) of WT and R1973P variant. Each point data represents the mean ± SEM of 14 experiments. d Representative ICa recorded in response to the voltage clamp protocol shown at the inset on top in TSA201 cells expressing WT or R1973P mutant in CACNA1C. Peak currents were normalized to their respective maximum values and plotted against the conditioning potential to obtain the steady-state inactivation curves. e R1973P mutant channels showed a significantly more negative mid-inactivation potential compared to WT channels. R1973P (red circle) V 1/2 = −34.1 ± 0.3 mV, K = 6.5 ± 0.3 vs; WT (black squares) V 1/2 = −30.3 ± 0.3 mV, K = 7.4 ± 0.2. Each point data represents the mean ± S.E.M of 4–6 experiments
Typical ICa tracings of voltage-dependent activation from CACNA1C-WT and R1937P mutation are shown in Fig. 4b with holding potential at −90 mV to various depolarization potentials (see inset). Analysis of the current–voltage relationship shows that the R1937P mutant dramatically decreased peak current density at −10 mV by 31.6% from −45.7 ± 3.6 pA/pF (WT, n = 14) to −14.45 ± 8.2 pA/pF (R1937P, n = 14, P = 0.001), indicating a marked loss-of-function electrophysiological phenotype (Fig. 4c).
Steady-state inactivation was assessed by a standard two-pulse voltage-clamp protocol. Typical tracings of inactivation from CACNA1C-WT and -R1937P mutation are shown in Fig. 4d. There was 3.8 negative shift in V1/2 of inactivation for R1937P containing channels (−34.1 ± 0.3 mV, n = 4) compared with CACNA1C-WT (−30.3 ± 0.3 mV, n = 6, P = 0.07, Fig. 4e). The respective k slope factor also remained unchanged at 7.4 ± 0.2 (CACNA1C-WT, n = 4) and at 6.5 ± 0.3 (CACNA1C-R1937P, n = 6; P = 0.23).
Discussion
Hypertrophic cardiomyopathy (HCM) is characterized by asymmetric hypertrophy of interventricular septum, disorganization of cardiomyocytes, as well as myocardial interstitial fibrosis. The basis of HCM has been ascribed to multiple etiologies; however, in 1989 researchers first mapped a genetic mutation for HCM to chromosome [12]. Subsequently, hundreds mutations have been found in HCM patients. Most mutations involve the myofilaments of the cardiac sarcomere (MYH7, MYBPC3, et al.); however, there is increasing awareness of non sarcomeric mutations as well, such as Z-disk or intracellular calcium modulators.
Comprehensive testing of five HCM genes is strongly recommended to assess patients with HCM by clinical guidelines [2]. Whereas Nextgen sequencing approaches have facilitated much broader testing panels to be widely available, such as the one we employed in this study (Table 2). In both the proband and his daughter, we simultaneously identified trigenic mutations of p.E234K in the DES, p.R989H in the MYPN, and p.R1973P in CACNA1C.
There was few reported trigenic mutation related with cardiomyopathy. One study described a case of HCM with trigenic mutations (LAMA4, PKP2 and TTN) [13], but the ECG characteristics of the patient was not available. On the relationship between gene mutation and QT interval, one study showed that patients with HCM-related gene mutation frequently exhibited QT interval disruption, which was related with increased occurrence of ventricular arrhythmias [14]. Although CACNA1C mutation very likely underlined the shortened QT intervals in our case, it was hard to determine the role of each individual mutation in the genesis of the clinical phenotypes, due to incomplete family history as well as lacking of previous references.
It is well known that QT intervals are slightly prolonged in most HCM patients due to the increased late sodium current and fibrosis [15]. However, the QTc of this proband is significantly short. The intervals from the J points to the peak of T waves were approximately 120 ms. Combined with typical ER in the inferioral-lateral leads, the characteristics of short QT interval might be attributed to the mutation p.R1973P in CACNA1C.
