

As a leading cause for morbidity and mortality in young adults, traumatic brain injury (TBI), along with the poorly understood TBI-related seizures inducing their predispositions, pose a major health and socioeconomic problem in the world (Huang, 2013). The post-traumatic seizures caused by TBI may occur either early (within 1 week of the injury) or late (after 1 week of the injury). The proper control of early post-traumatic seizures are of paramount importance because the seizure attacks within the acute stage may add secondary injury to the already damaged brain (Liesemer et al., 2011). However, little is known for the etiological mechanism for TBI-induced early post-traumatic brain injury seizures. In addition, despite the distinct differences between early and late post-traumatic brain injury seizures, the treatments for early post-traumatic brain injury seizures are still limited to traditional anticonvulsant and anti-epileptic drugs. Therefore, there is an urgent need for understanding the mechanisms of the early post-traumatic brain injury seizures. Based on previous findings that up-regulation of Na+-K+-Cl– cotransporter 1 (NKCC1) in neurons is involved in neonatal seizures and in ammonia toxicity-induced seizures (Noebels et al., 2012), we examined the role of NKCC1 in early post-TBI seizures. Our study (Wang et al., 2016) showed that TBI induces up-regulation of NKCC1 and increases intracellular Cl– concentration. Genetic depletion or pharmacological inhibition of NKCC1 suppresses TBI-induced seizures. Moreover, a novel transforming growth factor beta (TGF-β)-NKCC1 mechanistic axis was discovered by revealing that TGF-β expression was increased after TBI andcompetitive antagonism of TGF-β reduced NKKC1 expression, ameliorated reactive astrocytosis, and inhibited seizures. Our findings identified the NKCC1 and TGF-β as important functional mediators for TBI induced early post-traumatic seizures, and suggest the therapeutic potential of targeting this pathway.
Each year, over one and a half million people in the United States suffer from TBI (Huang, 2013). Apart from being a leading cause of death and disability, TBI also plays a critical role in early post-traumatic brain injury seizures within one week of assault which could transpire in as many as 53% of all TBI's (Oberheim et al., 2008). Inflammation, hemorrhage, edema, aberrant plasticity, and neurodegeneration all participate in the cerebral injury process. Early post-traumatic seizures are also a major cause for secondary brain injury through increasing cerebral blood flow and metabolic requirements, elevating intracranial pressure which causes cerebral hypoxia and finally ischemia, harmfully elevating brain temperature, and exacerbating indiscriminate neurotransmitter release (Algattas and Huang, 2013; Wang et al., 2016). In the long run, approximately 25% of patients with a history of early post-traumatic seizures will experience another episode in their later lives (Liesemer et al., 2011). Clinically, it is known that seizures are correlated to the patient's age, severe conditions such as depressed skull fracture, intracranial hematoma, and penetrating head injury. However, the mechanisms of the early post-traumatic seizures on the biomolecular level remain elusive, hindering the application of effective prophylactic and therapeutic managements. Thus, understanding the seizure-inducing mechanisms of TBI is of the utmost importance for both preventing and treating these seizures.
Currently, despite the distinct features of early post-traumatic seizures and epileptic seizures, treatment of early post-traumatic seizures is limited to traditional anticonvulsant and anti-epileptic medications designed for epileptic seizures. Many of these medications target on the receptors of gamma-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system. Depending on low concentration of intracellular Cl–, activation of this GABA receptor (GABAR) selectively conducts Cl– into the cell body, leading to hyperpolarization of the neurons. NKCC is a membrane symporter protein that transports one sodium ion, one potassium ion, and two chloride ions across the cell membrane from the blood into the cell body. The isoform NKCC1 encoded by SLC12A2 gene on chromosome 5 is widely distributed throughout the body and found in the central nervous system. K+-2Cl– cotransporter member 5 (KCC2) is a neuron-specific chloride potassium symporter encoded by SLC12A5 gene on chromosome 20 that functions to maintain low concentration of intracellular chloride. Several recent studies concluded that the modulation of intracellular Cl– occurs by opposing activity of NKCC1 and KCC2 in neurons (Fu et al., 2015; Mòdol et al., 2015). Up-regulation of NKCC1 and down-regulation of KCC2 results in increased intracellular Cl–, leading to less GABAergic inhibition and more seizure susceptibility (Figure 1). To further explore and investigate the relationship between early post-traumatic seizures and the expression/function of NKCC1 and KCC2, our laboratory performed a series of analyses using comprehensive approaches, and revealed the first evidence for the putative link between TBI, TGF-β, NKCC1, and physiological alterations in seizures (Figure 2).
Figure 1.
