

Abstract
Background
Numerous studies have highlighted the elevated degree of comorbidity associated with autism spectrum disorder (ASD). These comorbid conditions may add further impairments to individuals with autism and are substantially more prevalent compared to neurotypical populations. These high rates of comorbidity are not surprising taking into account the overlap of symptoms that ASD shares with other pathologies. From a research perspective, this suggests common molecular mechanisms involved in these conditions. Therefore, identifying crucial genes in the overlap between ASD and these comorbid disorders may help unravel the common biological processes involved and, ultimately, shed some light in the understanding of autism etiology.
Results
In this work, we used a two-fold systems biology approach specially focused on biological processes and gene networks to conduct a comparative analysis of autism with 31 frequently comorbid disorders in order to define a multi-disorder subcomponent of ASD and predict new genes of potential relevance to ASD etiology. We validated our predictions by determining the significance of our candidate genes in high throughput transcriptome expression profiling studies. Using prior knowledge of disease-related biological processes and the interaction networks of the disorders related to autism, we identified a set of 19 genes not previously linked to ASD that were significantly differentially regulated in individuals with autism. In addition, these genes were of potential etiologic relevance to autism, given their enriched roles in neurological processes crucial for optimal brain development and function, learning and memory, cognition and social behavior.
Conclusions
Taken together, our approach represents a novel perspective of autism from the point of view of related comorbid disorders and proposes a model by which prior knowledge of interaction networks may enlighten and focus the genome-wide search for autism candidate genes to better define the genetic heterogeneity of ASD.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3667-9) contains supplementary material, which is available to authorized users.
Keywords: Autism Spectrum Disorder, Autism sibling disorders, Gene set enrichment, Process enrichment, Comparative network analysis, Systems biology
Background
Autism spectrum disorder (ASD) encompasses a group of complex neurodevelopmental disorders characterized, in different ranges, by impaired social interaction, difficulties in verbal and non-verbal communication and restricted, stereotyped and repetitive behaviors. Its symptoms begin in early childhood and persist through adulthood, affecting daily functioning [1].
This lifelong condition, 4 times more common in males than females, is one of the fastest-growing developmental disorders worldwide and its prevalence continues to increase at an alarming rate. In fact, large-scale surveys estimated median rates of increase at 1–2% [1–8]. The US Center for Disease Control and Prevention (CDC) [9] now indicates that 1 in 68 American children have ASD. In addition, the 2014 National Health Interview Survey, conducted by the National Center for Health Statistics (NCHS) estimates that 1 in 45 children ages 3 through 17 have an autism diagnosis [10].
It is clear that ASD is a complex and heterogeneous disorder that arises from the interaction of genetic, neurologic, immunologic and environmental factors [11] with a high and complex heritability, as both rare and common genetic variants contribute to autism risk [12]. The great variation reported in behavioral traits and cognitive profiles make it challenging to define specific genetic risk components [13]. Therefore, despite recent scientific advances shedding light into the molecular agents and biological mechanisms responsible for ASD, contributing to the discovery and validation of its causative genes [14], the exact factors still remain elusive and no unifying hypothesis about the molecular pathology of autism has emerged.
Interestingly, several large-scale clinical studies have confirmed the high rate of comorbidity associated with ASD. These comorbid conditions represent an additional burden of illness [15]. Indeed, approximately more than 70% of individuals diagnosed with autism have concurrent medical conditions with significantly higher frequency than in neurotypical populations [12]. Some of these disorders, like epilepsy or depression, can first appear in puberty or even later in life, compounding lifelong impairment. Almost 45% of individuals with autism are also affected by intellectual disability, 28–44% have been diagnosed with attention deficit hyperactivity disorder, 12–70% have clinical depression, 8–30% of ASD individuals have epilepsy, 42–56% have anxiety and 9–70% of manifest gastrointestinal problems [12].
These high rates of comorbidity are not surprising considering the overlap of ASD symptoms with many other human disorders, either neurological in nature or not. This suggests a testable hypothesis: disorders with an elevated level of co-occurrence with autism may have many genes in common with ASD and therefore an overlap in the biological processes involved. Thus, the detection of key genes present in the intersection between ASD and several concurrent disorders (behaviorally related, comorbid or both) may help decipher common molecular mechanisms and/or a shared pathophysiology and, ultimately, yield powerful insights in the understanding of autism etiology.
In the present work, we performed a comprehensive cross-disorder analysis comparing autism with 31 comorbid conditions with the aim of quantifying their overlap at the level of molecular physiology, specifically focusing on biological processes and gene networks. We used a systems biology approach to robustly characterize disease genes, identify the comorbid disorders most closely related with autism and quantify and explore the intersection. By implementing a two pronged strategy that leverages both gene function and network connectivity [16] we took advantage of the prior knowledge from related conditions to predict new genes of possible relevance to ASD etiology. Finally, we utilized transcriptome expression profiling experiments to validate our predictions, by identifying significant differential expression of our novel candidate genes in these high-throughput studies.
Methods
Diseases and gene lists
To obtain a robust set of related conditions, we leveraged the results of research studies that investigated autism comorbidity occurring at a significantly higher frequency in ASD patients than in an age-matched control population, using a population- derived sample [17–19], electronic records [19–23] and review papers [19, 24]. We extracted all the ICD-9 codes of autism and its comorbid conditions in these studies and, when ICD-9 code lists were not directly available, we matched the co-occurring conditions mined from these sources to their corresponding codes and references under the ICD-9 system, broadly used in healthcare [25]; then, we mapped each ICD-9 code in our comorbid disorder list to MeSH (Medical Subject Headings form from U.S. National Library of Medicine) terms in order to facilitate the subsequent automated gene search. For instance, the MeSH Term “Anxiety disorders” was matched to the ICD-9 code 300.02 consistent with the ICD-9 reference “Generalized anxiety disorder”, while the MeSH Term “Depressive disorder” corresponded to the 296.3 ICD-9 code with expanded description of “major depressive disorder recurrent episode”.
Next, we generated lists of disorder-related genes by using two powerful text mining tools, Phenopedia and Genehawk, which text mine disease-to-gene relationships in the bibliome. Phenopedia [26], is a web-based application that gathers human genetic associations from literature through a database constantly updated from Pubmed, using either genes or diseases as the starting point. The complete method is described in Yu et al. [26]. Genehawk [27] is a gene-disorder-publication database that collects and ranks associations between genes and diseases, built on evidences from all publication abstracts available via PubMed, as well as the type of study itself. For a given disorder, Genehawk retrieves all related abstracts, filters out those with specific genetic test results and mines gene symbols and maps these to unique identifiers; finally, the obtained results are ranked to assess their significance taking into account the number of supporting evidences, the article structure (review or hypothesis) and the strength of the publication (journal impact factor and year of publication). A complete explanation of this method can be found in Jung et al. [27]. Since both sources, Phenopedia and Genehawk, employ MeSH terms for their automatic exploration of Pubmed, we matched our comorbid disorder ICD-9 code list to this controlled vocabulary thesaurus used for article indexing, as pointed out previously. For ASD, we completed our resulting list of associated genes by adding the autism genes included in SFARI gene [28], as well as those reported as candidates in Iossifov et al. [21] and De Rubeis et al. [29].
Disease-gene cluster and bootstrap validation
We then converted the obtained seed list into a matrix of binary gene presence/absence with respect to each disorder. The matrix was analyzed using the Jaccard coefficient in MATLAB® to build a gene-based dendrogram of all comorbid disorders. The Jaccard statistic, defined by the size of the intersection divided by the size of the union of sample sets, was originally conceived for pattern discovery with binary matrices and computes the similarity and diversity among sample sets without considering the shared absence of a characteristic as evidence for relatedness.
For assessing clusterwise stability and validity of the groupings within the disease relationship tree we used clusterboot(), an integrated function of the ‘fpc’ package in R [30]. We resampled with replacement from the original data by using a non-parametric bootstrapping method (B = 1000 runs) with the aim of generating bootstrap matrices and clusters and iteratively utilized the Jaccard coefficient to measure the structural similarity of the resampled trees with the tree derived from the original data. We considered the mean of the Jaccard coefficients, calculated per permutation as the overall similarity between the original and iterated data, as the index of cluster’s stability and validity. In order to match the total number of clusters obtained in the observed disease relationship tree, we set for each permutation the number of subsets, k, to 6. Then, those clusters supported by a Jaccard coefficient greater than 0.6 were considered robust and stable, while values approaching 1.0 exhibited the highest stability. Jaccard coefficient values equal or lower than 0.5 were considered not stable and, thus, not taken into account for the analysis. A complete explanation of this method can be found in this study by Hennig [31]. These cluster stability analyses were complemented with a classical multidimensional scaling approach that projects our dissimilarity data onto its first two principal dimensions, generated by the ‘showplots’ argument of Clusterboot() function.
Generation of molecular networks
We used STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) version 10 [32] to generate networks for the gene lists of the concurrent conditions most closely related with ASD. The networks were created using the default settings in STRING and the lists of edges were derived from all the available lines of evidence: Neighborhood, Gene Fusion, Co-occurrence, Co-expression, Experiments, Databases and Textmining. It is worth highlighting that, in STRING, every source of interaction evidence is benchmarked and calibrated against prior knowledge, according to the manually curated information provided by the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway maps [33]. The complete method has been described by Szklarczyk et al. [32]. The returned gene interactions were used for subsequent analysis in our network-driven search for autism candidate genes.
