


Abstract
The identification and study of evolutionarily conserved genomic sequences that surround disease-related genes is a valuable tool to gain insight into the functional role of these genes and to better elucidate the pathogenetic mechanisms of disease. We created the DG-CST (Disease Gene Conserved Sequence Tags) database for the identification and detailed annotation of human–mouse conserved genomic sequences that are localized within or in the vicinity of human disease-related genes. CSTs are defined as sequences that show at least 70% identity between human and mouse over a length of at least 100 bp. The database contains CST data relative to over 1088 genes responsible for monogenetic human genetic diseases or involved in the susceptibility to multifactorial/polygenic diseases. DG-CST is accessible via the internet at http://dgcst.ceinge.unina.it/ and may be searched using both simple and complex queries. A graphic browser allows direct visualization of the CSTs and related annotations within the context of the relative gene and its transcripts.
INTRODUCTION
Alignment of DNA sequences from different species provides an effective tool to decode genomic information, based on the assumption that functional sequences tend to evolve at a slower rate than non-functional sequences. The availability of the complete genomic sequences from a variety of species (1–4) allows to carry out these analyses very effectively and to identify, besides coding sequences, also non-coding sequences with either regulatory or structural functions (5–8).
A comparative analysis of the human and murine genomes revealed the presence of a surprisingly high number of sequence elements longer than 100 bp and displaying a sequence identity >70% between human and mouse (6). Interestingly, more than half of these conserved sequences do not represent known elements belonging to protein-coding genes and may therefore represent non-coding RNAs, expression control elements or chromosomal structural elements. Such sequences have been previously termed CNG (conserved non-genic sequences) (9,10) or CNS (conserved non-coding sequences) (2). Here, we use the more neutral and descriptive expression ‘conserved sequence tags’ (CST), which is appropriate also to describe exons.
To gain further insight into the biological role of these conserved sequences, we chose to identify and annotate CSTs belonging to a set of human genes involved in the pathogenesis of genetic diseases. These are among the best-studied human genes as they have been the objects of very detailed structural and functional characterization in the past 15–20 years. Furthermore, novel functional elements within these genes may be targets of yet unidentified mutations leading to genetic diseases. Information on CSTs related to human disease genes can also be gathered from reference genome sequence databases, i.e. Ensembl (11) and Genome Browsers (12), or from more specialized resources, i.e. Vista Browser (13) and GALA (14). However, these valuable resources are not specifically designed for the study of human disease genes and retrieval of CST data for these genes may turn out to be difficult and statistical analysis impossible since CSTs are not explicitly annotated. Therefore, we decided to build DG-CST (Disease Gene CST), a database of human–mouse conserved elements associated to disease genes. To this purpose, systematic identification of CSTs in human disease genes was carried out, followed by detailed bioinformatic analysis, aimed at identifying novel functional elements associated with these genes, either transcribed and possibly coding sequences or non-transcribed sequence elements with a hypothetical role in the control of gene expression. The DG-CST database is available to the scientific community through a Web interface at the address http://dgcst.ceinge.unina.it/. The annotation of CSTs related to disease genes will be valuable for the elucidation of the functional role of these conserved sequences and for a better understanding of the pathogenesis of human genetic disorders.
CONSTRUCTION AND ORGANIZATION OF THE DG-CST
Sequence acquisition and CST identification
A list of human genes involved in either the pathogenesis of monogenic human disorders or in the predisposition to multifactorial diseases was obtained by screening the Genecards (15) and the On-Line Mendelian Inheritance in Man (OMIM) (16) databases. We then searched the human Ensembl database (assembly release NCBI34) to retrieve the human genomic sequences spanning the selected transcripts as well as 250 additional kilobases of flanking sequence on both sides. The extent of the flanking sequence was reduced when known genes were annotated in proximity of the disease gene, but a minimum of 20 kb was taken in all cases. The Ensembl database was also used as the source of the corresponding murine sequences. Orthologous gene annotation was used, when available, to find the mouse counterparts; when more than one orthologous gene was found, sequences were manually selected, on the basis of overall sequence conservation and relationships with other neighboring sequences. Mouse sequence size was defined according to the length of the human sequence.
A total set of 1088 human genomic sequences was compared to the corresponding murine orthologous genomic sequences (the full list is available online). Overall, 193 million bp of human genomic sequences were analyzed, corresponding to 7% of the human genome. Human and mouse genomic sequences, prefiltered to mask all known repeated sequences, were compared using the BLASTZ program (17). Sequences showing at least 70% identity, over a region of at least 100 bp, were selected and further analyzed to eliminate redundancies, leading to the identification of 66 495 repeat-free, non-overlapping, human and mouse CST pairs. The CSTs were found to correspond or to overlap to known human exon sequences in about 32% of cases (n = 21 139) while they were located either in intronic or in intergenic region in the remaining 68% of cases (n = 45 356) (Table 1).