CACNA1C lie in chromosome 12, coding for α subunit of the L-type calcium channels. The genetic defects in ICa,L have been linked with LQTS [16], Brugada syndrome [17], ERS [8, 9] and SQTS [18]. Of the reported defects, loss-of-function mutations are extremely rare. In this case, the mutated site was located in the C-terminal of the channel protein (Fig. 4a), which is a “hot region” for loss-of-function mutations. It plays an important role in the Ca2+ influx and kinetics procedure, as well as intracellular signal transductions [19, 20]. In order to illuminate the disease-causing ability of the CACNA1C-R1973P mutation, heterologous expressions and patch clamping techniques were conducted. We found out that the R1937P mutant dramatically decreased peak current density by ~68%, while exerting no significant influences on the channel kinetics, which is consistent with the previously reported V2014I mutations in the vicinity area [8]. We speculated that the channel current reductions might be due to the trafficking deficiencies of the channel protein. Decreased ICa,L results in increased net outward currents and shortened cardiomyocyte repolarization period. Due to the transmural discrepancies of outward potassium currents (Ito, IK, etc.), the increase in net outward current result in partial or complete loss of the action potential dome, leading to a transmural voltage gradient that manifests as ER waves. Meanwhile, accelerated repolarization caused by the mutation could lead to subsequent short QT interval in the surface ECG. The SQT4-6 are linked to mutations in different subunits of calcium channel [8, 18]. There is a high prevalence of ER in SQTs [21], suggesting the genetic background of the two diseases may share common fields.
The CACNA1C mutation also linked with the slowed AV conduction and bundle branch block, because ICa,L is the main depolarization current in cardiac conduction cells. Decreased ICa,L hampers the depolarization of these cells and causes delayed conductions. However, it is also noteworthy that the DES mutations could cause atrioventricular blocks [22]. So the conduction delay is probably the outcome of compound genetic defects of both.
The daughter of the patient carries the CACNA1C mutation while her ECG presented far less prominent ER, and normal QTc interval. The incomplete penetrance of the CACNA1C mutation is probably caused by genetic background or certain factors, such as the gender protection. It is well known that sex hormones have significant influence on the ion channel functions as well as on the cardiac repolarization procedure. Testosterone can shorten cardiomyocyte APDs in guinea pigs, through inhibiting the ICa and enhancing IKs [23]. Thus, testosterone’s inhibiting effect on the ICa,L may worsen the effect of the CACNA1C mutation and cause the more severe clinical symptoms of the father. As a matter of fact, ER pattern are more common in male population [24]. Another possible explanation is the matter of age. Experiment on the animals show that the expression of the ICa,L is decreased with aging [25]. Aging has also been related to decreased expressions of potassium and sodium channels [26, 27]. Collectively, the net inward currents during cardiac repolarization decreased with age. The symptoms of ventricular tachycardia appeared when the patient was in his 50’s, indicating the importance of aging in the pathogenesis. Last but not the least, the degree of the myocardial hypertrophy is also a critical factor. The more severe myocardial hypertrophy of the father may increase the transmural dispersions in the cardiac repolarization and enhance the ST segment elevations, as well as the onset of ventricular tachycardia. Previous studies indicate that variants in sarcomere genes may individually or collectively affect cardiac morphology and function, even without causing overt HCM. HCM is transmitted as a dominant trait. However, penetrance is incomplete and lowest at young ages [11]. Identical HCM mutations in the same pedigree can produce distinct LVH morphologies, varying amounts of myocardial fibrosis, and differing susceptibility to arrhythmias. Genetic modifiers, epigenetic differences, and unique environmental factors are likely to influence these variables. But without long term follow-up and deeper