

Abstract
Menin up-regulates transcription of cell cycle inhibitors to suppress endocrine tumors, but it is poorly understood how menin suppresses non-endocrine tumors such as lung cancer. Here, we show that menin inhibits proliferation of human lung cancer cells and growth of lung cancer in mice. The menin-mediated tumor suppression requires repression of growth factor pleiotrophin (PTN), which binds to its cell surface receptor, anaplastic lymphoma kinase (ALK) that is activated in certain lung adenocarcinomas. Menin represses PTN transcription and PTN-induced proliferation of human lung cancer cells, and menin expression is substantially reduced in primary human lung adenocarcinomas. Notably, menin binds the PTN locus and enhances Polycomb gene EZH2-mediated histone H3 lysine 27 trimethylation (H3K27m3), a negative mark for gene transcription but does not affect histone H3K4 methylation that is usually up-regulated by menin in endocrine cells. Together, our findings indicate that menin suppresses lung cancer partly through increasing polycomb gene-mediated H3K27 methylation and repressing PTN transcription, unraveling a novel, epigenetically regulated PTN-ALK signaling pathway in suppressing lung cancer.
Keywords: lung adenocarcinoma, histone H3 lysine 27, Men1, pleiotrophin, Polycomb
Introduction
Lung cancer is a leading cause of cancer-related death over the world, and the 5-year survival rate remains poor despite aggressive chemotherapy (Jemal et al., 2002). Somatic mutations in proto-oncogenes, such as K-Ras, epidermal growth factor receptor (EGFR), and anaplastic lymphoma kinase (ALK), are often identified in lung adenocarcinoma (Herbst et al., 2008; Jemal et al., 2002; Soda et al., 2007). Pei et al had reported that non-small cell lung cancers (NSCLC) develop in both Men1+/- and p18-/-; Men1+/- mice at a high penetrance (Pei et al., 2007). However, little is known as to how menin suppresses development of NSCLC.
The Men1 gene, which encodes the nuclear protein menin, is mutated in patients with an inherited tumor syndrome, multiple endocrine neoplasia type 1 (MEN1) (Chandrasekharappa et al., 1997). In MEN1 endocrine tumors with a germline mutation in one of the MEN1 alleles, the remaining wild type MEN1 allele is often inactivated due to a somatic mutation (Loss of Heterozygocity, LOH), indicating MEN1 as a bona fide tumor suppressor gene in endocrine tumors (Lemos and Thakker, 2008).
Though menin's primary sequence does not reveal any paralogs, multiple lines of evidence suggest that one of the major functions of menin is to regulate gene transcription (Karnik et al., 2005; Milne et al., 2005; Yokoyama et al., 2004). Menin associates with chromatin and the nuclear matrix and exerts multiple biological functions including regulation of cell proliferation (Jin et al., 2003; Schnepp et al., 2006; Schnepp et al., 2004). These diverse menin functions may be largely attributed to the crucial role of menin as a scaffold protein in coordinately regulating transcription of various target genes.
Menin interacts with mixed lineage leukemia proteins (MLL), histone H3 methyltransferases that catalyze histone H3 lysine 4 (H3K4) methylation with their highly conserved SET domain (Milne et al., 2002). MLL is an orthologue of the Drosophila trithorax group (TrxG) genes (Yu et al., 1998), which interact with and antagonize another group of genes, the Polycomb group (PcG) genes (Ringrose and Paro, 2007; Schuettengruber et al., 2007). PcG genes form various protein complexes including Polycomb repressive complex 2 (PRC2), which contains Enhancer of Zeste homolog 2 (EZH2) and its regulatory protein SUZ12 (Ringrose and Paro, 2007; Schuettengruber et al., 2007; Sparmann and van Lohuizen, 2006). EZH2 is also a chromatin-associating protein with a conserved SET domain (Cao et al., 2002). However, unlike the SET domain in MLL that methylates H3K4 (Yokoyama et al., 2004), the EZH2 SET domain specifically methylates H3K27 and the methylated H3K27 can be recognized by other specific binding proteins to compress chromatin structure, leading to repression of gene transcription (Cao et al., 2002; Sparmann and van Lohuizen, 2006). Site-specific histone modifications are a major epigenetic mechanism for maintaining stable gene transcription or silencing in differentiated cells (Cao et al., 2002; Martin and Zhang, 2007; Sparmann and van Lohuizen, 2006). Though menin promotes H3K4 methylation, which is presumably catalyzed partly by MLL at loci of the cell cycle inhibitors p18Ink4c (p18) and p27Kip1 (p27) gene (Karnik et al., 2005; Milne et al., 2005), little is known as to whether PcG proteins ever participate in menin-regulated gene transcription.
Our current studies show that menin inhibits human lung cancer cells through repression of pleiotrophin (PTN) and its cell surface receptors including ALK (Stoica et al., 2001). Instead of working with its known partner MLL to methylate H3K4, a positive mark, menin bound the promoter of PTN and recruited the Polycomb group (PcG) complex to the locus, resulting in H3K27 trimethylation, PTN suppression, and inhibition of proliferation of lung cancer cells. These findings unravel a novel mean in suppressing lung cancer through epigenetically repressing the PTN and ALK via a menin-PcG complex.