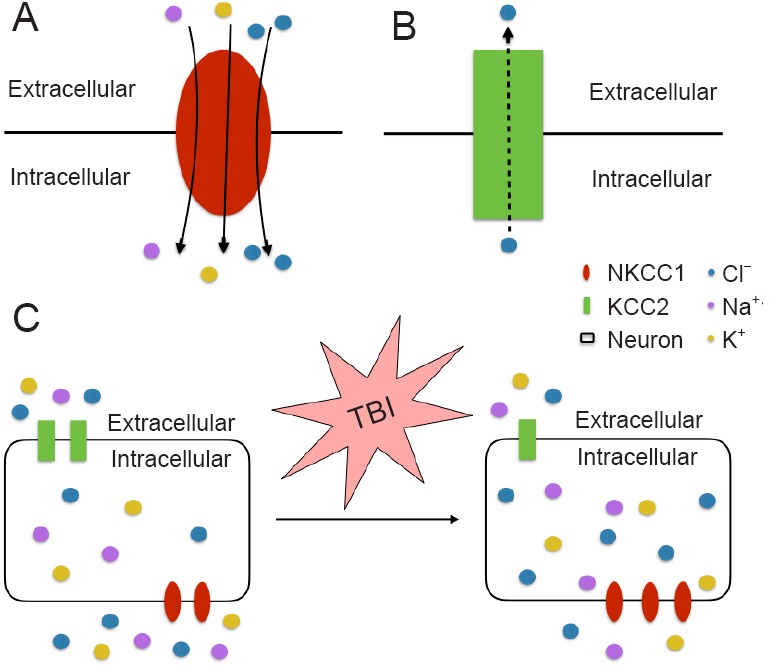
The functions of NKCC1 (A) and KCC2 (B).
In traumatic brain injury, up-regulation of NKCC1 and down-regulation of KCC2 leads to the increased intracellular concentrations of Cl– (C). KCC2: K+-2Cl– cotransporter member 5; NKCC1: Na+-K+-Cl– cotransporter 1; TBI: traumatic brain injury.
Figure 2.
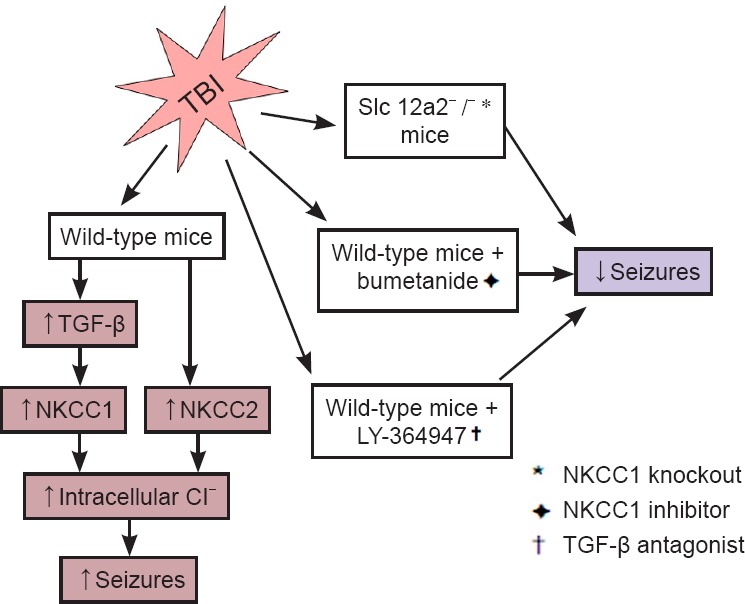
The schematic diagram for experiment design and our discovered mechanism for traumatic brain injury induced seizures.
KCC2: K+-2Cl– cotransporter member 5; NKCC1: Na+-K+-Cl– cotransporter 1; TBI: traumatic brain injury; TGF-β: transforming growth factor-β.
NKCC1 contributes to early post-traumatic seizures: The widely observed elevation of NKCC1 level and disruption of Cl– homeostasis after TBI (Fu et al., 2015) suggests an important role of NKCC1 for the neuronal hyper-excitability in the early post-traumatic seizures. In line with previous reports, using the single, closed-head, unilateral cortical injury TBI model adopted from a report by Petraglia et al. (2014), a significant increase of NKCC1+ neurons, as well as increased NKCC1 and decreased KCC2 expressions in both neocortex and archicortex were observed. Further investigations on NKCC1 knockout mice and pharmacological NKCC1 inhibitor bumetanide showed not only decreased seizure tendency after TBI but also reduced hyper-excitability supported by electrophysiological monitoring. Our study (Wang et al., 2016) clearly identified a role of NKCC1 in the GABA inhibitory pathway, which was suppressed by the elevated intracellular Cl– levels and the depolarized equilibrium potential. However, the increased intracellular Cl– levels may potentially affect the extracellular K+ buffering, which could also consequently increase seizure susceptibility. Similarly, based on this theory, a study performed by Dzhala and Staley (2015) also achieved reduced frequency and power of early post-traumatic epileptiform activities in mice by administering bumetanide. Future investigations to examine how the direct modulation of GABA currents and/or the indirect modulation via changes in Cl– and K+ by the NKCC1 increasing affect post-traumatic seizure susceptibility would be valuable.