Biological process enrichment
To identify the biological processes for which the comorbid disorders most closely related with autism were enriched, we utilized DAVID Bioinformatics Resources (Database for Annotation, Visualization and Integrated Discovery) version 6.7 [34], a high-throughput data-mining environment. This web-accessible functional annotation tool for gene ontology (GO) enrichment analysis embodies an integrated biological knowledge database and analytical implements to automatically extract biological features/meanings associated with large lists of genes. Further information regarding DAVID protocol is detailed in [34]. For our analysis, we employed the “Functional Annotation” tool that basically provides batch annotation and gene-GO term enrichment analysis to emphasize the most important GO terms related with a specified gene list. By choosing the GO fat categories,“GOTERM-BP-FAT” option, to report the enrichment results, we are selecting a subset of the more general GO term; hence, the broadest terms are filtered so that they will not overshadow the more specific ones. In order to evade over counting duplicated genes, DAVID performs Fisher Exact statistics on corresponding DAVID gene IDs by which all redundancies in original IDs are eliminated. All the results displayed in the Functional Chart Report did pass the established thresholds (by default, Max. Prob. < =0.1 and Min. Count > =2) so as to ensure only the statistically significant outcomes are showed. Finally, only those biological processes with a false discovery rate (FDR) score below 0.05 were selected as strongly enriched, according to their statistical significance after multiple test correction.
Expression analysis
From Gene Expression Omnibus (GEO) [35] we downloaded data from three independent experiments, GSE18123 (gpl570) [36], GSE25507 [37] and GSE42133 [38], in order to validate our autism candidate genes. Additional file 1 (Table S1) summarizes the information about the datasets selected. Raw data of Affymetrix datasets, GSE18123 (gpl570) and GSE25507, was preprocessed and RMA normalized using ‘affy’ package in R [39] and Bioconductor [40], while with the Illumina dataset GSE42133 we employed the preprocessed data provided in GEO database, Log2 transformed and quantile normalized using Illumina GenomeStudio® software (version 1.1.1) and ‘Lumi’ package in R and Bioconductor [41]. Additional file 2 (Figure S1) shows the distribution of the samples after preprocessing, median-centered values indicate that the data are normalized and cross-comparable. All expression analyses were done using mt.teststat function from “multtest” package in R and Bioconductor [42]; to increase the test power for samples with unequal sample size and variance, we performed a t-test based on two-sample Welch t-statistics to determine the difference in signal between the ASD and control group. Finally, we performed multiple test correction to the unadjusted p-values from the comparative analyses by calculating q-values, a measure of significance in terms of the FDR [43].
Functional analysis of ASD candidate genes
Our candidate genes identified to be differentially expressed in all three experiments were uploaded into the QIAGEN® Ingenuity® Pathway Analysis (IPA®) software, in order to explore gene connectivity and related biological functions both within and across disease. IPA® uses a human-curated pathways knowledge base containing genes, proteins and RNAs to retrieve biological interactions and associate biological functions and disorders with experimental results, providing statistical support for gene-to-gene associations. We generated networks for our ASD candidate genes using an edge rank score (p-score = −log10 (p-value)) that designated the likelihood of the concurrent or interacting genes by random chance. A rank score value greater than 3 (p < 0.001) denoted an edge linking two genes as a statistically relevant not random association, with more than 99.9% confidence. Additionally, we performed a “disease and function” analysis to test whether our ASD gene candidates were enriched in specific human disorders and investigated their role in the context of statistically significant biological processes, pathways and networks. IPA® performs a Fisher exact test to calculate p-values that define the significance of the association between a focus gene and a biological process or pathway; thus, those biological processes with p-values ≤0.05 are considered as statistically significantly enriched with genes of interest.
Results
The multi-disorder component of ASD
We retrieved from the literature [17–24] 132 medical conditions concurrent with autism that were matched with their corresponding codes and references under ICD-9 system [25] and later consolidated into a defined set of 31 disorders comorbid with ASD (See Additional file 3: Table S2). Using Phenopedia [26] and Genehawk [27], we generated lists of genes associated to each comorbid condition and, as pointed out previously, in the case of autism, we completed its gene list by adding the ASD candidate genes included in SFARI gene [28], Iossifov et al. [21] and De Rubeis et al. [29]. The total number of genes utilized for this study are detailed in Additional file 4: Table S3. By converting the retrieved gene lists into a binary matrix of gene presence/absence, we were able to generate a disorder phylogeny using the Jaccard Coefficient (Fig. 1). The tree obtained grouped autism with 13 disorders that we called “sibling” comorbid disorders of ASD, including epilepsy, intellectual disability, fragile X syndrome, schizophrenia, depressive disorder, bipolar disorder and attention deficit hyperactivity disorder (ADHD), among others. Cluster wise validity and stability within the tree was assessed by means of a non-parametric bootstrap procedure (1000 runs) that yielded a mean Jaccard value of 0.785 for our autism sibling comorbid disorders cluster (See Additional file 5: Table S4, also Additional file 6: Figure S2). Thus, we considered this sibling group as stable and statistically robust and focused on this group for subsequent analyses.
Fig. 1.
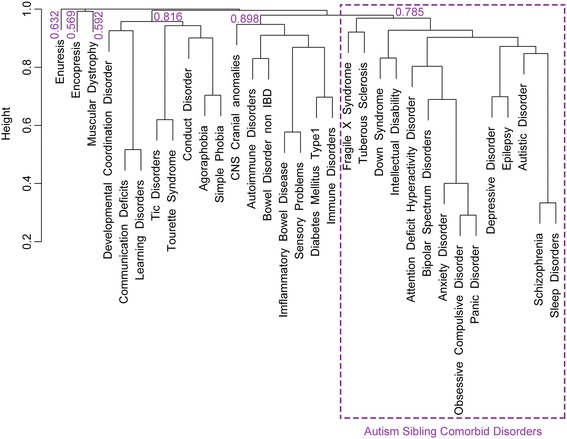
Gene-based phylogeny of autism and related co-ocurring conditions, generated using the Jaccard Coefficient. The group containing autism is highlighted and referred to in the text as “Autism sibling comorbid disorders”. Bootstrap stability indices are also provided for each subgroup
With the aim of exploring genetic overlap between autism and its sibling comorbid conditions, we used the tool STRING [32] to generate gene networks for each member of the ASD sibling group (edge summary included in Additional file 7: Table S5). Of the 1066 genes present in our seed list for ASD, 710 have also been linked to at least one other autism sibling disorder (the multi disorder autism gene set, and the sibling comorbid conditions where they are found, are detailed in Additional file 8: Table S6). This multi disorder autism gene set (MDAG) conforms a highly interconnected subcomponent of the ASD gene network (Additional file 9: Figure S3), suggesting common molecular mechanisms and shared biological functions among the MDAG members. To test this, we utilized DAVID [34] to identify significant enrichment of MDAG genes in biological processes (BP). A total of 378 BP had significant overrepresentation following FDR multiple test correction (top 30 BP are listed in the Table 1), significant if FDR < 0.05; the complete list of BP for which the MDAG genes are enriched can be found in Additional file 10: Table S7.
Table 1.
Top 30 biological processes for which the multi-disorder component of the autism gene set (MDAG) were enriched
| Biological process | # MDAG genes | p-Value | FDR |
|---|---|---|---|
| Transmission of nerve impulse | 119 | 1.18E-71 | 2.18E-68 |
| Synaptic transmission | 110 | 2.53E-70 | 4.67E-67 |
| Behavior | 123 | 1.07E-59 | 1.97E-56 |
| Cell-cell signaling | 137 | 7.08E-59 | 1.31E-55 |
| Regulation of system process | 84 | 2.00E-41 | 3.70E-38 |
| Neurological system process | 164 | 7.03E-39 | 1.30E-35 |
| Regulation of neurological system process | 57 | 4.56E-36 | 8.42E-33 |
| Learning or memory | 49 | 4.57E-35 | 8.43E-32 |
| Regulation of transmission of nerve impulse | 54 | 8.64E-34 | 1.59E-30 |
| Regulation of synaptic transmission | 51 | 2.12E-32 | 3.91E-29 |
| Neuron differentiation | 85 | 2.91E-30 | 5.38E-27 |
| Neuron development | 74 | 1.13E-29 | 2.08E-26 |
| Neuron projection development | 61 | 1.40E-26 | 2.59E-23 |
| Second-messenger-mediated signaling | 58 | 4.18E-26 | 7.71E-23 |
| Cyclic-nucleotide-mediated signaling | 44 | 7.72E-26 | 1.43E-22 |
| Cell morphogenesis involved in neuron differentiation | 54 | 2.48E-25 | 4.57E-22 |
| Cell morphogenesis involved in differentiation | 58 | 3.25E-25 | 5.99E-22 |
| G-protein signaling, coupled to cyclic nucleotide second messenger | 41 | 4.61E-25 | 8.51E-22 |
| Learning | 31 | 1.12E-24 | 2.07E-21 |
| Neuron projection morphogenesis | 53 | 4.86E-24 | 8.98E-21 |
| Response to endogenous stimulus | 73 | 6.40E-24 | 1.18E-20 |
| Regulation of secretion | 51 | 1.92E-23 | 3.54E-20 |
| Axonogenesis | 49 | 1.21E-22 | 2.23E-19 |
| Feeding behavior | 31 | 2.00E-22 | 3.69E-19 |
| Response to organic substance | 97 | 5.25E-22 | 9.70E-19 |
| Intracellular signaling cascade | 135 | 1.40E-21 | 2.59E-18 |
| Cell projection organization | 66 | 1.65E-21 | 3.04E-18 |
| Cell projection morphogenesis | 53 | 4.54E-21 | 8.39E-18 |
| Regulation of cellular localization | 53 | 8.10E-21 | 1.50E-17 |
| Cell motion | 74 | 2.12E-20 | 3.92E-17 |
Biological process-driven search for novel ASD candidates
The large extent of genetic overlap between autism and several of its sibling conditions may rely on specific dysregulations of any or all the biological processes for which the MDAG is enriched. Therefore, other genes associated to any of the 378 statistically significant processes that have not yet been linked to ASD could be regarded as possible novel candidates for autism. To address this premise, the gene lists of all the ASD sibling disorders were mined to identify and retrieve a non-redundant set of 1588 process-based candidates (PBC); 34 processes were not found among the genes in the autism sibling disorders (See Additional file 10: Table S7). All other enriched processes returned 2 or more predictions all of which are implicated in at least 2 autism sibling disorders, but not found in our original gene candidate list for ASD. The complete list of 1588 process-based candidates can be found in Additional file 11: Table S8.