translational research, especially more information form the pedigree, it is hard for us to deduce which assumption is the major cause of ventricular arrhythmia. The proband suffered from severe obstructive HCM and his daughter also showed mild myocardial hypertrophy. The CACNA1C mutations have been related to HCM in previous reports. However, there were two other mutations detected in this family, which might be part of the pathogenesis of the HCM. The DES gene is located in chromosome 2, coding for desmin protein. Desmin is the type III intermediate filament protein with the molecular weight of 53.5 kD of 470 residues, which is expressed in various types of muscle cells. In the heart, it integrates the Z disc, the sarcolemma and the myocyte nucleus. Mutations in the DES gene are the causes of the desmin-related myopathies (DRM) [28]. Recent reports linked DES mutations with arrhymogenic right ventricular cardiomyopathy (ARVC) [29] and dilated cardiomyopathy (DCM) [30]. DES mutations are rarely associated with HCM except in few sporadic cases [31]. The MYPN gene is a 145.2 kDa protein of 1320 residue for encoding myopalladin, which is located in chromosome 10. Myopalladin is the component of the myofilament, connecting the Z disc, the sarcolemma and the myocyte nucleus. The myopalladin participate in the regulations of the contraction and adhension of the cardiomyocytes, as well as the gene expressions. Also, the mutations in MYPN were linked with DCM, HCM and restrictive cardiomyopathy (RCM) [32]. Both desmin and myopalladin were important components of the cytoskeleton. The dual mutations cause instability of the sarcomeres and decreased contraction efficiencies. On the other hand, the Ca2+ cycling exert significant impact on the sarcomere contractions. Intracellular Ca2+ level is crucial for the normal contraction of cardiomyocytes [33]. The impaired function of ICa,L lead to decreased influx of Ca2+ ions, influencing the intracellular Ca2+ cycling and reduce the efficiency of sarcomere contraction. The impaired cytoskeletons caused by digenic DES/MYPN mutations may also hamper the CACNA1C expressions. Thus, the three mutations work together in the impairment of the sarcomere functions and subsequent onset of cardiomyopathy.
The managements of HCM include optimized drug therapy and septal reduction therapy, when the LVOT gradients exceed 50 mmHg with clinical symptoms like dyspnea or chest pain. Either surgical myectomy or septal ablation is generally effective in reducing LOVT obstructions and relieving clinical symptoms. The patient developed refractory LV hypertrophy and severe LVOT obstruction 3 years after the septal ablation, due to those mutations. Interestingly, the CRT significantly reduces the outflow tract gradient. The CRT was not considered as the standard therapy for HCM patients [2], but there are some reports on the effectiveness of resynchronization therapy on reducing the outflow tract gradients in HCM patients [34–36]. Although the mechanism is not clear yet, some proposes that the biventricular pacing changed the sequence of the ventricular excitation, which helps to reduce the outflow tract gradient and to reverse the remodeling of the left ventricles [35]. Our report adds clinical support to the effectiveness of the CRT on reducing the outflow tract gradients, which might be a promising strategy for the treatment of HCM patients.
The daughter gave birth to a boy recently. Unfortunately, she refused genetic screening for the baby boy. To observing the progressive nature of the disease, we plan to follow the daughter as well as her son continuously. Meanwhile, we plan to build human induced pluripotent stem cells (hiPSC) from skin tissue of the proband and family members, to replicating the situation and perform further research on stem cell level with all the genetic features in this pedigree.
In this study, we reported a family with ER, SQTSs and HCM. Novel CACNA1C mutation is the pathogenic substrate of the electrophysiological as well as structural abnormities. Meanwhile the rare trigenic mutations make the clinical manifestation complicated and aggravated in this HCM family. At last, the study also suggests the effectiveness of CRT-D on reducing the LVOTG of septal ablation refractory hypertrophic cardiomyopathy.
