


Abstract
The PEDANT genome database (http://pedant.gsf.de) contains pre-computed bioinformatics analyses of publicly available genomes. Its main mission is to provide robust automatic annotation of the vast majority of amino acid sequences, which have not been subjected to in-depth manual curation by human experts in high-quality protein sequence databases. By design PEDANT annotation is genome-oriented, making it possible to explore genomic context of gene products, and evaluate functional and structural content of genomes using a category-based query mechanism. At present, the PEDANT database contains exhaustive annotation of over 1 240 000 proteins from 270 eubacterial, 23 archeal and 41 eukaryotic genomes.
INTRODUCTION
Sequencing of a new genome is not a sensational event anymore: while the first genome sequences determined several years ago often made it to the front page of the New York Times, nowadays even getting them published in major scientific journals is becoming increasingly difficult. The novelty of genomic data as such is certainly passé, but their usefulness remains intact, and perhaps is even increasing owing to virtually unlimited opportunities for comparative genomic analysis and large-scale data mining. At the same time, due to the sheer amount of genomic data currently available, the task of maintaining an up-to-date and complete genome analysis database represents a significant challenge.
The MIPS group (now Institute for Bioinformatics) in Munich began to provide exhaustive automatic analysis of all publicly available genomes in 1996, when only five genomic sequences were published (1). The main mission of the PEDANT genome database is to fill the gap between manually curated high-quality protein sequence databases, such as SWISS-PROT (2) or PIR International (3), and the enormous amounts of other protein sequences produced by genome sequencing projects at an ever increasing pace. For example, release 44.0 of SWISS-PROT contains 153 871 manually annotated proteins, while the total number of currently known protein sequences stands at roughly 2 500 000. Since the aforementioned gap is quickly growing, it is probably safe to say that the majority of protein sequences will never be subjected to in-depth annotation by human experts.
IMPLEMENTATION
We use the PEDANT software suite (4) for annotation of large amounts of protein sequences by a carefully selected set of established bioinformatics methods. Exhaustive functional characterization of protein sequences includes similarity searches against the entire non-redundant sequence database, detection of motifs and patterns, automatic assignment of genes to functional categories and clusters of orthologous groups (5), similarity-based prediction of enzyme classification, and extraction of keywords and superfamily information. Structural characterization of gene products is based on similarity searches against the Protein Data Bank (PDB) (6) database, sensitive recognition of structural domains using profile searches, secondary structure prediction, detection of transmembrane regions, and prediction of low complexity and coiled coil regions. By design, PEDANT provides protein sequence annotation in genomic context. The PEDANT genome browser enables the user to select functional or structural categories of interest, obtain the list of gene products from a particular organism assigned to this category, and then view detailed information on each protein presented as an integrated report page. Advanced DNA and protein viewers allow visualizing the positions of genes and other genetic elements on the chromosome, and predicted structural and functional information about proteins, respectively. Facilities for searching the PEDANT annotation using text queries as well as BLAST (7) and pattern searches are provided.
The PEDANT genome database is produced by systematically applying the automatic annotation pipeline described above to all genomic sequences that are being released in the public domain. The major premises of the PEDANT database are as listed below:
Timeliness. The MIPS CPU resources make it possible to process a medium-size prokaryotic genome and make it available online essentially overnight.
Completeness. We seek to process all completely sequenced genomes as well as many incomplete genomes, which are being made available by sequencing centers. In many cases, PEDANT represents the only source of annotation for a given genome.
Standardization. Automatic annotation of sequences follows a clearly defined protocol in terms of the particular set of bioinformatics techniques applied to each sequence and the values of pre-determined recognition thresholds used for individual methods (e.g. BLAST E-values).
Documentation. Since the results of automatic sequence analyses are inevitably afflicted by a large number of false assignments, we make available the raw output of each bioinformatics method used. This allows the user to make his own judgment on the validity of functional predictions appearing on each protein's report page.
DATABASE CONTENT
Over the past eight years, the number of analyzed genomes in the PEDANT database has grown steadily (Figure 1) and stands at 334 at the time of writing, including 228 completely sequenced and 106 unfinished genomic sequences from all three kingdoms of life (Figure 2). Most of these genomes were annotated in a totally unsupervised fashion. However, the database also includes several genomes that were manually annotated and, in many cases, published by MIPS. Those are Saccharomyces cerevisiae (8), Thermoplasma acidophilum (9), Arabidopsis thaliana (10), Neurospora crassa (11), Parachlamydia UWE25 (12), Listeria monocytogenes EGD, Listeria innocuaClip 11262 and Helicobacter pylori KE26695. The total amount of data managed by PEDANT via a relational database system MySQL, is ∼360 GB, more than one gigabyte per genome on average.