To empirically test the importance of our process-based candidates, we checked whether they were significantly differentially regulated in autistic patients versus healthy controls, using the three independent GEO experiments described above (GSE18123gpl570, GSE25507 and GSE42133) [36–38]. Since our foremost interest was to confirm our PBC, we performed multiple test correction to the unadjusted p-values obtained from the analyses by calculating q-values (see Additional file 12: Figure S4), an FDR-based measure of significance. For each experiment, we considered the number of PBC present in the array as the total number of hypotheses, as previously done in [16]. The resultant number of differentially expressed PBC with q-value <0.05 are summarized in Table 2, where there are 1058 significant PBC for GSE18123gpl570, 626 for GSE25507 and 269 in the case of GSE42133; a total of number of 80 significant PBC constituted the overlap among the three datasets. The identities of the differentially expressed PBC in each experiment, along with their corresponding q-values, the biological processes where they are involved and the comorbid disorders where they are implicated, can be also found in Additional file 11: Table S8.
Table 2.
Number of significantly differentially expressed process and network based candidates in the datasets
Network-driven search for new autism genes
Using data derived from STRING [32], we constructed gene networks for each of the autism sibling disorders with the purpose of exploring the surrounding members of the MDAG genes, specifically focusing on their first neighbors that were not included in our original ASD candidate list. This analysis yielded 1794 network-based candidates (NBC), directly linked to a member of the MDAG but not known yet as relevant for autistic disorder. From the total set of genes constituting the NBC, 233 candidates occur in at least 5 sibling conditions, 74 are present in 7 or more siblings, 29 in 8 or more, 13 in 9 autism siblings, 3 in 10 autism sibling disorders (MAGI2, NR3C1, SLC1A2) and one (SLC1A2) present in 12 siblings. The complete list of 1794 network-based candidates can be found in Additional file 13: Table S9.
We leveraged the same mRNA expression datasets as before to calculate q-values and verify whether our network-based candidates exhibited significantly different gene expression in individuals with autism when compared to normal controls (see Additional file 12: Figure S4). We validated the NBC by testing for significant differential expression in each of the three separate microarray experiments, GSE18123gpl570, GSE25507 and GSE42133. We treated each test of the NBC as a separate experiment and adjusted for multiple testing each time by computing the q-value for the total number of NBC genes found on the separate arrays, 1210, 691, 298, respectively. Table 2 shows the number of NBC found to be significantly differentially regulated (q-value <0.05) in each experiment. A total of 91 significant NBC were found in common among the three gene expression datasets. Their identities, q-values, MDAG interactors and comorbid disorders in which they play a role, can be also found in Additional file 13: Table S9.
Intersection of PBC and NBC to prioritize autism candidate genes
We intersected our two computational strategies to triangulate on the set of genes that were independently predicted and verified by both approaches. A total of 1358 genes formed the overlap of PBC and NBC (PBC∩NBC); the total number of significant differentially expressed candidates, with q-value < 0.05, predicted in each experiment by both approaches is 925, 532 and 214 genes for GSE18123gpl570, GSE25507 and GSE42133 respectively (Table 2), with a total of 64 significant candidates overlapping across all three experiments. The identities of these candidate genes are detailed in Additional file 14: Table S10, along with the biological processes where they participate, their MDAG interactors and the comorbid disorders associated with them. Next, we cut down the size of the overlap by removing those genes that occur in 2 or fewer autism sibling disorders. This is based on the premise that genes with numerous independent associations to our sibling comorbid disorders are more likely to participate in typical neurodevelopmental processes and functions. From the 1358 genes present in the overlap (PBC∩NBC), only 489 candidates predicted by both strategies occurred in 3 or more siblings. Table 2 also shows, for each dataset, the number of differentially expressed candidates independently predicted and verified by both strategies occurring in 3 or more autism sibling disorders: 330 for GSE18123gpl570, 183 for GSE25507 and 69 in the case of GSE42133.
Finally, with the aim of obtaining a definitive set of candidates, we intersected the differentially expressed genes obtained from the analysis of the three GEO datasets occurring in 3 or more sibling comorbid disorders; this yielded an overlap of 19 genes (Table 3). The overlap across the three sets of differentially expressed genes is shown in Fig. 2.
Table 3.
List of the 19 candidate genes significantly differentially expressed in the three experiments and the disorders where they are implicated
| Genes | Sibling disorders | # Disorders |
|---|---|---|
| ADAM10 | Bipolar Spectrum Disorders, Down Syndrome, Sleep Disorders | 3 |
| ADCY9 | Bipolar Spectrum Disorders, Depressive Disorder, Epilepsy, Schizophrenia, Sleep Disorders | 5 |
| ADCYAP1R1 | Anxiety Disorder, Bipolar Spectrum Disorders, Obsessive Compulsive Disorder, Panic Disorder | 4 |
| AKT1 | Bipolar Spectrum Disorders, Depressive Disorder, Epilepsy, Fragile X Syndrome, Schizophrenia, Tuberous Sclerosis | 6 |
| ATN1 | Epilepsy, Fragile X Syndrome, Intellectual Disability, Schizophrenia, Sleep Disorders | 5 |
| DGCR8 | Depressive Disorder, Fragile X Syndrome, Schizophrenia, Sleep Disorders | 4 |
| DLGAP4 | Anxiety Disorder, Bipolar Spectrum Disorders, Obsessive Compulsive Disorder, Panic Disorder, Schizophrenia, Sleep Disorders | 6 |
| HSPA1L | Bipolar Spectrum Disorders, Depressive Disorder, Schizophrenia | 3 |
| KCNH2 | Epilepsy, Intellectual Disability, Schizophrenia, Sleep Disorders | 4 |
| MEGF10 | Bipolar Spectrum Disorders, Schizophrenia, Sleep Disorders | 3 |
| MMP2 | Epilepsy, Sleep Disorders, Tuberous Sclerosis | 3 |
| NDE1 | Bipolar Spectrum Disorders, Epilepsy, Intellectual Disability, Schizophrenia | 4 |
| NPPB | Anxiety Disorder, Bipolar Spectrum Disorders, Obsessive Compulsive Disorder, Panic Disorder, Sleep Disorders | 5 |
| NRP1 | Anxiety Disorder, Bipolar Spectrum Disorders, Obsessive Compulsive Disorder, Panic Disorder, Sleep Disorders | 5 |
| PPP3CB | Attention Deficit Hyperactivity Disorder, Schizophrenia, Sleep Disorders | 3 |
| PRKG1 | Attention Deficit Hyperactivity Disorder, Fragile X Syndrome, Schizophrenia, Sleep Disorders | 4 |
| SLC29A2 | Depressive Disorder, Epilepsy, Sleep Disorders | 3 |
| SMARCA2 | Epilepsy, Intellectual Disability, Schizophrenia, Sleep Disorders | 4 |
| VIPR2 | Anxiety Disorder, Bipolar Spectrum Disorders, Depressive Disorder, Down Syndrome, Epilepsy, Intellectual Disability, Obsessive Compulsive Disorder, Panic Disorder, Schizophrenia | 9 |
Fig. 2.
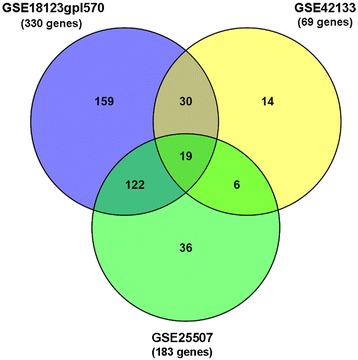
Venn diagram showing the overlap in the number of significant differentially regulated process and network based candidate genes (PBC∩NBC) occurring in 3 or more autism sibling comorbid disorders for the three datasets. Only 19 candidate genes are present in all three transcriptome experiments
Additionally, this final set of 19 candidates was loaded into IPA® to explore connectivity and biological function. The analysis performed associated our 19 candidate genes to four Top Canonical Pathways (Table 4), related to brain development and function, neurodegeneration and behavior and known to be relevant in the molecular pathology of ASD. We also conducted a “Diseases & Function” analysis for the 19 genes that linked them with several significant annotations; Table 5 shows the most compelling examples and the candidate genes involved in each biological function.
Table 4.
Top canonical pathways for which the 19 candidates genes are enriched, according to Ingenuity® Pathway Analysis (IPA®)
| Canonical pathway | p-value | Overlap |
|---|---|---|
| eNOS signaling | 9.45–06 | 3.0% 4/135 |
| Gap junction signaling | 1.47–05 | 2.6% 4/151 |
| Axonal guidance signaling | 5.55–05 | 1.2% 5/427 |
| Glucocorticoid receptor signaling | 1.46–04 | 1.5% 4/272 |
Table 5.