Table 1. Classification of human CSTs present in DG-CST.
| CST type | Number | % | Length (bp) | Length (%) |
|---|---|---|---|---|
| Exonic | 21 139 | 31.8 | 5 247 362 | 34.9 |
| Intronic | 18 390 | 27.7 | 3 832 169 | 25.5 |
| Intergenic | 26 966 | 40.5 | 5 962 769 | 39.6 |
| Total | 66 495 | 100 | 15 042 300 | 100 |
CST annotation
The identified CSTs are collected in the DG-CST database, together with a large number of annotations including:
species;
genomic location, i.e. chromosome, position, relationship with the closest gene and with the selected disease gene (often coincident);
sequence content, i.e. sequence, length, GC percentage;
identity between human and mouse sequences, number of gaps, polarity;
BLAST matches with other CSTs, as well as with other human genomic sequences;
BLAST matches versus non-redundant nucleotide databases;
conservation in other species, as assessed by BLAST analysis versus the drafts of fugu (3), chicken (11), rat (4) and zebrafish (11) genome sequences;
classification of CSTs in ‘intronic’, ‘intergenic’, ‘exonic’ based on Ensembl gene annotations;
potential of CSTs of representing transcribed/coding elements based on a number of different tests, including determination of maximum ORF size, presence of putative splice sites, exonic splicing enhancers (18), exon predictions based on GENSCAN (19), BLAST matches with expressed sequence tags (ESTs) and non-redundant protein databases, word frequencies, determination of the coding potential score (c.p.s.) according to the CSTMiner algorithm (20,21), a recently developed software based on pairwise genome comparison;
presence of single nucleotide polymorphisms (SNPs), as reported in Ensembl;
presence of palindromes, tandem repeats, putative RNA secondary structures as predicted by using the ddbRNA software (22);
presence of putative transcription factor (TF) binding sites, as assessed using BID, a newly developed algorithm (A. Ambesi, M. Bansal and D. di Bernardo, unpublished data).
DATABASE SEARCH
The DG-CST database contains all the annotations and is designed to allow easy retrieval of CST information. Searching is supported in a number of different ways. A graphic browser allows direct visualization of the CSTs, within the context of the relative gene and its transcripts. Briefly, CST information can be accessed in the following ways:
By choosing from a list of all analyzed disease genes available in the home page (Figure 1A and B).
By selecting one or more genes either as a quick search option from the home page (Figure 1A, black box) or following the ‘gene’ link. Gene selection may be carried out by gene symbol, disease name and several other criteria, also in combination (Figure 1E).
By querying the database for CSTs selected according to a large number of annotated features, alone or in combination, in the ‘Advanced’ section (Figure 1D). To facilitate the search, reduced feature sets are available where CSTs can be searched by (a) DNA-based features such as presence of tandem repeats, palindromes, SNPs (Figure 1C); (b) RNA-based features such as presence of putative secondary structures, matches with ESTs, GENSCAN predicted exons; (c) protein-coding features, such as exon annotation, coding potential, BLAST matches with proteins; (d) CSTs localized to selected chromosomal regions.
Finally, CSTs can be searched by BLAST sequence analysis from the home page (Figure 1A, red box).
Each CST entry present in DG-CST is assigned a unique identifier (CST ID) that can also be used to quickly find the CST from different sections of the database, including the home page.
Figure 1.