Materials and methods
Cell culture
A549 (lung adenocarcinoma), NCI-H157 (NSCLC), and NCI-H446 (SCLC) human lung cancer cell lines were purchased from the American Type Culture Collection (ATCC, Philadelphia, PA, U.S.A.). A549 and NCI-H446 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Hyclone, Utah, USA) or RPMI 1640 (Hyclone), respectively, and supplemented with 10% (v/v) fetal bovine serum (FBS) (Hyclone), and 1×Penicillin-Streptomycin (Pen-Strep, 100 U/ml-100 μg/ml) (Invitrogen, Carlsbad CA, USA). NCI-H157 cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 1 mM Sodium Pyruvate (Hyclone), 10 mM Hepes (Hyclone) and 1×Pen-Strep. The Men1+/+ and Men1-/- MEFs were described previously (Schnepp et al., 2006) and cultured in DMEM supplemented with 10 % FBS, 1× Pen-Strep, 2 mM L-glutamine (Invitrogen), and 0.1 mM Non-essential amino acids (Hyclone).
Immunohistochemistry (IHC) and immunofluorescent (IF) staining
IHC and IF detection was performed using an affinity-purified anti-menin antibody (Jin et al., 2003), the specificity of the anti-menin antibody was verified in menin-null and menin-expressing cells (Suppl Fig. 1), anti-PTN (Abnova) or anti-ALK (Cell Signaling) antibodies. Nuclei were counterstained with DAPI, and the stained cells were analyzed and photographed under a confocal microscope (OLYMPUS FV1000, ×400).
Western blotting and enzyme-linked immunosorbent assay (ELISA)
For Western blotting, cells were lysed in 20 mM Tris-HCl (pH 8.0), 5% glycerol, 138 mM NaCl, 2.7 mM KCl, 1% NP-40, 20 mM NaF, 5 mM EDTA, 1 mM sodium orthovanadate, 5μg /ml leupeptin, 1μg /ml pepstatin, and 1 mM DTT. The extracted proteins were resolved by SDS-PAGE before transfer onto PVDF membrane, followed by incubation with the anti-menin (Bethyl Laboratories, Montgomery, USA), anti-PTN (Abnova, Walnut, USA), or anti-beta-actin antibodies (Santa Cruz biotechnology, Santa Cruz, USA). ELISA for PTN was performed by using a PTN ELISA kit (Uscn Life Science& Technology, Wuhan, China), according to the manufacturer's instructions.
Chromatin immunoprecipitation assay (ChIP)
ChIP assays were performed as described with certain modifications (Chen et al., 2006). Briefly, 1×106 Men1-/- and Men1+/+MEFs were treated with 1 % formaldehyde for 10 min, followed by pulsed sonication to shear cellular DNA. ChIP assays were then carried out with indicated antibodies according to the protocol of the ChIP Assay kit (Millipore Billerica, USA). Antibodies used for ChIP assays were: anti-menin (Bethyl Laboratories), anti-trimethyl-Histone H3 Lys27 (Millipore), anti-trimethylated histone H3 Lys 4 (Abcam, Cambridge, UK), anti-acetyl-Histone H3 (Millipore), anti-EZH2 (Cell Signaling, Danvers, USA), anti-SUZ12 (Cell Signaling), anti-RNA polymerase II (Millipore), or control IgG (Santa Cruz). After overnight incubation with the antibodies, the crosslinks between nuclear proteins and genomic DNA were reversed, and the antibody pulled down DNA was purified by phenol/chloroform extraction. Quantitative PCR was performed using primers specific for menin target genes or GAPDH. Primer pair (PP) sequences were shown in Suppl Tab 1.
Results
Menin inhibits lung cancer cell proliferation through repression of PTN expression
p18, an inhibitor of cyclin-dependent kinase, suppresses lung cancer in collaboration with Men1 in mice (Pei et al., 2007). Our earlier work showed that loss of menin in MEFs increases expression of PTN 11 fold (La et al., 2004). PTN is a heparin-binding growth factor that is highly expressed in certain solid cancers such as breast and lung cancer (Jager et al., 2002; Perez-Pinera et al., 2007a). Though it is still relatively unclear how PTN transduces its pro-proliferation signal into cells and several cell surface receptors have been proposed to bind PTN (Lu et al., 2005; Stoica et al., 2001), a prevalent model suggests that PTN binds to its cell surface receptor, protein tyrosine phosphatase receptor Z1 (PTPRZ1), inhibiting its phosphatase activity towards another oncogenic kinase, ALK (Lu et al., 2005; Meng et al., 2000; Powers et al., 2002; Stoica et al., 2001). To explore whether menin affects proliferation and PTN expression of lung cancer cells, we stably transfected A549 cells with either control or menin-expressing construct. Our results show that ectopic menin expression reduced the number of the menin cDNA-transfected cells (Fig. 1A). Ectopic menin expression was confirmed by RT-PCR and Western blotting (Fig. 1B and C). PTN expression was reduced in A549 cells with ectopic menin expression at the mRNA level (Fig. 1B, lane 2), as well as at the intracellular and secreted PTN protein level (Fig. 1C, D, respectively). No obvious apoptosis was observed in menin-transfected cells, using Annexin V staining and flow cytometry analysis (Suppl Fig. 2).
Figure 1.