Significant functional annotations of our final set of candidate genes according to Ingenuity® Pathway Analysis (IPA®)
| Diseases or functions annotation | p-value | Candidate genes involved |
|---|---|---|
| Proliferation of nervous tissue cell lines | 2.07E-05 | AKT1,NRP1 |
| Action potential of embryonic stem cell lines | 1.02E-03 | KCNH2 |
| Arrest in growth of nervous tissue cell lines | 1.02E-03 | NRP1 |
| Formation of cranium | 1.02E-03 | MMP2 |
| Quiescence of nervous tissue cell lines | 1.02E-03 | NRP1 |
| Schizophrenia | 1.55E-03 | AKT1,KCNH2,PPP3CB,SMARCA2 |
| Generation of plasmacytoid dendritic cells | 4.08E-03 | AKT1 |
| Induction of CD4+ T-lymphocytes | 5.10E-03 | PRKG1 |
| Induction of Th17 cells | 5.10E-03 | PRKG1 |
| Binding of cells | 5.40E-03 | MMP2,NPPB,NRP1 |
| Permeability of blood–brain barrier | 6.11E-03 | MMP2 |
| Gene silencing | 8.14E-03 | SMARCA2 |
| Loss of neurons | 1.32E-02 | ATN1 |
| Rasmussen’s encephalitis | 1.42E-02 | PPP3CB |
| Cognition | 1.52E-02 | AKT1 |
| Neuropathic pain | 2.12E-02 | KCNH2 |
| Apoptosis of dendritic cells | 2.22E-02 | AKT1 |
| Release of nitric oxide | 2.32E-02 | AKT1 |
| Quantity of neurons | 2.42E-02 | ATN1 |
| Transcription of RNA | 2.84E-02 | AKT1,ATN1,SMARCA2 |
| Epilepsy | 3.13E-02 | HSPA1L,NPPB |
Regarding gene connectivity, IPA® provided a statistically robust network (score = 31), shown in Fig. 3, where 14 genes from the original 19 candidates are interacting with other molecules in several significant neurological processes involved in normal brain growth and development, such as proliferation of neuronal cells, formation and branching of neurites, migration of neurons, among others (see Table 6). Dysregulation of any of these candidates may affect crucial brain processes since many of them interact with genes already included in our original seed list for autism (APP, CYP19A1, ESR1, MAPK1, SETD2, SHANK2, TRPV1), some of them being highly interconnected nodes within the network (ESR1, APP, MAPK1). In addition, important neurological processes such as cognition, learning and memory may be altered since several of our candidates are linked to key network genes (ESR1, Pkcs, AKT1), implicated in postsynaptic density and glutamatergic synapses, and, hence, in synaptic plasticity. Furthermore, our candidate genes also seem to be involved in pathways where central genes within the network, such as MAPK1, ESR1, Pkcs, TP53, APP and EGFR, are thought to regulate molecular functions associated with multiple aspects of social and anxiety-related behaviors, mood outcomes and impaired long-term memory, cognitive degeneration and neurological dysfunction. This network also showed an interesting connection between genes linked to ASD and other neurological conditions and endocrine hormones of the hypothalamic-pituitary-gonadal axis, such as the luteinizing hormone (Lh). The neurological functions statistically significantly enriched in the network are described in Table 6, along with the genes implicated in each process and their corresponding p-values.
Fig. 3.
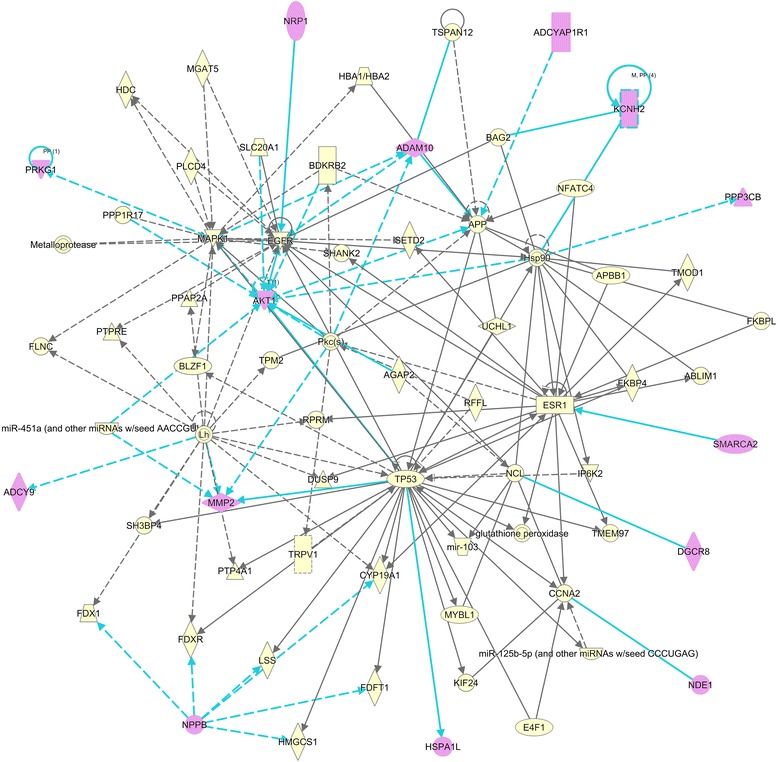
Statistically significant biological network obtained through Ingenuity Pathway Analysis (IPA®). 14 of our 19 candidate genes are tightly connected and interact in neurological processes and conditions detailed in Table 6. Our candidate genes are highlighted in purple and their interactions in turquoise
Table 6.
Significant diseases and functions enriched in the biological network (Fig. 3) obtained through Ingenuity® Pathway Analysis (IPA®)
| Diseases or functions annotation | p-Value | Molecules |
|---|---|---|
| Proliferation of neuronal cells | 1.95E-06 | ADAM10, ADCYAP1R1, AKT1, APBB1, APP, CYP19A1, EGFR, ESR1, FKBP4, HBA1/HBA2, MAPK1, MMP2, NDE1, NFATC4, NRP1, Pkc(s), TP53 |
| Growth of neurites | 3.24E-06 | ADAM10, AKT1, APBB1, APP, CYP19A1, EGFR, ESR1, FKBP4, HBA1/HBA2, MAPK1, MMP2, NFATC4, NRP1, pkc(s), TP53 |
| Interphase of brain cells | 8.22E-06 | ADCYAP1R1, APP, TP53 |
| Outgrowth of neurites | 1.78E-05 | ADAM10, AKT1, APBB1, APP, EGFR, ESR1, FKBP4, HBA1/HBA2, MAPK1, NFATC4, NRP1, Pkc(s), TP53 |
| Behavior | 1.98E-05 | ADAM10, AGAP2, APBB1, APP, CYP19A1, ESR1, HBA1/HBA2, HDC, MAPK1, mir-103, MYBL1, NFATC4, NPPB, Pkc(s), PPP3CB, PRKG1, SHANK2, TP53, TRPV1, UCHL1 |
| Alzheimer’s disease | 5.88E-05 | ADAM10, APBB1, APP, ESR1, FDFT1, HBA1/HBA2, LSS, mir-103, miR-125b-5p (and other miRNAs w/seed CCUGAG), MMP2, NFATC4, Pkc(s), TP53, UCHL1 |
| Microtubule dynamics | 7.64E-05 | ABLIM1, ADAM10, AGAP2, AKT1, APP, CYP19A1, DUSP9, EGFR, ESR1, FKBP4, Hsp90, KIF24, MAPK1, NDE1, NFATC4, NRP1, Pkc(s), PPP3CB, PRKG1, PTPRE, TP53, UCHL1 |
| Organization of cytoskeleton | 7.78E-05 | ABLIM1, ADAM10, AGAP2, AKT1, APP, CYP19A1, DUSP9, EGFR, ESR1, FKBP4, FLNC, Hsp90, KIF24, MAPK1, MGAT5, NDE1, NFATC4, NRP1, Pkc(s), PPP3CB, PRKG1, PTPRE, TP53, UCHL1 |
| Branching of neurites | 7.87E-05 | ADAM10, AGAP2, AKT1, APP, CYP19A1, NFATC4, NRP1, PRKG1, TP53 |
| Entry into S phase of cerebral cortex cells | 1.18E-04 | ADCYAP1R1, APP |
| Anxiety | 1.65E-04 | ADCYAP1R1, APP, HDC, MAPK1, NFATC4, SHANK2, TRPV1 |
| Branching of cells | 1.86E-04 | ADAM10, AGAP2, AKT1, APP, BDKRB2, CYP19A1, NFATC4, NRP1, Pkc(s), PRKG1, TP53 |
| Hyperactive behavior | 1.92E-04 | ADCYAP1R1, AKT1, APP, ESR1, PPP3CB, SHANK2 |
| Development of central nervous system | 2.07E-04 | ADAM10, ADCYAP1R1, AKT1, APBB1, APP, CYP19A1, EGFR, MAPK1, NDE1, PRKG1, SETD2, TP53, TRPV1 |
| Interphase of neural precursor cells | 2.67E-04 | ADCYAP1R1, TP53 |
| Conditioning | 2.87E-04 | ADCYAP1R1, APP, ESR1, MAPK1, MMP2, TRPV1, UCHL1 |
| Firing of neurons | 2.97E-04 | APP, MAPK1, NPPB, TRPV1 |
| Locomotion | 3.09E-04 | AGAP2, APP, CYP19A1, ESR1, NFATC4, PPP3CB, TMOD1, TP53, UCHL1 |
| Formation of brain | 3.37E-04 | ADAM10, ADCYAP1R1, APBB1, APP, CYP19A1, EGFR, NDE1, PRKG1, SETD2, TP53, TRPV1 |
| Cell viability of neuroglia | 3.48E-04 | AKT1, APP, EGFR, TP53 |
| Cell death of sympathetic neuron | 3.96E-04 | AGAP2, AKT1, APP, Pkc(s), TP53 |
| Morphogenesis of neurites | 4.00E-04 | ADAM10, AGAP2, AKT1, APP, CYP19A1, EGFR, NFATC4, NRP1, PRKG1, TP53 |
| Neuritogenesis | 4.44E-04 | ADAM10, AGAP2, AKT1, APP, CYP19A1, EGFR, NFATC4, NRP1, PRKG1, PTPRE, TP53, UCHL1 |
| Formation of forebrain | 5.97E-04 | ADAM10, ADCYAP1R1, APBB1, APP, NDE1, PRKG1, SETD2 |
| Migration of neurons | 9.72E-04 | ADAM10, APBB1, DGCR8, EGFR, NDE1, NRP1, PRKG1 |
| Emotional behavior | 1.06E-03 | APP, CYP19A1, ESR1, MAPK1, NPPB, SHANK2, TRPV1 |
| Schizophrenia spectrum disorder | 1.21E-03 | AKT1, APP, EGFR, ESR1, KCNH2, mir-103, Pkc(s), PPP3CB, SHANK2, SMARCA2 |
| Long-term potentiation | 1.22E-03 | ADCYAP1R1, APP, CYP19A1, EGFR, MAPK1, Pkc(s), SHANK2, TRPV1 |
| Cognition | 1.52E-02 | AKT1 |
Our candidate genes are highlighted in bold
Using this novel two-pronged computational approach, we were able to discover a final set of 19 ASD candidate genes that have been predicted by both strategies (network and process-based) that occur in 3 or more autism siblings and that were found to be significantly differentially regulated in three independent mRNA expression experiments, lending support to the hypothesis of common molecular mechanisms between autism and other comorbid disorders.