Authors’ contributions
DH and WXY designed the study. CYH, ZDX, WXH, CCH, ZRJ, SJJ, WXM, TYJ, JWD, WXY, and DH performed clinical evaluations. CYH, HBM and DH coordinated the pathological phenotyping of study subjects. DH supervised and coordinated the laboratory work. CYH, HBM and DH constructed plasmids, and DH and HBM performed the electrophysiology laboratory work. CYH, DH and HBM analyzed, organized and summarized the data. DH and WXY developed the conceptual approaches to data analysis. CYH, DH, HBM and WXY wrote the manuscript. All co-authors contributed to editing of manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors are grateful to Judy Hefferon for technical assistance.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All available to access.
Consent for publication
Yes.
Ethics approval and consent to participate
Approved and available.
Funding
This study was supported by grants from National Natural Science Foundation of China (No. 81670304), China; Independent Research Project of Wuhan University (No. 2042016kf0138), China; Innovation fund from Wuhan Asia Heart Hospital, China; Health and Family Planning Commission of Wuhan (No. WX16A09), China; the Science and Technology Department of Hubei Province (No. 2016CFB476), China; CONACYT #FM201866, Mexico; and the Masons of New York, Florida, Massachusetts, Connecticut, Maryland, Wisconsin, and Rhode Island, USA.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- CACNA1C
α1C subunit of calcium voltage-gated channel
- CLBBB
complete left bundle branch block
- DES
desmin
- ECG
electrocardiogram
- ER
early repolarization
- HCM
hypertrophic cardiomyopathy
- hiPSC
human induced pluripotent stem cells
- MYPN
myopalladin
- SCD
sudden cardiac death
- SQTS
short QT syndrome
- UCG
ultrasonic cardiogram
Additional file
Additional file 1: Table S1. List of genes sequenced for the proband.
Contributor Information
Yanhong Chen, Email: sailorsdog@gmail.com.
Hector Barajas-Martinez, Email: barajash@mmrl.edu.
Dongxiao Zhu, Email: 617785712@qq.com.
Xihui Wang, Email: wangxihui0807@126.com.
Chonghao Chen, Email: wxcch357@163.com.
Ruijuan Zhuang, Email: zhuangrj@126.com.
Jingjing Shi, Email: mao970202@163.com.
Xueming Wu, Email: 1419129406@qq.com.
Yijia Tao, Email: yijiatao@163.com.
Weidong Jin, Email: jinwd.md@163.com.
Xiaoyan Wang, Phone: +86(510)82607391, Email: 1807250234@qq.com.
Dan Hu, Phone: +1(315)7352217, Email: dianah@mmrl.edu.
References
- 1.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH, 3rd, Spirito P, Ten Cate FJ, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for practice guidelines. J Am Coll Cardiol. 2003;42:1687–1713. doi: 10.1016/S0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 2.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Thorac Cardiovasc Surg. 2011;142:e153–e203. doi: 10.1016/j.jtcvs.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 3.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Europace. 2011;13(1077–1109):4. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- 4.Furushima H, Chinushi M, Iijima K, Sanada A, Izumi D, Hosaka Y, Aizawa Y. Ventricular tachyarrhythmia associated with hypertrophic cardiomyopathy: incidence, prognosis, and relation to type of hypertrophy. J Cardiovasc Electrophysiol. 2010;21:991–999. doi: 10.1111/j.1540-8167.2010.01769.x. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–1963. doi: 10.1016/j.hrthm.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 6.Tikkanen JT, Anttonen O, Junttila MJ, Aro AL, Kerola T, Rissanen HA, Reunanen A, Huikuri HV. Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med. 2009;361:2529–2537. doi: 10.1056/NEJMoa0907589. [DOI] [PubMed] [Google Scholar]