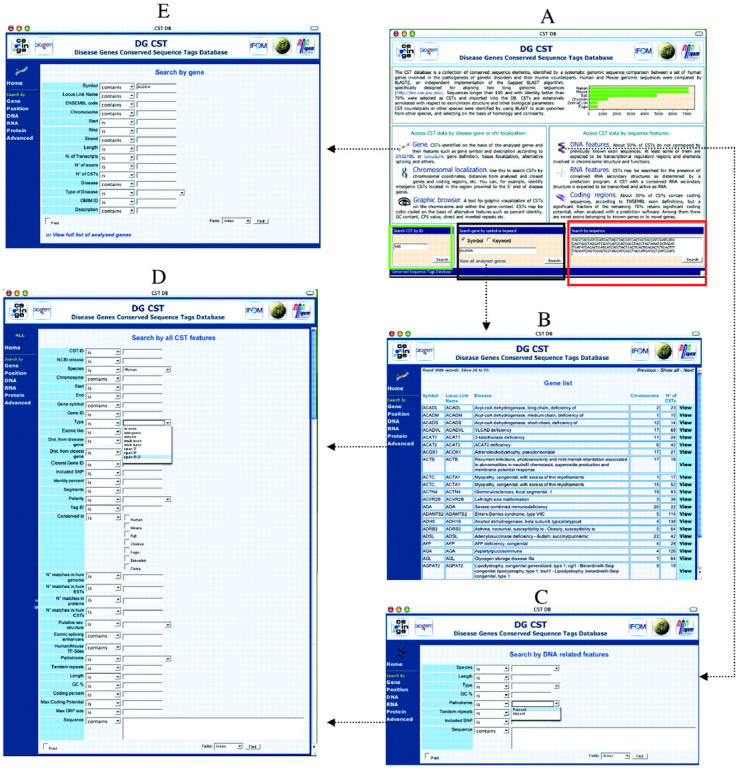
The DG-CST database: examples of query interfaces. (A) The DG-CST home page. The quick search boxes are highlighted in color: the CST ID box in green; the gene box in black; and the BLAST box in red. (B) The list of all analyzed genes obtained following the link on the home page. (C) The DNA-based feature search page. (D) The advanced CST search page, where all annotated features may be used in combination or alone to query the database. (E) The gene-based CST search page, which allows a more detailed gene search.
DATA DISPLAY
When searching DG-CST using the previously described ‘DNA-based’, ‘RNA-based’, ‘protein-based’, ‘advanced’ and ‘localization’ features, a list of CST entries that meet the search criteria can be accessed. Individual CSTs may be visualized in a specific page where all annotations available on that particular CST are displayed (Figure 2C). Matching CSTs from other species may be seen and compared in a multi-sequence alignment (Figure 2D). Matches found for each CST in a number of BLAST searches, pre-run against collections of genomic, EST or protein databases, may also be displayed starting from the CST page.
Figure 2.

The DG-CST database: data display. (A) Example of a gene entry (A2M) and the related CST list. (B) Graphical representation of the selected gene, accessible via the map link in (A). On mouse over, details of CST #250083 are displayed as an example. In this representation, CSTs are color-coded based on the number of matches with human ESTs. (C) Example of a CST entry with all annotations and the list of the corresponding CSTs conserved in other species. CST details are accessible either from the CST list of the gene page (A) or by clicking on the interactive graphical browser in (B). (D) Graphical representation of the sequence alignment of the orthologous CSTs shown in (C).
On the other hand, when searching by gene/disease name and/or symbol, it is possible to obtain a list of gene entries that meet the search criteria. Each gene entry, in addition to links to external resources such as LocusLink, ENSEMBL and OMIM, provides a ‘CST list’ link that gives access to the list of all CSTs found by analyzing the selected disease gene region, as shown in Figure 2A. By clicking on each entry, it is possible to access all the data pertaining to a given human CST, as described above.
Graphical representation is accessible through a ‘map’ link, where CSTs and related annotations are shown within the context of the relative gene and its transcripts (Figure 2B). Moving through the genomic region and zooming to various levels of detail are supported. CSTs may be labeled by a color code on the basis of several quantitative parameters such as degree of human–mouse sequence identity, GC content, number of gaps, putative RNA secondary structures, palindromes and tandem repeats. To avoid an exceedingly crowded map, the graphic visualization tool allows the user to display selected CST subsets, such as:
intergenic, intronic or exonic CSTs;
CSTs containing putative TF binding sites;
CSTs with matches to ESTs;
CSTs conserved in additional species, besides human and mouse, such as chicken, fugu, zebrafish. These CSTs have a higher probability of representing functional elements playing a basic role in vertebrates as suggested by recent reports (23).
CONCLUSIONS
DG-CST is an annotated collection of conserved sequences related to genes involved in genetic diseases and may represent a valuable resource for investigators interested in studying the molecular mechanisms that underlie genetic diseases. The database will be updated on a regular basis to include information on newly identified human disease genes as well as on new genomic data (e.g. sequences from additional organisms). DG-CST may help in deciphering the spectrum of pathogenetic mutations that determine genetic diseases. Mutations are usually searched for in the coding regions of a gene, but may easily occur in other areas. CSTs provide a vast library of putative novel functional sites, such as non-previously described exons and/or elements possibly playing a role in regulating the level of gene expressions, which may be functionally tested as well as screened for mutations in patients, particularly in diseases where the analysis of the known functional elements of the disease gene failed so far in identifying a relevant number of causative mutations (24–27). There are a number of evidences that point to the direct involvement of regulatory control elements in the pathogenesis of human disorders, both due to chromosomal rearrangements (28,29) and to point mutations (30–32). However, the recognition of pathogenic mutations leading to genetic disorders in regulatory elements has been so far hampered by our limited knowledge of the structure and function of the elements associated to disease genes. The availability of the DG-CST database should be a valuable resource in order to fill this gap of information and to facilitate the efforts aimed at both elucidating the function of disease genes and at better understanding the pathogenetic mechanisms of genetic diseases.