Discussion
In this study, we conducted a comparative analysis of autism and 31 comorbid conditions mined and retrieved from bibliome. By focusing on a set of 13 disorders that appeared to be most closely related to ASD (autism sibling comorbid conditions, see Fig. 1), we discovered that more than half of the autism genes included in our ASD seed list are also associated to related comorbid conditions. This finding supports our hypothesis, confirming the existence of molecular overlap and suggesting that these autism sibling comorbid disorders may share molecular mechanisms that could be enlightening for our understanding of the genetic etiology of ASD. Moreover, the multi-disorder component of the autism network (MDAG) is highly interconnected and significantly enriched for relevant and informative biological processes, such as synaptic transmission, neuron development, axonogenesis, transmission of nerve impulse and learning or memory, among others.
Motivated by these findings, we devised two analytical approaches to verify whether information from concurrent conditions could yield meaningful focus to the genome-wide search for ASD gene candidates. Our first approach, a process-based strategy, was grounded on the premise that processes for which the MDAG genes were enriched are generally relevant for neurological dysfunction. It is further predicated on the assumption that genes implicated in these processes that have been tied to one or more autism sibling comorbid disorders, but still have not yet been associated to ASD, should be autism gene candidates. To test this hypothesis we used available whole-genomic expression data from three independent experiments and found that 80 genes from our process-based candidate list were under significant differential expression in individuals with autism in the three datasets. The fact that they have been linked to neurological dysfunction together with having been implicated in biological processes that seem to play a role in autism makes these genes appealing new leads that may shed light in elucidating the molecular pathology of ASD.
The second approach, a network-based strategy, was based on the mainstream conception that protein interaction networks could give relevant and sometimes fortuitous leads for disease causative agents, suggesting potential points for biomarkers or drug targets and helping in elucidating the biological mechanisms involved [44–52]. In our approach, instead of looking at the whole protein interaction network, we took the set of all genes directly interacting with MDAG genes such that they contained only those proteins present in the list of autism sibling comorbid conditions, but absent from our seed list of published ASD candidates. Several genes within these network-based candidates have been previously related to neurological dysfunction. For instance, rare genetic variation in SLC1A2, necessary for proper synaptic activation and neurotransmission, has been associated with a wide range of neurological conditions including bipolar disorder, schizophrenia and autism [53]. Methylation of the glucocorticoid receptor gene NR3C1 through epigenetic processes, crucial in the hypothalamic-pituitary-adrenal axis modulation, our primary stress response system, has been linked to psychopathological conditions such as anxiety and depression [54, 55]. Variations in MAGI2, a synaptic scaffolding molecule with an essential role in synaptic transmission, are known to be related to epilepsy and cognitive impairment in patients with schizophrenia [56, 57]. In addition, mutations in CTNND2, a gene that plays a key role in neuronal development, particularly in the formation and maintenance of dendritic spines and synapses, have also been recently associated to autism [58]. Moreover, other candidate genes such as GRIA1, GRIA2, GABBR1, GABRG2, GABRR2, NRG2, NRG3, GRIK1, GRIK4, GRIN3A and GRM3, with functions that comprise formation of synapse, transmission of nerve impulse, behavior, learning or memory, are among families of genes that have been shown to have roles in neurological dysfunction jointly impacted in disorders like autism, schizophrenia and bipolar disorder [59–64]. Overall, the network-driven strategy yielded 91 genes found to be differentially expressed in individuals with autism when compared to healthy controls in all three experiments. This approach revealed the existence of a significant signal in the protein interaction networks of these related comorbid conditions, even one step removed from those genes that are shared among them (the MDAG). Even though these variations may represent real mechanistic differences between ASD and its sibling comorbid conditions, the overlap of 91 candidates found to be differentially regulated in autistic individuals from three independent datasets makes more likely that at least some reflect key holes in our understanding of autism.
In both analytical approaches, we were able to leverage the prior knowledge from two different sources, in this case from biological processes and protein interaction networks, to provide focused sets of candidates hypothesized to be under differential regulation in individuals with autism. From a methodological point of view, it is worth highlighting that in the absence of such prior knowledge several of the genes measured in the autistic patients included in all the three experiments would have had false discovery rate (FDR) values above the 0.05 threshold. In fact, this is a common circumstance in cases of weak signals and large background noise in several transcriptome-level experiments [65–67]. Conversely, with the utilization of prior knowledge, the major part of the candidate genes tested showed an FDR < 0.05. This turnabout of the frequent specificity problem at the genome-scale points towards a promising merging between knowledge and data-driven methodologies.
Finally, although the results evaluated herein should be considered preliminary, how the related comorbid disorder networks overlap with ASD have proved to be useful in enlightening important disease related biological processes and discovering potential autism candidate genes. In this work, by combining our process and network-based strategies, we were able to algorithmically assemble 19 candidate genes confirmed to be significantly differentially expressed in individuals with autism from three independent experiments. Moreover, to better understand the biological significance of our final set of candidates, we tested their enrichment in signaling pathways and specific biological processes and whether they were interconnected within a biological network. Our analysis revealed that our predicted genes were implicated in 4 canonical pathways associated with brain structure and functioning, neuroinflammation, neurodegeneration, cognition and behavior [68–77]; alteration in these signaling pathways may play an important role in the pathophysiology of ASD.
Fourteen of these candidates interact with other molecules conforming a network significantly enriched in relevant biological processes related to normal brain growth and development. Dysregulation of any of these candidates may cause relevant disruptions in these fundamental processes altering neural outcomes and affecting cognition, learning and memory, especially since many of them interact with genes already associated to autism (APP, CYP19A1, ESR1, MAPK1, SETD2, SHANK2, TRPV1). In addition, some of the most connected nodes within the network (ESR1, TP53, AKT1, MAPK1, Pkcs, EGFR and APP) may support molecular mechanisms implicated in neuronal connectivity and synaptic plasticity; dysfunction in these neurological pathways have been linked to social and anxiety-related behaviors, mood conditions, cognitive degeneration and loss of neurological function, characteristic features observed in many neurological conditions, including ASD [78–90]. Finally, a remarkable connection was observed in this network between genes associated to ASD and other comorbid disorders and Luteinizing hormone (Lh), an endocrine hormone of the hypothalamic-pituitary-gonadal axis that acts in synergy with follicle-stimulating hormone (FSH), with roles in brain development and neuron differentiation [91]. Moreover, the regulation of these hormones release in blood is controlled by oxytocin, a neurohypophysial hormone that also operates as a brain neurotransmitter and that have been implicated in social behavior, recognition and bonding [92–96] and, therefore, alterations of its neuromodulatory activity have been associated to several mental disorders including autism [97–100]. Interestingly, dysregulation of the endocrine activity, particularly an interaction between potential ASD candidate genes and endocrine hormones of the hypothalamic-pituitary-gonadal axis was also found in our previous study [101]. Overall, these results lend additional support to the hypothesis that prior knowledge leveraged from comorbid conditions may contribute significantly to the progress in the genome wide search for autism candidate genes.
Conclusion
A number of large-scale clinical studies have shown the high rates of comorbidity linked to autism that suggest the existence of an overlap in genes and biological processes in common between ASD and its co-occurring conditions. In the present work, we used a twofold systems biology approach to conduct a comparative analysis of autism and 31 comorbid disorders, with the aim of using the prior knowledge from these related conditions to predict 19 novel ASD gene candidates validated through transcriptome expression profiling experiments. This new set of genes appeared to be of potential etiologic relevance to ASD, as most of its members have been implicated in neurological processes critical for optimal brain growth and function, and have confirmed roles in neurological disease. Future work, including the evaluation of more comorbid conditions clustered in Fig. 1 and disorders neurological in nature or nor (for instance, autoimmune disorders), may be useful in the effort to arrange and reorder genes that have been associated to autism so far, and possibly unveil new genes worth investigating for our understanding of the pathophysiology of autism.