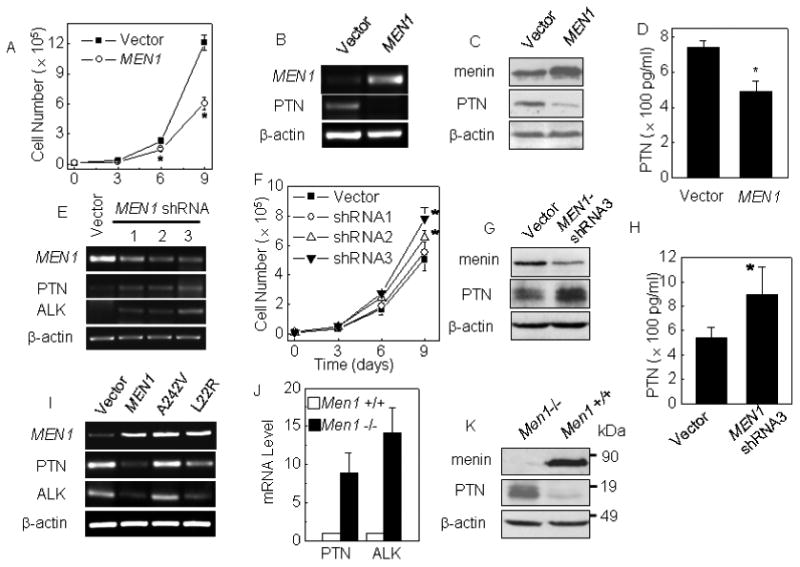
Menin inhibits proliferation of lung cancer cells and PTN expression. (A) The growth curves of A549 cells transfected with either pMX-puro or pMX-menin. A549 cells were transfected with either empty vector or vector expressing the MEN1 gene and selected by puromycin. (B-D) Down-regulated PTN expression was determined by RT-PCR, Western blot and ELISA, respectively. A549 cells transfected with a vector expressing shRNA against either Luc or menin. (E) MEN1, PTN and ALK mRNA level were detected by RT-PCR. (F) The growth curves of A549 cells with MEN1 knockdown. (G-H) Impact of menin silencing on PTN expression is determined by Western blotting and ELISA, respectively. (I) A549 cells were transfected with empty vector, wild-type menin, point mutation A242V or L22R, and selected by puromycin. The MEN1, PTN and ALK expression were detected by RT-PCR. (J) Increased PTN and ALK mRNA levels in Men1-/- MEFs were detected by real-time qRT-PCR. (K) Western blotting detection the impact of menin silencing or expression on PTN expression. * p<0.05 vs. control group.
Next, we examined whether knockdown of MEN1 (human gene encoding menin) using shRNAs affects proliferation of the cells. Control vector and constructs expressing each of the three distinct shRNAs that specifically target MEN1 were generated and stably transfected into A549 cells. MEN1 shRNAs 2-3 substantially reduced menin expression, but shRNA1 only mildly reduced menin expression, as shown by RT-PCR (Fig. 1E). Correlated with the levels of menin knockdown, shRNA 2-3 significantly increased cell proliferation(p<0.05), but control vector and menin shRNA1, which were unable to substantially reduce menin expression, did not significantly alter proliferation of A549 cells (Fig. 1F, p<0.05). Notably, MEN1 knockdown also increased mRNA and the intracellular and secreted PTN (Fig. 1E, G and H), but did not affect expression of a PTN paralog, MK (Suppl Fig. 3). Menin also suppresses proliferation of two additional human lung cancer cell lines, NCI-H157 and NCI-H446 (Suppl Fig. 4B and C). We also examined the effect of MEN1 point mutations, A242V and L22R, which were identified from inherited MEN1 patients (Milne et al., 2005), on expression of PTN and ALK, a receptor of PTN. We found that L22R and A242V point mutants lost or partially lost the ability to repress PTN and ALK expression (Fig. 1I). Together, these results indicate that menin is required for inhibiting PTN expression and proliferation of the human lung cancer cells.
To further reinforce the role of menin in repressing PTN expression, we next evaluated the impact of loss of menin on expression of PTN and PTN downstream regulator, ALK, in Men1+/+ and Men1-/- MEFs. Men1 excision substantially increased expression of PTN and ALK (Fig. 1J and K). The role of menin in repressing PTN and ALK expression was also confirmed in a separate pair of Men1-expressing and Men1-null MEFs. Consistent with this observation, ectopic menin expression also repressed proliferation of MEFs. (Suppl Fig. 5). Together, these data indicate that menin is required for repressing PTN expression in human A549 lung cancer cells and MEFs.
PTN expression was essential for Menin -inhibited lung cancer cell proliferation
We further examined if PTN is crucial for menin-mediated repression of proliferation of human A549 lung cancer cells. To this end, we generated three shRNAs that specifically target the PTN gene. Transfection of A549 cells with each of the three PTN shRNAs reduced the PTN mRNA (Fig. 3H) and the secreted PTN in culture medium (Fig. 2A), with PTN shRNAs 1-2 more effective. Notably, PTN knockdown by PTN shRNA1 significantly reduced proliferation of A549 cells (p<0.05), but PTN shRNA3, which was less effective in knocking down PTN, was also less potent in repressing proliferation of A549 cells (Fig. 2B), suggesting that it is PTN shRNA-targeted PTN knockdown, but not their off-targeting effect, that led to repressing proliferation of the lung cancer cells. Consistently, stably PTN-transfected cells (Fig.2C) proliferated ∼40% faster than the control vector cells at day 6 of culture (Fig. 2D, p<0.05), indicating that ectopic PTN expression is sufficient to increase proliferation of A549 cells. To further decipher the relationship between menin and PTN in controlling proliferation of A549 cells, these cells were transfected with either menin or menin and PTN, and their overexpression was confirmed by Western blotting (Fig. 2E). As expected, on day 6 of culture, menin reduced proliferation of the cells by over 30% (Fig. 2F, p<0.05). Notably, co-expression with PTN neutralized menin-induced repression of A549 cell proliferation (Fig. 2F). Collectively, these data demonstrate that menin-regulated PTN as a crucial target of menin in regulating proliferation of the lung cancer cells.