Acknowledgments
ACKNOWLEDGEMENTS
We thank A. Ambesi, G. Borsani, A. Ciccodicola, G. Diez-Roux, R. Di Lauro, B. Franco, P. Gasparini, C. Missero and M. Zollo for helpful discussion. This work was supported by the Telethon Foundation, the Associazione Italiana per la Ricerca sul Cancro (AIRC), the Federazione Italiana per la Ricerca sul Cancro (FIRC), the Italian Ministry for Research (MURST), the National Council for Research (CNR) and Regione Campania. A. Boccia was also supported by BioGeM for this project.
REFERENCES
- 1.Lander E.S., Linton,L.M., Birren,B., Nusbaum,C., Zody,M.C., Baldwin,J., Devon,K., Dewar,K., Doyle,M., FitzHugh,W. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 2.Waterston R.H., Lindblad-Toh,K., Birney,E., Rogers,J., Abril,J.F., Agarwal,P., Agarwala,R., Ainscough,R., Alexandersson,M., An,P. et al. (2002) Initial sequencing and comparative analysis of the mouse genome. Nature, 420, 520–562. [DOI] [PubMed] [Google Scholar]
- 3.Aparicio S., Chapman,J., Stupka,E., Putnam,N., Chia,J.M., Dehal,P., Christoffels,A., Rash,S., Hoon,S., Smit,A. et al. (2002) Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science, 297, 1301–1310. [DOI] [PubMed] [Google Scholar]
- 4.Gibbs R.A., Weinstock,G.M., Metzker,M.L., Muzny,D.M., Sodergren,E.J., Scherer,S., Scott,G., Steffen,D., Worley,K.C., Burch,P.E. et al. (2004) Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature, 428, 493–521. [DOI] [PubMed] [Google Scholar]
- 5.Couronne O., Poliakov,A., Bray,N., Ishkhanov,T., Ryaboy,D., Rubin,E., Pachter,L. and Dubchak,I. (2003) Strategies and tools for whole-genome alignments. Genome Res., 13, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pennacchio L.A. and Rubin,E.M. (2001) Genomic strategies to identify mammalian regulatory sequences. Nature Rev. Genet., 2, 100–109. [DOI] [PubMed] [Google Scholar]
- 7.Thomas J.W., Touchman,J.W., Blakesley,R.W., Bouffard,G.G., Beckstrom-Sternberg,S.M., Margulies,E.H., Blanchette,M., Siepel,A.C., Thomas,P.J., McDowell,J.C. et al. (2003) Comparative analyses of multi-species sequences from targeted genomic regions. Nature, 424, 788–793. [DOI] [PubMed] [Google Scholar]
- 8.Boffelli D., Nobrega,M.A. and Rubin,E.M. (2004) Comparative genomics at the vertebrate extremes. Nature Rev. Genet., 5, 456–465. [DOI] [PubMed] [Google Scholar]
- 9.Dermitzakis E.T., Reymond,A., Lyle,R., Scamuffa,N., Ucla,C., Deutsch,S., Stevenson,B.J., Flegel,V., Bucher,P., Jongeneel,C.V. et al. (2002) Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature, 420, 578–582. [DOI] [PubMed] [Google Scholar]
- 10.Dermitzakis E.T., Reymond,A., Scamuffa,N., Ucla,C., Kirkness,E., Rossier,C. and Antonarakis,S.E. (2003) Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs). Science, 302, 1033–1035. [DOI] [PubMed] [Google Scholar]
- 11.Birney E., Andrews,D., Bevan,P., Caccamo,M., Cameron,G., Chen,Y., Clarke,L., Coates,G., Cox,T., Cuff,J. et al. (2004) Ensembl 2004. Nucleic Acids Res., 32, D468–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kent W.J., Sugnet,C.W., Furey,T.S., Roskin,K.M., Pringle,T.H., Zahler,A.M. and Haussler,D. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brudno M., Poliakov,A., Salamov,A., Cooper,G.M., Sidow,A., Rubin,E.M., Solovyev,V., Batzoglou,S. and Dubchak,I. (2004) Automated whole-genome multiple alignment of rat, mouse, and human. Genome Res., 14, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giardine B., Elnitski,L., Riemer,C., Makalowska,I., Schwartz,S., Miller,W. and Hardison,R.C. (2003) GALA, a database for genomic sequence alignments and annotations. Genome Res., 13, 732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rebhan M., Chalifa-Caspi,V., Prilusky,J. and Lancet,D. (1997) GeneCards: integrating information about genes, proteins and diseases. Trends Genet., 13, 163. [DOI] [PubMed] [Google Scholar]