Additional files
Information summary of the datasets selected. (DOCX 12 kb)
Box plots showing the distribution of the samples of each dataset after preprocessing; median-centered values indicate that the data are normalized and cross-comparable. (PDF 5181 kb)
Complete list of comorbid conditions to autism. Autism sibling disorders are highlighted in blue. (XLSX 16 kb)
Total number of genes of each comorbid condition utilized for this study. Highlighted in blue are ASD sibling disorders. (XLSX 135 kb)
Comorbid disorders integrating each group generated by the bootstrap analysis (Additional file 6: Figure S2), along with their Mean Jaccard Coefficient value. The different groups of disorders generated by our bootstrap procedure corresponds to the disorder clusters obtained in our original gene-based dendrogram (Fig. 1). Groups 1, 2 and 3 have the highest Mean Jaccard values meaning they are the most robust and stable groupings of the tree. Group 2 coincides with the cluster conformed by the autism sibling disorders with a highly significant Mean Jaccard value of approximately 0.785. (DOCX 13 kb)
First two Multidimensional Scaling (MDS) dimensions of our dataset generated by MDS on a dissimilarity matrix using Jaccard Coefficient when k = 6. Each group is highlighted in a different color and the disorders conforming them are detailed in Additional file 5: Table S4, along with their corresponding mean Jaccard Coefficient value. The autism sibling comorbid disorders are clustered together in group 2 (PDF 180 kb)
STRING edge summary for each member of the ASD sibling group. (XLSX 8411 kb)
The multi disorder autism gene set (MDAG) and the sibling comorbid conditions where these genes are found. (XLSX 25 kb)
A. The complete network of autism candidate genes. The MDAG genes are highlighted in yellow and their interactions in red; these are the genes that occur in one or more of the autism sibling comorbid disorders, circumscribed in Fig. 1. B. The highly interconnected subcomponent conformed by the MDAG genes, separated from the autism network. (TIF 406700 kb)
Biological processes for which the Multi-disorder component of the autism gene set (MDAG) were enriched. Identities of the MDAG genes overrepresented in the processes as well as the corrected p-values for the enrichment scores are provided. Enrichment was calculated using the biological processes only found among the MDAG genes and not found among the sibling disorders. (XLSX 50 kb)
Identities of the differentially expressed PBC, along with their corresponding q-values, the biological processes where they are involved and the comorbid disorders where they are implicated. Also, PBC significantly differentially expressed in each dataset and in all the three datasets. (XLSX 210 kb)
P-value and q-value histograms and q-plots from the multiple test correction analyses performed on the PBC and NBC to verify whether they were significantly differentially regulated in autistics in comparison to controls. (PDF 11411 kb)
Complete list of NBC, along with their q-values, MDAG interactors and comorbid disorders where they are present. Also, NBC significantly differentially expressed in each dataset and in all the three datasets. (XLSX 185 kb)
Complete list of genes present in the intersection of PBC∩NBC, along with the biological processes where they are involved, MDAG interactors and comorbid disorders where they are present. Also, PBC∩NBC significantly differentially expressed in each dataset and in all the three datasets. (XLSX 226 kb)
Acknowledgements
We are grateful to all the members of the Wall Lab for helpful discussions, their time and assistance. We also would like to thank all the families who contributed the data that made this study possible.
Funding
This work was supported by funds to DPW, from grant 1R01MH090611 from the National Institutes of Health and the Hartwell Foundation’s Autism Research and Technology Initiative (iHART), and to FJE from Junta de Andalucía (BIO-302) and MEIC (Systems Medicine Excellence Network SAF2015-70270-REDT).
Availability of data and materials
Supporting data are contained in the Additional files.
Authors’ contributions
LDB, FJE and DPW conceived of the study and designed the research. LDB performed the analyses. LDB, FJE and DPW interpreted the data and results. LDB wrote the initial manuscript. DPW, FJE provided oversight and revisions. LDB, MV, AO and MD participated in the data acquisition and preprocessing. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ADHD
Attention Deficit Hyperactivity Disorder
- ASD
Autism Spectrum Disorder
- BP
Biological Processes
- CDC
Center for Disease Control and Prevention
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- FDR
False Discovery Rate
- FSH
Follicle-Stimulating Hormone
- GEO
Gene Expression Omnibus
- GO
Gene Ontology
- ICD-9
International Classification of Diseases, 9th version
- IPA®
Ingenuity® Pathway Analysis software
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- Lh
Luteinizing hormone
- MATLAB®
MATrix LABoratory software
- MDAG
Multi Disorder Autism Gene Set
- MeSH
Medical Subject Headings
- mRNA
Messenger Ribonucleic Acid
- NBC
Network-based Candidates
- NCHS
National Center for Health Statistics
- PBC
Process-based Candidates
- RMA
Robust Multiarray Average
- RNA
Ribonucleic acid
- SFARI
Simons Foundation Autism Research Initiative
- STRING
Search Tool for the Retrieval of Interacting Genes/Proteins
References
- 1.Mattila M-L, Kielinen M, Linna S-L, Jussila K, Ebeling H, Bloigu R, et al. Autism spectrum disorders according to DSM-IV-TR and comparison with DSM-5 draft criteria: an epidemiological study. J Am Acad Child Adolesc Psychiatry. 2011;50(6):583–92. doi: 10.1016/j.jaac.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Kim YS, Leventhal BL, Koh Y-J, Fombonne E, Laska E, Lim E-C, et al. Prevalence of Autism Spectrum Disorders in a Total Population Sample. Am J Psychiatry. 2011;168(9):904–12. doi: 10.1176/appi.ajp.2011.10101532. [DOI] [PubMed] [Google Scholar]
- 3.Baron-Cohen S, Scott FJ, Allison C, Williams J, Bolton P, Matthews FE, et al. Prevalence of autism-spectrum conditions: UK school-based population study. Br J Psychiatry. 2009;194(6):500–9. doi: 10.1192/bjp.bp.108.059345. [DOI] [PubMed] [Google Scholar]
- 4.Hsu S-W, Chiang P-H, Lin L-P, Lin J-D. Disparity in autism spectrum disorder prevalence among Taiwan National Health Insurance enrollees: Age, gender and urbanization effects. Res Autism Spectr Disord. 2012;6(2):836–41. [Google Scholar]
- 5.Idring S, Rai D, Dal H, Dalman C, Sturm H, Zander E, et al. Autism Spectrum Disorders in the Stockholm Youth Cohort: Design, Prevalence and Validity. PLoS One. 2012;7(7):e41280. doi: 10.1371/journal.pone.0041280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell G, Rodgers LR, Ukoumunne OC, Ford T. Prevalence of Parent-Reported ASD and ADHD in the UK: Findings from the Millennium Cohort Study. J Autism Dev Disord. 2013;44(1):31–40. doi: 10.1007/s10803-013-1849-0. [DOI] [PubMed] [Google Scholar]
- 7.Saemundsen E, Magnússon P, Georgsdóttir I, Egilsson E, Rafnsson V. Prevalence of autism spectrum disorders in an Icelandic birth cohort. BMJ Open. 2013;3(6):e002748. doi: 10.1136/bmjopen-2013-002748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zablotsky B, Black LI, Maenner MJ, Schieve LA, Blumberg SJ. Estimated Prevalence of Autism and Other Developmental Disabilities Following Questionnaire Changes in the 2014 National Health Interview Survey. Natl Health Stat Report. 2015;87:1–21. [PubMed] [Google Scholar]
- 9.Blumberg SJ BM, Kogan MD, Schieve LA, Jones JR, Lu MC. Changes in Prevalence of Parent-reported Autism Spectrum Disorder in School-aged U.S. Children: 2007 to 2011–2012. Natl Health Stat Report. 2013;65:1–7. [PubMed] [Google Scholar]
- 10.Christensen DL, Baio J, Braun KVN, Bilder D, Charles J, Constantino JN, et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill Summ. 2016;65(3):1–23. doi: 10.15585/mmwr.ss6503a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matelski L, Van de Water J. Risk factors in autism: Thinking outside the brain. J Autoimmun. 2016;67:1–7. doi: 10.1016/j.jaut.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai M-C, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383(9920):896–910. doi: 10.1016/S0140-6736(13)61539-1. [DOI] [PubMed] [Google Scholar]
- 13.Yoo H. Genetics of Autism Spectrum Disorder: Current Status and Possible Clinical Applications. Exp Neurobiol. 2015;24(4):257–72. doi: 10.5607/en.2015.24.4.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geschwind DH, State MW. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 2015;14(11):1109–20. doi: 10.1016/S1474-4422(15)00044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leyfer OT, Folstein SE, Bacalman S, Davis NO, Dinh E, Morgan J, et al. Comorbid Psychiatric Disorders in Children with Autism: Interview Development and Rates of Disorders. J Autism Dev Disord. 2006;36(7):849–61. doi: 10.1007/s10803-006-0123-0. [DOI] [PubMed] [Google Scholar]
- 16.Wall DP, Esteban FJ, DeLuca TF, Huyck M, Monaghan T, Velez de Mendizabal N, et al. Comparative analysis of neurological disorders focuses genome-wide search for autism genes. Genomics. 2009;93(2):120–9. doi: 10.1016/j.ygeno.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Simonoff E, Pickles A, Charman T, Chandler S, Loucas T, Baird G. Psychiatric Disorders in Children With Autism Spectrum Disorders: Prevalence, Comorbidity, and Associated Factors in a Population-Derived Sample. J Am Acad Child Adolesc Psychiatry. 2008;47(8):921–9. doi: 10.1097/CHI.0b013e318179964f. [DOI] [PubMed] [Google Scholar]
- 18.Wingate M, Kirby RS, Pettygrove S, Cunniff C, Schulz E, Ghosh T, et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years-Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2010. MMWR Surveill Summ. 2014;63:21. [PubMed] [Google Scholar]
- 19.Kohane IS, McMurry A, Weber G, MacFadden D, Rappaport L, Kunkel L, et al. The Co-Morbidity Burden of Children and Young Adults with Autism Spectrum Disorders. PLoS One. 2012;7(4):e33224. doi: 10.1371/journal.pone.0033224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Autism and Developmental Disabilities Monitoring Network . Prevalence of Autism Spectrum Disorders Surveillance Year 2008 Principal Investigators-Autism and Developmental Disabilities Monitoring Network, 14 Sites, United States, 2008. 2012. pp. 1–24. [PubMed] [Google Scholar]
- 21.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–21. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuen RKC, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21:185–91. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
- 23.Jiang Y-H, Yuen RKC, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93:249–63. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matson JL, Nebel-Schwalm MS. Comorbid psychopathology with autism spectrum disorder in children: An overview. Res Dev Disabil. 2007;28(4):341–52. doi: 10.1016/j.ridd.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 25.World Health Organization . International Statistical Classification of Diseases and Related Health Problems: 10th Revision. 2007. [Google Scholar]
- 26.Yu W, Clyne M, Khoury MJ, Gwinn M. Phenopedia and Genopedia: disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics. 2010;26(1):145–6. doi: 10.1093/bioinformatics/btp618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jung J-Y, DeLuca TF, Nelson TH, Wall DP. A literature search tool for intelligent extraction of disease-associated genes. J Am Med Inform Assoc. 2014;21(3):399–405. doi: 10.1136/amiajnl-2012-001563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs) Mol Autism. 2013;4(1):1–3. doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional, and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–15. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hennig C. Flexible procedures for clustering. R package Version 2.1–9. 2014. (https://cran.r-project.org/web/packages/fpc/index.html).