Figure 1.

Growth of the number of annotated genomes in the PEDANT database since 1998. *Number as of September 1, 2004.
Figure 2.
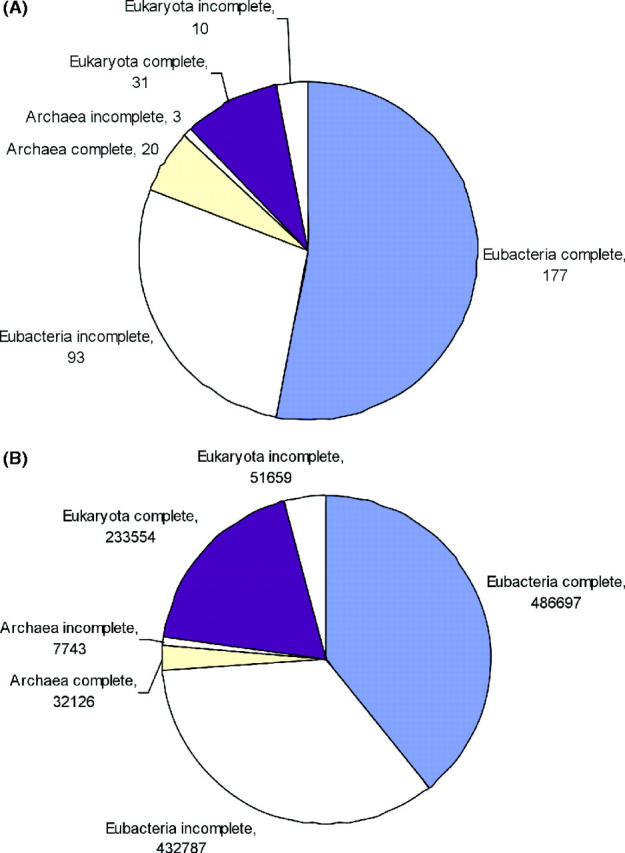
Number of annotated genomes (A) and protein sequences (B) in different genome categories.
To illustrate the functional and structural content of the PEDANT database, we calculated the coverage of all 1 240 000 annotated protein sequences by three selected popular categories: PFAM sequence motifs (13), SCOP structural domains (14) and MIPS functional role categories (15). As seen in Figure 3, the coverage varies in a wide range—from 64.3% by PFAM to 34.5% by SCOP. Only 15.2% of proteins possess all three attributes emphasizing the usefulness of applying many complementary bioinformatics techniques. The total number of all attributes computed by PEDANT for each sequence exceeds 20. The PEDANT database thus represents a valuable resource for large-scale association rule mining in automatically generated protein annotation.
Figure 3.

An illustration of the functional and structural content of the PEDANT database. The figure shows the percentage of protein sequences associated with PFAM sequence motifs, SCOP structural domains and MIPS functional categories, as well as any combinations of these three attributes.
AUTOMATIC FUNCAT
The MIPS Functional Catalogue (FunCat) was developed in 1996 and used in the annotation of S.cerevisiae (8). It comprises a hierarchically structured classification system, which at first only contained categories describing yeast biology. Since then, it has been extended and used to annotate the following genomes: T.acidophilum, Bacillus subtilis 168, L.monocytogenes EGD, L.innocuaClip 11262, H.pylori KE26695, N.crassa, A.thaliana and H.sapiens. The most recent version of the FunCat (v. 2.0; 16) is organism independent and consists of 28 main categories, covering features such as metabolism and cellular transport, as well as some more recently introduced categories (e.g. development and organ localization). The main categories are assigned a unique two-digit number e.g. 01. metabolism, which appears as the first two digits of the FunCat number. The main categories are branched into more specific categories, with up to six levels of increasing specificity (e.g. 01.01.06.05.01.01 biosynthesis of homocysteine).
The PEDANT software calculates automatic FunCat numbers based on a gene product's similarity to proteins in the manually annotated protein FunCat database. Although assignment of FunCat numbers by homology alone is not always reliable, it may provide useful information in the absence of manual annotation. The automatic FunCat tables for all PEDANT databases were recalculated using the new FunCat version and updated manually annotated FunCat database. Figure 4 shows the FunCat distribution of all 334 genomes in PEDANT.