Figure 3.
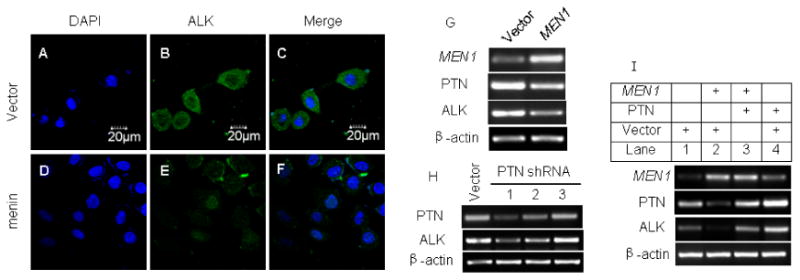
Menin indirectly down-regulates ALK expression by repressing PTN expression. A549 cells were stably transfected with pMX-puro or pMX-menin. (A-F) IF detection of ALK (green), DAPI (blue), and merge in the A549 cells. (G) Over-expression of menin and down-regulation of PTN and ALK expression were detected by RT-PCR. (H) A549 cells stably transfected with either control vector or vector expressing one of three distinct PTN shRNAs were analyzed by RT-PCR for mRNA level. (I) A549 cells were stably transfected with either control vector (30μg) or construct expressing either menin (MEN1 15μg+ vector 15μg) or PTN (PTN 15μg+ vector 15μg) or co-transfected with menin and PTN-expressing constructs (MEN1 15μg+ PTN 15μg). The resulting cells were processed to determine the menin, PTN and ALK mRNA level by RT-PCR.
Figure 2.
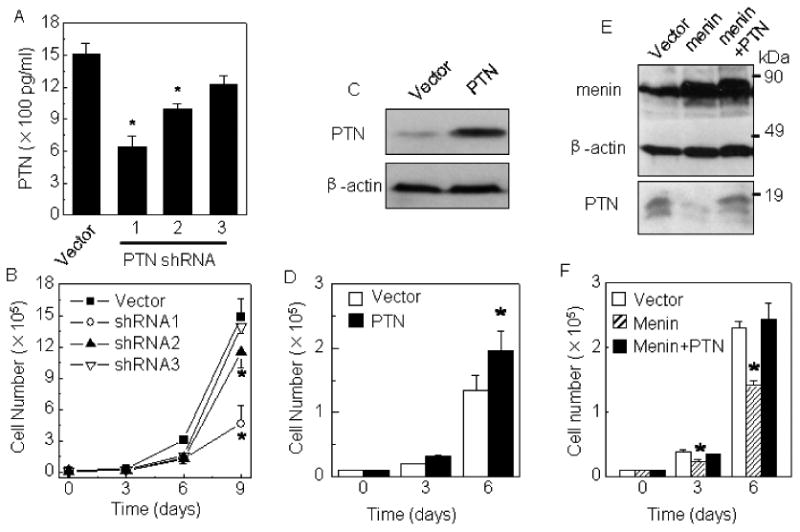
Menin inhibits A549 lung cancer cell proliferation partly through down-regulating PTN expression. (A) A549 cells were stably transfected with either empty vector or vector expressing one of three distinct PTN shRNAs. The efficiency of PTN knockdown was determined by measuring the secreted PTN by ELISA. (B) The cells generated above were evaluated for their proliferation at indicated on days of culture. (C) Growth curve of A549 cells stably transfected with either empty vector (pcDNA3.1) or pcDNA3.1-PTN. (D) A549 cells were stably transfected with either vector or vector expressing either menin or PTN, or cotransfected with menin and PTN-expressing constructs. (E,F) The over-expression of menin and its effect on down-regulation on PTN. * p<0.05 vs. control group.
Menin downregulates ALK expression partly through repressing PTN
MEN1 knockdown upregulates ALK as well as PTN in A549 cells (Fig. 1E), but ectopic menin expression reduced ALK expression (Fig. 3E and G). To further dissect the potential relationship between PTN and ALK, we stably transfected A549 cells with either control vector or one of the three PTN shRNAs. PTN shRNAs 1-2, which reduced PTN expression, also decreased the ALK mRNA level (Fig. 3H). Conversely, PTN shRNA3 that failed to knock down PTN also failed to reduce the ALK mRNA level (Fig. 3H). Though it is possible that menin represses PTN and ALK separately, it could not be ruled out that PTN also up-regulates ALK expression. Thus, we ectopically expressed menin and/or PTN to determine their effect on the ALK mRNA level in A549 cells. Notably, PTN expression abrogated menin-induced reduction of the ALK mRNA level (Fig. 3I, lane 3), and ectopic PTN expression alone also increased ALK expression (Fig. 3I, lane 4). Together, these findings argue that PTN up-regulates ALK expression, but menin represses ALK expression indirectly through inhibiting PTN expression, downregulating two crucial components of the PTN pathway, PTN and the downstream receptor ALK. These findings highlight the importance of menin in controlling this signaling pathway.