- 31.Hennig C. Cluster-wise assessment of cluster stability. Comput Stat Data Anal. 2007;52:258–71. [Google Scholar]
- 32.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2008;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 35.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong SW, Collins CD, Shimizu-Motohashi Y, Holm IA, Campbell MG, Lee I-H, et al. Characteristics and Predictive Value of Blood Transcriptome Signature in Males with Autism Spectrum Disorders. PLoS One. 2012;7(12):e49475. doi: 10.1371/journal.pone.0049475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alter MD, Kharkar R, Ramsey KE, Craig DW, Melmed RD, Grebe TA, et al. Autism and Increased Paternal Age Related Changes in Global Levels of Gene Expression Regulation. PLoS One. 2011;6(2):e16715. doi: 10.1371/journal.pone.0016715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pramparo T, Lombardo MV, Campbell K, Barnes CC, Marinero S, Solso S, et al. Cell cycle networks link gene expression dysregulation, mutation, and brain maldevelopment in autistic toddlers. Mol Syst Biol. 2015;11(12):841. doi: 10.15252/msb.20156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 40.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Meth. 2015;12(2):115–21. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–8. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 42.Pollard KS, Dudoit S, Laan MJ. Multiple Testing Procedures: the multtest Package and Applications to Genomics. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York: Springer New York; 2005. pp. 249–71. [Google Scholar]
- 43.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci. 2003;100(16):9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, Shen C, Sivachenko A. Mining Alzheimer Disease Relevant Proteins from Integrated Protein Interactome Data. Pac Symp Biocomput. 2006;11:367–78. [PubMed] [Google Scholar]
- 45.Cusick ME, Klitgord N, Vidal M, Hill DE. Interactome: gateway into systems biology. Hum Mol Genet. 2005;14(suppl 2):R171–81. doi: 10.1093/hmg/ddi335. [DOI] [PubMed] [Google Scholar]
- 46.Giorgini F, Muchowski PJ. Connecting the dots in Huntington’s disease with protein interaction networks. Genome Biol. 2005;6(3):210. doi: 10.1186/gb-2005-6-3-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol Cell. 2004;15(6):853–65. doi: 10.1016/j.molcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 48.Humbert S, Saudou F. The Ataxia-ome: Connecting Disease Proteins of the Cerebellum. Cell. 2006;125(4):645–7. doi: 10.1016/j.cell.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Kann MG. Protein interactions and disease: computational approaches to uncover the etiology of diseases. Brief Bioinform. 2007;8(5):333–46. doi: 10.1093/bib/bbm031. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Aronow BJ, Jegga AG. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics. 2009;10(1):1–14. doi: 10.1186/1471-2105-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu C, Zhu J, Zhang X. Integrating gene expression and protein-protein interaction network to prioritize cancer-associated genes. BMC Bioinformatics. 2012;13(1):1–10. doi: 10.1186/1471-2105-13-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo J, Liang S. Prioritization of potential candidate disease genes by topological similarity of protein-protein interaction network and phenotype data. J Biomed Inform. 2015;53:229–36. doi: 10.1016/j.jbi.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Fiorentino A, Sharp SI, McQuillin A. Association of rare variation in the glutamate receptor gene SLC1A2 with susceptibility to bipolar disorder and schizophrenia. Eur J Hum Genet. 2015;23(9):1200–6. doi: 10.1038/ejhg.2014.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palma-Gudiel H, Córdova-Palomera A, Leza JC, Fañanás L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: A critical review. Neurosci Biobehav Rev. 2015;55:520–35. doi: 10.1016/j.neubiorev.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 55.van der Knaap LJ, Riese H, Hudziak JJ, Verbiest MMPJ, Verhulst FC, Oldehinkel AJ, et al. Glucocorticoid receptor gene (NR3C1) methylation following stressful events between birth and adolescence. The TRAILS study. Transl Psychiatry. 2014;4(4):e381. doi: 10.1038/tp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marshall CR, Young EJ, Pani AM, Freckmann M-L, Lacassie Y, Howald C, et al. Infantile Spasms Is Associated with Deletion of the MAGI2 Gene on Chromosome 7q11.23-q21.11. Am J Hum Genet. 2008;83(1):106–11. doi: 10.1016/j.ajhg.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koide T, Banno M, Aleksic B, Yamashita S, Kikuchi T, Kohmura K, et al. Common Variants in MAGI2 Gene Are Associated with Increased Risk for Cognitive Impairment in Schizophrenic Patients. PLoS One. 2012;7(5):e36836. doi: 10.1371/journal.pone.0036836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner TN, Sharma K, Oh EC, Liu YP, Collins RL, Sosa MX, et al. Loss of delta catenin function in severe autism. Nature. 2015;520(7545):51–6. doi: 10.1038/nature14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collins AL, Ma D, Whitehead PL, Martin ER, Wright HH, Abramson RK, et al. Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics. 2006;7(3):167–74. doi: 10.1007/s10048-006-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hedges DJ, Hamilton-Nelson KL, Sacharow SJ, Nations L, Beecham GW, Kozhekbaeva ZM, et al. Evidence of novel fine-scale structural variation at autism spectrum disorder candidate loci. Mol Autism. 2012;3:2–2. doi: 10.1186/2040-2392-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong L, Piton A, et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry. 2011;1(11):e55. doi: 10.1038/tp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawakubo Y, Suga M, Tochigi M, Yumoto M, Itoh K, Sasaki T, et al. Effects of Metabotropic Glutamate Receptor 3 Genotype on Phonetic Mismatch Negativity. PLoS One. 2011;6(10):e24929. doi: 10.1371/journal.pone.0024929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ginsberg SD, Hemby SE, Smiley JF. Expression profiling in neuropsychiatric disorders: emphasis on glutamate receptors in bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):705–11. doi: 10.1016/j.pbb.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mei L, Xiong W-C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9(6):437–52. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alter O, Brown PO, Botstein D. Singular value decomposition for genome-wide expression data processing and modeling. Proc Natl Acad Sci U S A. 2000;97(18):10101–6. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Butte A, Ye J, Haring H, Stumvoll M, White M, Kohane I. Determining significant fold differences in gene expression analysis. Pac Symp Biocomput. 2001;6:6–17. doi: 10.1142/9789814447362_0002. [DOI] [PubMed] [Google Scholar]
- 67.Kuo WP, Jenssen T-K, Butte AJ, Ohno-Machado L, Kohane IS. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics. 2002;18(3):405–12. doi: 10.1093/bioinformatics/18.3.405. [DOI] [PubMed] [Google Scholar]
- 68.Steinert JR, Chernova T, Forsythe ID. Nitric Oxide Signaling in Brain Function, Dysfunction, and Dementia. Neuroscientist. 2010;16(4):435–52. doi: 10.1177/1073858410366481. [DOI] [PubMed] [Google Scholar]
- 69.Yuste JE, Tarragon E, Campuzano CM, Ros-Bernal F. Implications of glial nitric oxide in neurodegenerative diseases. Front Cell Neurosci. 2015;9:322. doi: 10.3389/fncel.2015.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Skafidas E, Testa R, Zantomio D, Chana G, Everall IP, Pantelis C. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol Psychiatry. 2014;19(4):504–10. doi: 10.1038/mp.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nualart-Marti A, Solsona C, Fields RD. Gap Junction Communication in Myelinating Glia. Biochim Biophys Acta. 2013;1828(1):69–78. doi: 10.1016/j.bbamem.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grubišić V, Parpura V. The second brain in autism spectrum disorder: could connexin 43 expressed in enteric glial cells play a role? Front Cell Neurosci. 2015;9:242. doi: 10.3389/fncel.2015.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suda S, Iwata K, Shimmura C, Kameno Y, Anitha A, Thanseem I, et al. Decreased expression of axon-guidance receptors in the anterior cingulate cortex in autism. Mol Autism. 2011;2(1):1–5. doi: 10.1186/2040-2392-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robichaux MA, Cowan CW. Signaling Mechanisms of Axon Guidance and Early Synaptogenesis. In: Andersen LS, Pine SD, editors. The Neurobiology of Childhood. Heidelberg: Springer Berlin Heidelberg; 2014. pp. 19–48. [DOI] [PubMed] [Google Scholar]
- 75.Bakos J, Bacova Z, Grant SG, Castejon AM, Ostatnikova D. Are Molecules Involved in Neuritogenesis and Axon Guidance Related to Autism Pathogenesis? Neuromolecular Med. 2015;17(3):297–304. doi: 10.1007/s12017-015-8357-7. [DOI] [PubMed] [Google Scholar]
- 76.Raison CL, Miller AH. When Not Enough Is Too Much: The Role of Insufficient Glucocorticoid Signaling in the Pathophysiology of Stress-Related Disorders. Am J Psychiatry. 2003;160(9):1554–65. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- 77.Godavarthi SK, Dey P, Maheshwari M, Ranjan JN. Defective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndrome. Hum Mol Genet. 2012;21(8):1824–34. doi: 10.1093/hmg/ddr614. [DOI] [PubMed] [Google Scholar]