- 16.Hamosh A., Scott,A.F., Amberger,J., Bocchini,C., Valle,D. and McKusick,V.A. (2002) Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res., 30, 52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz S., Kent,W.J., Smit,A., Zhang,Z., Baertsch,R., Hardison,R.C., Haussler,D. and Miller,W. (2003) Human–mouse alignments with BLASTZ. Genome Res., 13, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cartegni L., Wang,J., Zhu,Z., Zhang,M.Q. and Krainer,A.R. (2003) ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res., 31, 3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burge C. and Karlin,S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol., 268, 78–94. [DOI] [PubMed] [Google Scholar]
- 20.Mignone F., Grillo,G., Liuni,S. and Pesole,G. (2003) Computational identification of protein coding potential of conserved sequence tags through cross-species evolutionary analysis. Nucleic Acids Res., 31, 4639–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castrignano T., Canali,A., Grillo,G., Liuni,S., Mignone,F. and Pesole,G. (2004) CSTminer: a web tool for the identification of coding and noncoding conserved sequence tags through cross-species genome comparison. Nucleic Acids Res., 32, W624–W627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Bernardo D., Down,T. and Hubbard,T. (2003) ddbRNA: detection of conserved secondary structures in multiple alignments. Bioinformatics, 19, 1606–1611. [DOI] [PubMed] [Google Scholar]
- 23.Bejerano G., Pheasant,M., Makunin,I., Stephen,S., Kent,W.J., Mattick,J.S. and Haussler,D. (2004) Ultraconserved elements in the human genome. Science, 304, 1321–1325. [DOI] [PubMed] [Google Scholar]
- 24.Weston M.D., Eudy,J.D., Fujita,S., Yao,S., Usami,S., Cremers,C., Greenburg,J., Ramesar,R., Martini,A., Moller,C. et al. (2000) Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am. J. Hum. Genet., 66, 1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schiaffino M.V., Bassi,M.T., Galli,L., Renieri,A., Bruttini,M., De Nigris,F., Bergen,A.A.B., Charles,S.J., Yates,J.R.W., Meindl,A. et al. (1995) Analysis of the OA1 gene reveals mutations in only one-third of patients with X-linked ocular albinism. Hum. Mol. Genet., 4, 2319–2325. [DOI] [PubMed] [Google Scholar]
- 26.Krantz I.D., Colliton,R.P., Genin,A., Rand,E.B., Li,L., Piccoli,D.A. and Spinner,N.B. (1998) Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am. J. Hum. Genet., 62, 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowe P.S., Oudet,C.L., Francis,F., Sinding,C., Pannetier,S., Econs,M.J., Strom,T.M., Meitinger,T., Garabedian,M., David,A. et al. (1997) Distribution of mutations in the PEX gene in families with X-linked hypophosphataemic rickets (HYP). Hum. Mol. Genet., 6, 539–549. [DOI] [PubMed] [Google Scholar]
- 28.Lettice L.A., Horikoshi,T., Heaney,S.J., van Baren,M.J., van der Linde,H.C., Breedveld,G.J., Joosse,M., Akarsu,N., Oostra,B.A., Endo,N. et al. (2002) Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc. Natl Acad. Sci. USA, 99, 7548–7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleinjan D.J. and van Heyningen,V. (1998) Position effect in human genetic disease. Hum. Mol. Genet., 7, 1611–1618. [DOI] [PubMed] [Google Scholar]
- 30.Plenge R.M., Hendrich,B.D., Schwartz,C., Arena,J.F., Naumova,A., Sapienza,C., Winter,R.M. and Willard,H.F. (1997) A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nature Genet., 17, 353–356. [DOI] [PubMed] [Google Scholar]
- 31.Berry M., Grosveld,F. and Dillon,N. (1992) A single point mutation is the cause of the Greek form of hereditary persistence of fetal haemoglobin. Nature, 358, 499–502. [DOI] [PubMed] [Google Scholar]
- 32.Lettice L.A., Heaney,S.J., Purdie,L.A., Li,L., De Beer,P., Oostra,B.A., Goode,D., Elgar,G., Hill,R.E. and De Graaff,E. (2003) A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet., 12, 1725–1735. [DOI] [PubMed] [Google Scholar]