Menin inhibits growth of cancer xenograft derived from A549 cells
To explore whether menin affects growth of A549 cell-derived tumors in nude mice, A549 cells stably transfected with either control or menin-expressing constructs were subcutaneously transplanted into nude mice (n=10 per group). The size of the solid tumor was measured after various periods of time following transplantation. Menin expression resulted in smaller tumors in the nude mice after transplantation (Fig. 4A and B, p< 0.05). To determine if menin and PTN affect the growth of the established tumors in nude mice, A549 cells were transplanted to nude mice. When visible tumors were formed (day 6 post-transplantation), control DNA or construct expressing either menin or PTN shRNA was injected into the tumor mass. Both ectopic menin expression and PTN knockdown significantly reduced the xenograft tumor sizes (Fig. 4C, p< 0.05, respectively) and weight (Fig. 4D, p< 0.05) at day 8 and 10 post transplantation respectively. Both ectopic menin expression and PTN knockdown reduced PTN expression at the mRNA level (Fig. 4E) and protein level, as shown by ELISA assay (Fig. 4F). Ectopic menin expression in the tumor was confirmed by IHC staining (Fig. 4 G and H). These results indicate that menin represses, but PTN promotes, growth of xenograft of human lung adenocarcinoma in mice, highlighting the crucial role of menin and PTN in controlling growth of the tumors in vivo.
Figure 4.
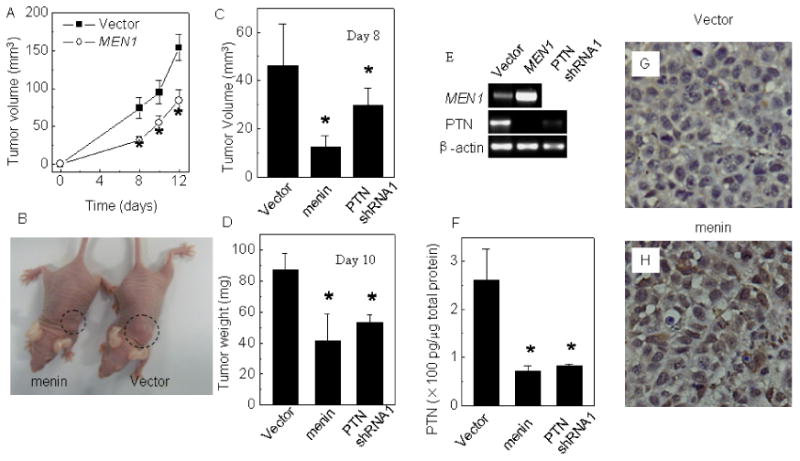
Menin over-expression significantly inhibits growth of A549 cell-derived tumor and PTN expression in vivo. (A) Menin-expressing vector or empty vector were stably transfected into A549 cells and injected subcutaneously into nude mice, and tumor formation was examined day 8-post transplantation. The tumor growth rate of recipient mice (n=10 per group) is shown. (B) A representative pair of nude mice transplanted with either menin-expressing or vector control A549 cells (day 12). (C) A549 cells were transplanted to nude mice and when visible tumors were formed (day 6 post-transplantation), control vector or vector expressing menin cDNA or PTN shRNA1, which were treated with PEI, were injected into the tumor mass. The tumor volume in mice was measured on day 8 after transplantation. (D) The recipient mice were sacrificed on day 10 post transplantation; the weight of tumors was measured. (E) Both ectopic menin expression and PTN knockdown reduced PTN expression at the mRNA level, as determined by qRT-PCR. (F) PTN protein from menin cDNA or PTN shRNA-transfected tumors was detected by ELISA assay. (G, H) Immunohistochemical staining of the above various tumors with antibodies against menin. Original magnification, ×400. * p<0.05 vs. control group.
Menin expression is reduced in certain primary human lung cancer
We have shown a crucial role of menin in repressing a human lung cancer cell line and MEFs, and we wonder if the menin protein level is altered in patients' primary lung cancers. Thus, we examined 39 adenocarcinoma samples and 9 squamous cell carcinomas, all with adjacent normal tissues. Sections from paraffin-embedded samples were stained with affinity-purified anti-menin antibody for IHC staining. Menin was easily detectable in the nucleus of the normal alveolar epithelial cells (Fig. 5A Right and C). In contrast, in certain tumors, staining for menin was much weaker or undetectable, as compared to that in the adjacent normal epithelial cells (Fig. 5A, Left). In the same section from a lung cancer sample, in normal bronchiolo epithelial cells abundant menin was detected in the nucleus (Fig. 5C), but was barely visible in the nucleus of the cancer cells (Fig. 5B). It is also obvious that the nucleus of the cancer cells tended to be larger than the normal epithelial cells.
Figure 5.
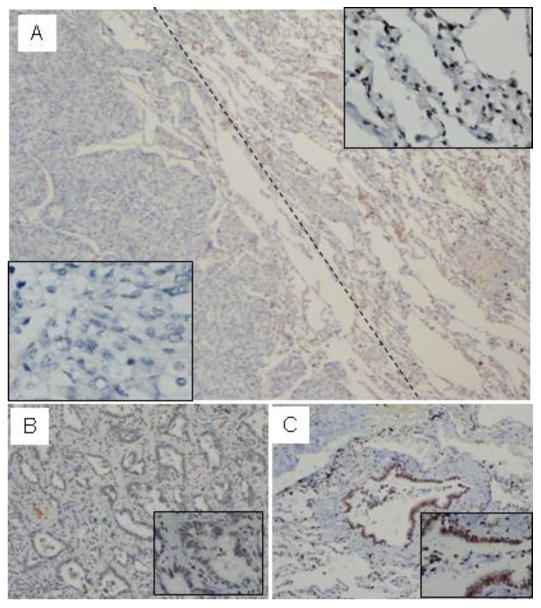
Menin expression is reduced in certain primary human lung cancer cells. Sections from paraffin-embedded lung cancer samples were stained with affinity-purified anti-menin antibody for immunohistochemistry staining. (A) Menin was easily detectable in the nucleus of the normal alveolar (Right) and in certain tumors (Left), where staining for menin was not detectable or markedly reduced (40×). (B) The menin staining was markedly reduced in lung adenocarcinoma, but in normal bronchiolo epithelial cells, the staining for menin was easily detectable, (C, 20×).