Figure 4.

The FunCat distribution of all 334 genomes in PEDANT. Here, the relative amounts of proteins that are assigned to one or more of six general FunCat classes are shown. Since proteins can be assigned to more than one functional category, the total fraction exceeds 100%.
REFERENCES
- 1.Frishman D. and Mewes,H.W. (1997) PEDANTic genome analysis. Trends Genet., 13, 415–416. [Google Scholar]
- 2.Boeckmann B., Bairoch,A., Apweiler,R., Blatter,M.C., Estreicher,A., Gasteiger,E., Martin,M.J., Michoud,K., O'Donovan,C., Phan,I. et al. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res., 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu C.H., Nikolskaya,A., Huang,H., Yeh,L.S., Natale,D.A., Vinayaka,C.R., Hu,Z.Z., Mazumder,R., Kumar,S., Kourtesis,P. et al. (2004) PIRSF: family classification system at the Protein Information Resource. Nucleic Acids Res., 32, D112–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frishman D., Mokrejs,M., Kosykh,D., Kastenmuller,G., Kolesov,G., Zubrzycki,I., Gruber,C., Geier,B., Kaps,A., Albermann,K. et al. (2003) The PEDANT genome database. Nucleic Acids Res., 31, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tatusov R.L., Fedorova,N.D., Jackson,J.D., Jacobs,A.R., Kiryutin,B., Koonin,E.V., Krylov,D.M., Mazumder,R., Mekhedov,S.L., Nikolskaya,A.N., et al. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics, 4, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourne P.E., Addess,K.J., Bluhm,W.F., Chen,L., Deshpande,N., Feng,Z., Fleri,W., Green,R., Merino-Ott,J.C., Townsend-Merino,W. et al. (2004) The distribution and query systems of the RCSB Protein Data Bank. Nucleic Acids Res., 32, D223–D225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mewes H.W., Albermann,K., Bahr,M., Frishman,D., Gleissner,A., Hani,J., Heumann,K., Kleine,K., Maierl,A., Oliver,S.G. et al. (1997) Overview of the yeast genome. Nature, 387, 7–65. [DOI] [PubMed] [Google Scholar]
- 9.Ruepp A., Graml,W., Santos-Martinez,M.L., Koretke,K.K., Volker,C., Mewes,H.W., Frishman,D., Stocker,S., Lupas,A.N. and Baumeister,W. (2000) The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature, 407, 508–513. [DOI] [PubMed] [Google Scholar]
- 10.Arabidopsis Genome Initiative. (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408, 796–815. [DOI] [PubMed] [Google Scholar]
- 11.Galagan J.E., Calvo,S.E., Borkovich,K.A., Selker,E.U., Read,N.D., Jaffe,D., FitzHugh,W., Ma,L.J., Smirnov,S., Purcell,S. et al. (2003) The genome sequence of the filamentous fungus Neurospora crassa. Nature, 422, 859–868. [DOI] [PubMed] [Google Scholar]
- 12.Horn M., Collingro,A., Schmitz-Esser,S., Beier,C.L., Purkhold,U., Fartmann,B., Brandt,P., Nyakatura,G.J., Droege,M., Frishman,D. et al. (2004) Illuminating the evolutionary history of chlamydiae. Science, 304, 728–730. [DOI] [PubMed] [Google Scholar]
- 13.Bateman A., Coin,L., Durbin,R., Finn,R.D., Hollich,V., Griffiths-Jones,S., Khanna,A., Marshall,M., Moxon,S., Sonnhammer,E.L. et al. (2004) The Pfam protein families database. Nucleic Acids Res., 32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreeva A., Howorth,D., Brenner,S.E., Hubbard,T.J.P., Chothia,C. and Murzin,A.G. (2004) SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res., 32, D226–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mewes H.W., Frishman,D., Guldener,U., Mannhaupt,G., Mayer,K., Mokrejs,M., Morgenstern,B., Munsterkotter,M., Rudd,S. and Weil,B. (2002) MIPS: a database for genomes and protein sequences. Nucleic Acids Res., 30, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruepp A., Zollner,A., Maier,D., Albermann,K., Hani,J., Mokrejs,M., Tetko,I., Guldener,U., Mannhaupt,G., Munsterkotter,M. et al. (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res., 32, 5539–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]