- 7.Noseworthy PA, Tikkanen JT, Porthan K, Oikarinen L, Pietila A, Harald K, Peloso GM, Merchant FM, Jula A, Vaananen H, et al. The early repolarization pattern in the general population: clinical correlates and heritability. J Am Coll Cardiol. 2011;57:2284–2289. doi: 10.1016/j.jacc.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpon E, Hu D, Desai M, Borggrefe M, Haissaguerre M, Kanter R, Pollevick GD, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7:1872–1882. doi: 10.1016/j.hrthm.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haissaguerre M, Chatel S, Sacher F, Weerasooriya R, Probst V, Loussouarn G, Horlitz M, Liersch R, Schulze-Bahr E, Wilde A, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 2009;20:93–98. doi: 10.1111/j.1540-8167.2008.01326.x. [DOI] [PubMed] [Google Scholar]
- 11.Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol. 2016;68:2871–2886. doi: 10.1016/j.jacc.2016.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenzweig A, Watkins H, Hwang DS, Miri M, McKenna W, Traill TA, Seidman JG, Seidman CE. Preclinical diagnosis of familial hypertrophic cardiomyopathy by genetic analysis of blood lymphocytes. N Engl J Med. 1991;325:1753–1760. doi: 10.1056/NEJM199112193252501. [DOI] [PubMed] [Google Scholar]
- 13.Waldmuller S, Schroeder C, Sturm M, Scheffold T, Imbrich K, Junker S, Frische C, Hofbeck M, Bauer P, Bonin M, et al. Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies. Mol Cell Probes. 2015;29:308–314. doi: 10.1016/j.mcp.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Uchiyama K, Hayashi K, Fujino N, Konno T, Sakamoto Y, Sakata K, Kawashiri MA, Ino H, Yamagishi M. Impact of QT variables on clinical outcome of genotyped hypertrophic cardiomyopathy. Ann Noninvasive Electrocardiol. 2009;14:65–71. doi: 10.1111/j.1542-474X.2008.00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray B, Ingles J, Medi C, Semsarian C. Prolongation of the QTc interval predicts appropriate implantable cardioverter-defibrillator therapies in hypertrophic cardiomyopathy. JACC Heart Fail. 2013;1:149–155. doi: 10.1016/j.jchf.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 17.Hedley PL, Jorgensen P, Schlamowitz S, Moolman-Smook J, Kanters JK, Corfield VA, Christiansen M. The genetic basis of Brugada syndrome: a mutation update. Hum Mutat. 2009;30:1256–1266. doi: 10.1002/humu.21066. [DOI] [PubMed] [Google Scholar]
- 18.Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, Sticht H, Rauch A, Puleo C, Hu D, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6) Eur Heart J. 2011;32:1077–1088. doi: 10.1093/eurheartj/ehr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobrinsky E, Schwartz E, Abernethy DR, Soldatov NM. Voltage-gated mobility of the Ca2+ channel cytoplasmic tails and its regulatory role. J Biol Chem. 2002;278:5021–5028. doi: 10.1074/jbc.M211254200. [DOI] [PubMed] [Google Scholar]
- 20.Kobrinsky E, Tiwari S, Maltsev VA, Harry JB, Lakatta E, Abernethy DR, Soldatov NM. Differential role of the 1C subunit tails in regulation of the Cav1.2 channel by membrane potential, subunits, and Ca2+ ions. J Biol Chem. 2005;280:12474–12485. doi: 10.1074/jbc.M412140200. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe H, Makiyama T, Koyama T, Kannankeril PJ, Seto S, Okamura K, Oda H, Itoh H, Okada M, Tanabe N, et al. High prevalence of early repolarization in short QT syndrome. Heart Rhythm. 2010;7:647–652. doi: 10.1016/j.hrthm.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Cao L, Hong D, Zhu M, Li X, Wan H, Hong K. A novel heterozygous deletion–insertion mutation in the desmin gene causes complete atrioventricular block and mild myopathy. Clin Neuropathol. 2013;32:9–15. doi: 10.5414/NP300514. [DOI] [PubMed] [Google Scholar]