- 78.Rosen TE, Connell JE, Kerns CM. A Review of Behavioral Interventions for Anxiety-Related Behaviors in Lower-Functioning Individuals with Autism. Behav Interv. 2016;31(2):120–43. [Google Scholar]
- 79.Sundermann EE, Maki PM, Bishop JR. A Review of Estrogen Receptor α Gene (ESR1) Polymorphisms, Mood, and Cognition. Menopause. 2010;17(4):874–86. doi: 10.1097/gme.0b013e3181df4a19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zettergren A, Jonsson L, Johansson D, Melke J, Lundström S, Anckarsäter H, et al. Associations between polymorphisms in sex steroid related genes and autistic-like traits. Psychoneuroendocrinology. 2013;38(11):2575–84. doi: 10.1016/j.psyneuen.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 81.Chang JR, Ghafouri M, Mukerjee R, Bagashev A, Chabrashvili T, Sawaya BE. Role of p53 in Neurodegenerative Diseases. Neurodegener Dis. 2012;9(2):68–80. doi: 10.1159/000329999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hallmayer J. Getting our AKT together in schizophrenia? Nat Genet. 2004;36(2):115–6. doi: 10.1038/ng0204-115. [DOI] [PubMed] [Google Scholar]
- 83.Balu DT, Carlson GC, Talbot K, Kazi H, Hill-Smith TE, Easton RM, et al. Akt1 Deficiency in Schizophrenia and Impairment of Hippocampal Plasticity and Function. Hippocampus. 2012;22(2):230–40. doi: 10.1002/hipo.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kitagishi Y, Minami A, Nakanishi A, Ogura Y, Matsuda S. Neuron Membrane Trafficking and Protein Kinases Involved in Autism and ADHD. Int J Mol Sci. 2015;16(2):3095–115. doi: 10.3390/ijms16023095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, et al. Deletion of ERK2 Mitogen-Activated Protein Kinase Identifies Its Key Roles in Cortical Neurogenesis and Cognitive Function. J Neurosci Off J Soc Neurosci. 2008;28(27):6983–95. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wen Y, Alshikho MJ, Herbert MR. Pathway Network Analyses for Autism Reveal Multisystem Involvement, Major Overlaps with Other Diseases and Convergence upon MAPK and Calcium Signaling. PLoS One. 2016;11(4):e0153329. doi: 10.1371/journal.pone.0153329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Russo AJ. Increased Epidermal Growth Factor Receptor (EGFR) Associated with Hepatocyte Growth Factor (HGF) and Symptom Severity in Children with Autism Spectrum Disorders (ASDs) J Cent Nerv Syst Dis. 2014;6:79–83. doi: 10.4137/JCNSD.S13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ray B, Long JM, Sokol DK, Lahiri DK. Increased Secreted Amyloid Precursor Protein-α (sAPPα) in Severe Autism: Proposal of a Specific, Anabolic Pathway and Putative Biomarker. PLoS One. 2011;6(6):e20405. doi: 10.1371/journal.pone.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sokol DK, Demao C, Farlow MR, Dunn DW, Maloney B, Zimmer JA, et al. High Levels of Alzheimer Beta-Amyloid Precursor Protein (APP) in Children With Severely Autistic Behavior and Aggression. J Child Neurol. 2006;21(6):444–9. doi: 10.1177/08830738060210062201. [DOI] [PubMed] [Google Scholar]
- 90.Lahiri DK, Sokol DK, Erickson C, Ray B, Ho CY, Maloney B. Autism as early neurodevelopmental disorder: evidence for an sAPPα-mediated anabolic pathway. Front Cell Neurosci. 2013;7:94. doi: 10.3389/fncel.2013.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vadakkadath Meethal S, Atwood CS. Alzheimer’s disease: the impact of age-related changes in reproductive hormones. Cell Mol Life Sci. 2005;62(3):257–70. doi: 10.1007/s00018-004-4380-4. [DOI] [PubMed] [Google Scholar]
- 92.Kosfeld M, Heinrichs M, Zak PJ, Fischbacher U, Fehr E. Oxytocin increases trust in humans. Nature. 2005;435(7042):673–6. doi: 10.1038/nature03701. [DOI] [PubMed] [Google Scholar]
- 93.Baumgartner T, Heinrichs M, Vonlanthen A, Fischbacher U, Fehr E. Oxytocin Shapes the Neural Circuitry of Trust and Trust Adaptation in Humans. Neuron. 2008;58(4):639–50. doi: 10.1016/j.neuron.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 94.Wittfoth-Schardt D, Gründing J, Wittfoth M, Lanfermann H, Heinrichs M, Domes G, et al. Oxytocin Modulates Neural Reactivity to Children’s Faces as a Function of Social Salience. Neuropsychopharmacology. 2012;37(8):1799–807. doi: 10.1038/npp.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ross HE, Young LJ. Oxytocin and the Neural Mechanisms Regulating Social Cognition and Affiliative Behavior. Front Neuroendocrinol. 2009;30(4):534–47. doi: 10.1016/j.yfrne.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peñagarikano O, Lázaro MT, Lu X-H, Gordon A, Dong H, Lam HA, et al. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci Transl Med. 2015;7(271):271ra278. doi: 10.1126/scitranslmed.3010257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jacobson JD, Ellerbeck KA, Kelly KA, Fleming KK, Jamison TR, Coffey CW, et al. Evidence for Alterations in Stimulatory G proteins and Oxytocin Levels in Children with Autism. Psychoneuroendocrinology. 2014;40:159–69. doi: 10.1016/j.psyneuen.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marazziti D, Catena D’o M. The role of oxytocin in neuropsychiatric disorders. Curr Med Chem. 2008;15(7):698–704. doi: 10.2174/092986708783885291. [DOI] [PubMed] [Google Scholar]
- 99.Gordon I, Vander Wyk BC, Bennett RH, Cordeaux C, Lucas MV, Eilbott JA, et al. Oxytocin enhances brain function in children with autism. Proc Natl Acad Sci U S A. 2013;110(52):20953–8. doi: 10.1073/pnas.1312857110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Damiano CR, Aloi J, Dunlap K, Burrus CJ, Mosner MG, Kozink RV, et al. Association between the oxytocin receptor (OXTR) gene and mesolimbic responses to rewards. Mol Autism. 2014;5:7–7. doi: 10.1186/2040-2392-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Diaz-Beltran L, Esteban FJ, Wall DP. A common molecular signature in ASD gene expression: following Root 66 to autism. Transl Psychiatry. 2016;6:e705. doi: 10.1038/tp.2015.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Information summary of the datasets selected. (DOCX 12 kb)
Box plots showing the distribution of the samples of each dataset after preprocessing; median-centered values indicate that the data are normalized and cross-comparable. (PDF 5181 kb)
Complete list of comorbid conditions to autism. Autism sibling disorders are highlighted in blue. (XLSX 16 kb)
Total number of genes of each comorbid condition utilized for this study. Highlighted in blue are ASD sibling disorders. (XLSX 135 kb)
Comorbid disorders integrating each group generated by the bootstrap analysis (Additional file 6: Figure S2), along with their Mean Jaccard Coefficient value. The different groups of disorders generated by our bootstrap procedure corresponds to the disorder clusters obtained in our original gene-based dendrogram (Fig. 1). Groups 1, 2 and 3 have the highest Mean Jaccard values meaning they are the most robust and stable groupings of the tree. Group 2 coincides with the cluster conformed by the autism sibling disorders with a highly significant Mean Jaccard value of approximately 0.785. (DOCX 13 kb)
First two Multidimensional Scaling (MDS) dimensions of our dataset generated by MDS on a dissimilarity matrix using Jaccard Coefficient when k = 6. Each group is highlighted in a different color and the disorders conforming them are detailed in Additional file 5: Table S4, along with their corresponding mean Jaccard Coefficient value. The autism sibling comorbid disorders are clustered together in group 2 (PDF 180 kb)
STRING edge summary for each member of the ASD sibling group. (XLSX 8411 kb)
The multi disorder autism gene set (MDAG) and the sibling comorbid conditions where these genes are found. (XLSX 25 kb)
A. The complete network of autism candidate genes. The MDAG genes are highlighted in yellow and their interactions in red; these are the genes that occur in one or more of the autism sibling comorbid disorders, circumscribed in Fig. 1. B. The highly interconnected subcomponent conformed by the MDAG genes, separated from the autism network. (TIF 406700 kb)
Biological processes for which the Multi-disorder component of the autism gene set (MDAG) were enriched. Identities of the MDAG genes overrepresented in the processes as well as the corrected p-values for the enrichment scores are provided. Enrichment was calculated using the biological processes only found among the MDAG genes and not found among the sibling disorders. (XLSX 50 kb)
Identities of the differentially expressed PBC, along with their corresponding q-values, the biological processes where they are involved and the comorbid disorders where they are implicated. Also, PBC significantly differentially expressed in each dataset and in all the three datasets. (XLSX 210 kb)
P-value and q-value histograms and q-plots from the multiple test correction analyses performed on the PBC and NBC to verify whether they were significantly differentially regulated in autistics in comparison to controls. (PDF 11411 kb)
Complete list of NBC, along with their q-values, MDAG interactors and comorbid disorders where they are present. Also, NBC significantly differentially expressed in each dataset and in all the three datasets. (XLSX 185 kb)
Complete list of genes present in the intersection of PBC∩NBC, along with the biological processes where they are involved, MDAG interactors and comorbid disorders where they are present. Also, PBC∩NBC significantly differentially expressed in each dataset and in all the three datasets. (XLSX 226 kb)
Data Availability Statement
Supporting data are contained in the Additional files.