Menin expression in tumors, as compared to that in the adjacent normal epithelial cells, was markedly reduced or not detectable in 9 out of 39 adenocarcinomas, accounting for 23 % of the tumors we examined (Suppl Table 2). On the other hand, in 9 squamous cell cancers, we failed to detect an obvious change of menin expression between the tumor cells and the adjacent normal lung tissues. Among 10 cases with loss of menin expression in lung cancers, 4 cases showed lymph node metastasis (40%) (Suppl. Table 2). In contrast, only 5 cases had lymph node metastasis out of 29 cases that still express menin in the tumors (17%), correlated with a reduction of lymph node metastasis in menin-expressing adenocarcinoma. While the number of cases that were analyzed is moderate, our findings suggest that menin expression was markedly reduced in 23% of lung adenocarcinomas, which was correlated with increased lymph node metastasis.
Menin binds to the PTN locus and affects the H3K27 trimethylation(H3K27m3)
To determine how menin regulates PTN expression, we performed ChIP assays with Men1-/- and Men1+/+ MEFs, using an anti-menin antibody. We designed two primers used for ChIP assays at PTN promoter loci (Fig.6A). ChIP assays showed that menin bound to the PTN locus (PP1) in the Men1+/+ cells but not in Men1-/- cells (Fig. 6B, lanes 3 and 6). These results provide the first direct evidence that menin regulates PTN expression by binding to the PTN locus. To elucidate how menin represses PTN expression, we turned our attention to the impact of menin on histone modification of PTN promoter locus. We first detected whether menin affects histone H3 lysine 4 trimethylation (H3K4m3) or acetyl-histone H3, which are correlated with positive gene transcription induced by menin and MLL in leukemia cells (Chen et al., 2006; Yan et al., 2006) at the PTN locus in Men1 knockout MEF cells, but we failed to observe an impact of Men1 excision on the level of H3K4m3 (Fig. 6C) or acetyl-histone H3 (Suppl Fig. 6A) at the PTN locus. We also failed to detect histone deacetylase 1 (HDAC1) at the PTN locus (Suppl Fig. 6B). On the other hand, histone H3 lysine 27 trimethylation (H3K27m3), which is catalyzed by EZH2 from transcription-repressing Polycomb group (PcG) genes, can be recognized by some PcG proteins to compress chromatin structure, leading to repression of gene transcription (Cao et al., 2002). We thus further determined if menin affects H3K27 trimethylation (H3K4m3). Notably, H3K27m3 was reduced in Men1 null cells at the PTN locus, but not at the GAPDH locus (Fig. 6C, and D lanes 3 and 6). Together, these findings suggest that menin represses PTN transcription at least in part through H3K27m3 at the PTN locus.
Figure 6.
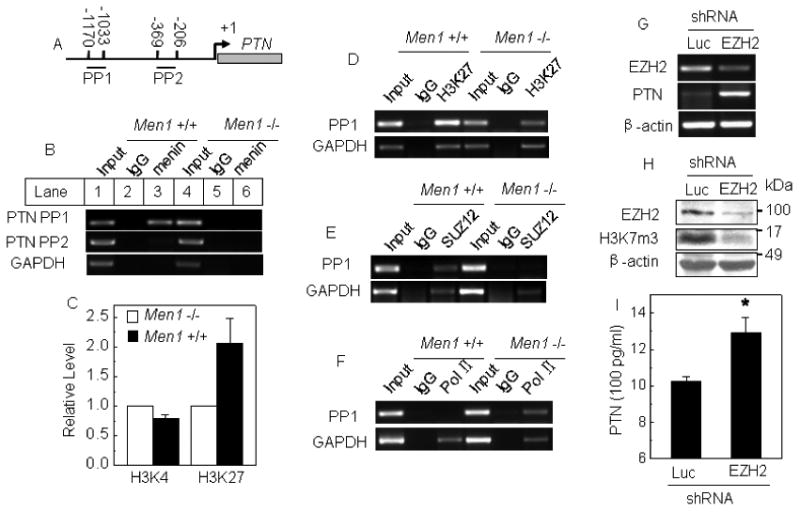
Menin binds the PTN promoter and increases H3K27 trimethylation. (A) A schematic representation of the PTN gene loci and amplicons used for ChIP assays. (B-F) ChIP assays using one of the antibodies against menin, H3K4m3, H3K27m3, SUZ12, RNA pol II, and control IgG in Men1-/- and Men1+/+ MEF cells. PCR was carried out using primers for each amplicon. (G) A549 cells were stably transfected with vector expressing a shRNA against either Luc or EZH2. EZH2 knockdown and PTN expression were determined by RT-PCR. (H-I) EZH2 knockdown and total H3K27m3 level were detected by Western blot, and the effect of EZH2 knockdown on secreted PTN protein was determined by ELISA.