TGF-β-NKCC1 axis as a novel potential target for early post- traumatic brain injury seizures: TGF-β is a multifunctional cytokine that plays fundamental roles in intercellular communication and regulates cellular processes, such as cell growth, migration, wound healing, apoptosis, and inflammation (Clark and Coker, 1998). Previous studies have shown that TGF-β interacts with WNK lysine deficient protein kinase 1 (with no lysine (K)) and modulates NKCC1 and KCC2 activities. In addition, TGF-β was found to be up-regulated in the cerebrospinal fluid of patients following TBI (Dohgu et al., 2005). However, the functional and mechanistic role of TGF-β in TBI-induced alterations in NKCC1 remained unexplored. In our study (Wang et al., 2016), we demonstrated the increase of TGF-β in both cortex and hippocampus by TBI. By injecting the TGF-β blocker LY-364947 in mice models, we demonstrated a significant reduction in PTZ-induced seizures after TBI, with both the latency and duration of seizures ameliorated. More importantly, the competitive antagonism of TGF-β significantly suppressed the expression of NKCC1 in the brain, indicating TGF-β as a critical modulator of NKCC1 and its associated regulatory machinery in the early post-traumatic brain injury seizures. This newly identified link between TBI, TGF-β, NKCC1, and early post-traumatic brain injury seizures revealed a potential and important novel mechanism for early post-traumatic brain injury seizures. Future investigations on the molecular details for bridging TGF-β signaling and TGF-β-NKCC1 axis to seizures are warranted.
Conclusions and perspectives: This novel discovery regarding the seizure-promoting role of NKCC1 at the early post-traumatic brain injury period and the implication of TGF-β in this function indicates that NKCC1 and TGF-β may be candidate targets for the development of novel prophylactic and therapeutic management of TBI and its related complications, including early post-traumatic seizures.
References
- Algattas H, Huang JH. Traumatic brain injury pathophysiology and treatments: early, intermediate, and late phases post-injury. Int J Mol Sci. 2013;15:309–341. doi: 10.3390/ijms15010309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DA, Coker R. Transforming growth factor-beta (TGF-beta) Int J Biochem Cell Biol. 1998;30:293–298. doi: 10.1016/s1357-2725(97)00128-3. [DOI] [PubMed] [Google Scholar]
- Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M, Tsuruo T, Sawada Y, Niwa M, Kataoka Y. Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res. 2005;1038:208–215. doi: 10.1016/j.brainres.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Dzhala V, Staley KJ. Acute and chronic efficacy of bumetanide in an in vitro model of posttraumatic epileptogenesis. CNS Neurosci Ther. 2015;21:173–180. doi: 10.1111/cns.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu P, Tang R, Yu Z, Huang S, Xie M, Luo X, Wang W. Bumetanide-induced NKCC1 inhibition attenuates oxygen-glucose deprivation-induced decrease in proliferative activity and cell cycle progression arrest in cultured OPCs via p-38 MAPKs. Brain Res. 2015;1613:110–119. doi: 10.1016/j.brainres.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Huang JH. Traumatic brain injury. Neurol Res. 2013;35:221–222. doi: 10.1179/1743132813Y.0000000178. [DOI] [PubMed] [Google Scholar]
- Liesemer K, Bratton SL, Zebrack CM, Brockmeyer D, Statler KD. Early post-traumatic seizures in moderate to severe pediatric traumatic brain injury: rates, risk factors, and clinical features. J Neurotrauma. 2011;28:755–762. doi: 10.1089/neu.2010.1518. [DOI] [PubMed] [Google Scholar]
- Mòdol L, Santos D, Cobianchi S, González-Pérez F, López-Alvarez V, Navarro X. NKCC1 activation is required for myelinated sensory neurons regeneration through JNK-dependent pathway. J Neurosci. 2015;35:7414–7427. doi: 10.1523/JNEUROSCI.4079-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, Kahle KT, Staley KJ. Neonatal Seizures and Neuronal Transmembrane Ion Transport. In: Noebels JL, editor. SourceJasper's Basic Mechanisms of the Epilepsies [Internet] 4th edition. Bethesda (MD): National Center for Biotechnology Information (US); 2012. [PubMed] [Google Scholar]
- Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M. Loss of astrocytic domain organization in the epileptic brain. J Neurosci. 2008;28:3264–3276. doi: 10.1523/JNEUROSCI.4980-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petraglia AL, Plog BA, Dayawansa S, Chen M, Dashnaw ML, Czerniecka K, Walker CT, Viterise T, Hyrien O, Iliff JJ, Deane R, Nedergaard M, Huang JH. The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. J Neurotrauma. 2014;31:1211–1224. doi: 10.1089/neu.2013.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Wang X, Shapiro LA, Cotrina ML, Liu W, Wang EW, Gu S, Wang W, He X, Nedergaard M, Huang JH. NKCC1 up-regulation contributes to early post-traumatic seizures and increased post-traumatic seizure susceptibility. Brain Struct Funct. 2016 doi: 10.1007/s00429-016-1292-z. doi: 10.1007/s00429-016-1292-z. [DOI] [PMC free article] [PubMed] [Google Scholar]