- 23.Bai CX, Kurokawa J, Tamagawa M, Nakaya H, Furukawa T. Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation. 2005;112:1701–1710. doi: 10.1161/CIRCULATIONAHA.104.523217. [DOI] [PubMed] [Google Scholar]
- 24.Sinner MF, Reinhard W, Muller M, Beckmann BM, Martens E, Perz S, Pfeufer A, Winogradow J, Stark K, Meisinger C, et al. Association of early repolarization pattern on ECG with risk of cardiac and all-cause mortality: a population-based prospective cohort study (MONICA/KORA) PLoS Med. 2010;7:e1000314. doi: 10.1371/journal.pmed.1000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu GJ, Gan TY, Tang BP, Chen ZH, Jiang T, Song JG, Guo X, Li JX. Age-related changes in cellular electrophysiology and calcium handling for atrial fibrillation. J Cell Mol Med. 2013;17:1109–1118. doi: 10.1111/jcmm.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ocorr K, Reeves NL, Wessells RJ, Fink M, Chen HS, Akasaka T, Yasuda S, Metzger JM, Giles W, Posakony JW, Bodmer R. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc Natl Acad Sci USA. 2007;104:3943–3948. doi: 10.1073/pnas.0609278104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeevaratnam K, Poh Tee S, Zhang Y, Rewbury R, Guzadhur L, Duehmke R, Grace AA, Lei M, Huang CL. Delayed conduction and its implications in murine Scn5a(±) hearts: independent and interacting effects of genotype, age, and sex. Pflugers Arch. 2011;461:29–44. doi: 10.1007/s00424-010-0906-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–780. doi: 10.1056/NEJM200003163421104. [DOI] [PubMed] [Google Scholar]
- 29.Lorenzon A, Beffagna G, Bauce B, De Bortoli M, Li Mura IE, Calore M, Dazzo E, Basso C, Nava A, Thiene G, Rampazzo A. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:400–405. doi: 10.1016/j.amjcard.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, Haubold K, Boucek MM, Ferguson D, Graw SL, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. 2007;115:1244–1251. doi: 10.1161/CIRCULATIONAHA.106.644013. [DOI] [PubMed] [Google Scholar]
- 31.He Y, Zhang Z, Hong D, Dai Q, Jiang T. Myocardial fibrosis in desmin-related hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2010;12:68. doi: 10.1186/1532-429X-12-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Purevjav E, Arimura T, Augustin S, Huby AC, Takagi K, Nunoda S, Kearney DL, Taylor MD, Terasaki F, Bos JM, et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet. 2012;21:2039–2053. doi: 10.1093/hmg/dds022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sequeira V, Nijenkamp LL, Regan JA, van der Velden J. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta. 2014;1838:700–722. doi: 10.1016/j.bbamem.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 34.Berruezo A, Vatasescu R, Mont L, Sitges M, Perez D, Papiashvili G, Vidal B, Francino A, Fernandez-Armenta J, Silva E, et al. Biventricular pacing in hypertrophic obstructive cardiomyopathy: a pilot study. Heart Rhythm. 2011;8:221–227. doi: 10.1016/j.hrthm.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Lenarczyk R, Wozniak A, Kowalski O, Sokal A, Pruszkowska-Skrzep P, Sredniawa B, Szulik M, Zielinska T, Kukulski T, Stabryla J, et al. Effect of cardiac resynchronization on gradient reduction in patients with obstructive hypertrophic cardiomyopathy: preliminary study. Pacing Clin Electrophysiol. 2011;34:1544–1552. doi: 10.1111/j.1540-8159.2011.03193.x. [DOI] [PubMed] [Google Scholar]
- 36.Berruezo A, Penela D, Burgos F, Evertz R, Fernandez-Armenta J, Roca J, Doltra A, Acosta J, Francino A, Sitges M, et al. Optimized pacing mode for hypertrophic cardiomyopathy: impact of ECG fusion during pacing. Heart Rhythm. 2015;12:909–916. doi: 10.1016/j.hrthm.2015.01.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All available to access.