To assess the scope of PcG proteins involved in menin-dependent repression of PTN, we examined the impact of Men1 excision on another PcG protein that associates with the H3K27-methylating complex, SUZ12, using ChIP assay. We found that SUZ12 bound the PTN locus in MEFs (Fig. 6E, Lane 3). Notably, loss of menin abrogated SUZ12 binding to the PTN locus (Fig. 6E, Lane 6), while the SUZ12 and EZH2 protein level in cells was not affected by menin excision (Supp Fig. 6C). These findings suggest that part of menin's role is to recruit the PcG complex including EZH2 and SUZ12 to the PTN locus to methylate H3K27 and then silence PTN expression. In addition, loss of menin enhanced detection of RNA pol II at the PTN locus (Fig. 6F, lane 6). EZH2 knockdown reduced the EZH2 mRNA and protein levels (Fig. 6G-H) but increased the PTN mRNA level (Fig. 6G) and secreted PTN for ∼30% (Fig. 6I). As EZH2-mediated H3K27 represses gene transcription, these findings demonstrate that menin cross-talks with EZH2 and SUZ12 to enhance H3K27m3 at the PTN locus to actively repress PTN transcription. Together, our findings are consistent with a model that menin represses PTN transcription in part by functionally interacting with the PcG proteins in H3K27m3 as shown in (Fig. 6H). These findings have unraveled a previously unrecognized connection between menin and Polycomb group proteins in H3K27 methylation mediated repression of gene transcription and cell proliferation.
Discussion
Although much has been learnt as to how active mutations in proto-oncogenes such as K-Ras and EGFR facilitate the development of lung adenocarcinoma (Herbst et al., 2008; Jemal et al., 2002; Soda et al., 2007), little is known about how mutations in tumor suppressor menin affect development of lung cancer (Pei et al., 2007). Menin suppresses endocrine tumors partly through enhancing MLL-mediated H3K4 methylation and upregulation of p18 and p27 transcription (Karnik et al., 2005), but it is poorly understood how menin suppresses other types of tumors. Our findings demonstrate that menin potently represses proliferation of the lung cancer cells and growth of the cancer cell-derived tumors at least partly through epigenetically repressing PTN transcription via PcG gene-mediated H3K27 methylation. This mechanism is quite distinct from menin suppression of endocrine tumors through upregulating a positive histone mark, H3K4 methylation (Karnik et al., 2005). PTN, a heparin-binding growth factor that binds its cell surface receptors PTPRZ1 and ALK, is a major effector of menin in repressing human lung adenocarcinoma cells. Our findings have linked the tumor suppressing function of menin to the PTN-ALK pathway that is known to be active in certain lung adenocarcinomas (Jager et al., 2002). This link is mediated partly through regulating EZH2-mediated H3K27 and gene silencing.
The relationship between menin and repression of PTN is disrupted in certain lung adenocarcinoma cells, as menin expression is abrogated or substantially diminished in certain primary lung cancer cells. This conclusion is supported by several lines of evidence. First, menin potently inhibited proliferation of several lung cancer cell lines and PTN expression. Second, ectopic PTN expression increased cell proliferation, while PTN knockdown reduced proliferation of the cancer cells as well as growth of lung cancer xenograft. Third, ectopic expression of PTN abrogated the menin-mediated inhibition of the cancer cells. Fourth, menin expression was markedly reduced in 23% of primary human lung adenocarcinomas. PTN possesses a pleiotrophic role in regulating neurite growth, mitogenesis, cell migration, and angiogenesis (Choudhuri et al., 1997). PTN is highly expressed in a number of cancers including lung cancer, and PTN concentrations in serum were over 10-fold higher in lung cancer patients as compared to the control group (Jager et al., 2002). Moreover, Men1 mutation coupled with mutation in the p18 gene leads to the development of NSCLC in certain mice (Pei et al., 2007). Thus, our findings are consistent with the notion that menin-mediated repression of PTN contributes to repression of growth of lung cancer cells and in particular, lung adenocarcinoma cells.
Menin appears to preferentially suppress development of a subtype of NSCLC, lung adenocarcinoma, because reduction of menin expression was detected in more primary lung adenocarinomas but not in squamous cell cancers (Suppl Table 2). As the EGFR and K-Ras pathway is often excessively upregulated in adenocarcinomas but not in squamous cell cancers (Herbst et al., 2008), it is possible that one of menin's functions is to repress the Ras pathway, which could in part be up-regulated by PTN or EGFR pathways. From the cultured A549 cells and human lung adenocarcinoma cell-derived xenograft, we have observed tight menin-mediated repression of PTN and the crucial role of PTN in proliferation of cancer cells. These results strongly suggest that the menin and PTN pathway plays a crucial role in suppressing lung adenocarcinoma. Further work still remains to elucidate how menin preferentially suppresses lung adenocarcinoma.
Our findings have uncovered the menin, PTN, and ALK pathway in controlling proliferation of lung cancer cells, and this pathway may serve as a target for therapy against lung adenocarcinoma. Though still lacking of a well-established and unified model to account for PTN signaling and tumor promotion, an attractive model suggests that PTN enhances cell proliferation and cell migration by binding to its cell surface receptor PTPRZ1 and inhibits its phosphatase activity toward its coreceptor, ALK (Perez-Pinera et al., 2007b). As a result, the phosphorylation of ALK and hence the ALK activity is increased (Perez-Pinera et al., 2007b). Interestingly, ALK has been reported to fuse with other partner proteins in ∼7% of human lung adenocarcinoma cases, and the mutated ALK has enhanced kinase and transforming ability (Soda et al., 2007). Therefore, it is likely that menin normally helps maintain the repressed status of the PTN and ALK transcription, but downregulation of the MEN1 gene in lung cancer cells leads to enhanced expression of PTN and ALK. These findings suggest a previously unrecognized pathway in controlling proliferation of lung cancer, i.e, the menin, PTN, and ALK pathway, therefore implying that targeting the PTN-ALK pathway may prove effective in treating lung adenocarcinomas that express a low level of menin. In addition, our findings may also help subdivide the lung adenocarcinoma with menin-positive and menin-negative cancers for diagnosis, and this diagnosis may help in choosing the therapy by effectively treating the menin-negative cancers with the ALK kinase inhibitors.
In summary, we have unraveled a novel mechanism whereby menin represses gene transcription by regulating the PcG gene-mediated repression of gene transcription. Unlike the classic role of menin in enhancing MLL-mediated H3K4 methylation and transcription of p18 and p27 (Karnik et al., 2005; Milne et al., 2005), menin potently inhibits transcription of PTN by recruiting PcG proteins such as SUZ12 and enhancing trimethylation of H3K27. These findings have expanded the role of menin-mediated suppression of endocrine tumors to suppression of human lung cancer. These results not only highlight menin as a potential diagnostic marker to classify a subtype of human lung adenocarcinoma with negative menin expression, but also unravel the PTN-ALK pathway as a potential target for treating the menin-negative lung adenocarcinoma with the ALK inhibitor.
Supplementary Material
Acknowledgments
This work is in partly supported by NFSC grants (No. 30701003, GH.Jin), the Natural Science Foundation of Fujian Province of China (No. C0710044, GH.Jin), National Cancer Institute Short-Term Scientist Exchange Program (GH.Jin) and National Cancer Institute grants (R01CA100912 and R01CA113962, XH). We appreciate the valuable comments from other members of our laboratories.
References
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–7. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, et al. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci U S A. 2006;103:1018–23. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri R, Zhang HT, Donnini S, Ziche M, Bicknell R. An angiogenic role for the neurokines midkine and pleiotrophin in tumorigenesis. Cancer Res. 1997;57:1814–9. [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager R, List B, Knabbe C, Souttou B, Raulais D, Zeiler T, et al. Serum levels of the angiogenic factor pleiotrophin in relation to disease stage in lung cancer patients. Br J Cancer. 2002;86:858–63. doi: 10.1038/sj.bjc.6600202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin. 2002;52:23–47. doi: 10.3322/canjclin.52.1.23. [DOI] [PubMed] [Google Scholar]
- Jin S, Mao H, Schnepp RW, Sykes SM, Silva AC, D'Andrea AD, et al. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003;63:4204–10. [PubMed] [Google Scholar]
- Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–64. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La P, Schnepp RW, C DP, A CS, Hua X. Tumor suppressor menin regulates expression of insulin-like growth factor binding protein 2. Endocrinology. 2004;145:3443–50. doi: 10.1210/en.2004-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29:22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- Lu KV, Jong KA, Kim GY, Singh J, Dia EQ, Yoshimoto K, et al. Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J Biol Chem. 2005;280:26953–64. doi: 10.1074/jbc.M502614200. [DOI] [PubMed] [Google Scholar]
- Martin C, Zhang Y. Mechanisms of epigenetic inheritance. Curr Opin Cell Biol. 2007;19:266–72. doi: 10.1016/j.ceb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, et al. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A. 2000;97:2603–8. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–17. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–54. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei XH, Bai F, Smith MD, Xiong Y. p18Ink4c collaborates with Men1 to constrain lung stem cell expansion and suppress non-small-cell lung cancers. Cancer Res. 2007;67:3162–70. doi: 10.1158/0008-5472.CAN-06-4517. [DOI] [PubMed] [Google Scholar]
- Perez-Pinera P, Chang Y, Deuel TF. Pleiotrophin, a multifunctional tumor promoter through induction of tumor angiogenesis, remodeling of the tumor microenvironment, and activation of stromal fibroblasts. Cell Cycle. 2007a;6:2877–83. doi: 10.4161/cc.6.23.5090. [DOI] [PubMed] [Google Scholar]
- Perez-Pinera P, Zhang W, Chang Y, Vega JA, Deuel TF. Anaplastic lymphoma kinase is activated through the pleiotrophin/receptor protein-tyrosine phosphatase beta/zeta signaling pathway: an alternative mechanism of receptor tyrosine kinase activation. J Biol Chem. 2007b;282:28683–90. doi: 10.1074/jbc.M704505200. [DOI] [PubMed] [Google Scholar]
- Powers C, Aigner A, Stoica GE, McDonnell K, Wellstein A. Pleiotrophin signaling through anaplastic lymphoma kinase is rate-limiting for glioblastoma growth. J Biol Chem. 2002;277:14153–8. doi: 10.1074/jbc.M112354200. [DOI] [PubMed] [Google Scholar]
- Ringrose L, Paro R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development. 2007;134:223–32. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- Schnepp RW, Chen YX, Wang H, Cash T, Silva A, Diehl JA, et al. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–15. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnepp RW, Mao H, Sykes SM, Zong WX, Silva A, La P, et al. Menin induces apoptosis in murine embryonic fibroblasts. J Biol Chem. 2004;279:10685–91. doi: 10.1074/jbc.M308073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–45. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–56. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- Stoica GE, Kuo A, Aigner A, Sunitha I, Souttou B, Malerczyk C, et al. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J Biol Chem. 2001;276:16772–9. doi: 10.1074/jbc.M010660200. [DOI] [PubMed] [Google Scholar]
- Yan J, Chen YX, Desmond A, Silva A, Yang Y, Wang H, et al. Cdx4 and menin co-regulate Hoxa9 expression in hematopoietic cells. PLoS ONE. 2006;1:e47. doi: 10.1371/journal.pone.0000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–49. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci U S A. 1998;95:10632–6. doi: 10.1073/pnas.95.18.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.