

Abstract
The Gene Expression Omnibus (GEO) at the National Center for Biotechnology Information (NCBI) is the largest fully public repository for high-throughput molecular abundance data, primarily gene expression data. The database has a flexible and open design that allows the submission, storage and retrieval of many data types. These data include microarray-based experiments measuring the abundance of mRNA, genomic DNA and protein molecules, as well as non-array-based technologies such as serial analysis of gene expression (SAGE) and mass spectrometry proteomic technology. GEO currently holds over 30 000 submissions representing approximately half a billion individual molecular abundance measurements, for over 100 organisms. Here, we describe recent database developments that facilitate effective mining and visualization of these data. Features are provided to examine data from both experiment- and gene-centric perspectives using user-friendly Web-based interfaces accessible to those without computational or microarray-related analytical expertise. The GEO database is publicly accessible through the World Wide Web at http://www.ncbi.nlm.nih.gov/geo.
INTRODUCTION
Since 2000, the Gene Expression Omnibus (GEO) has served as a public repository for high-throughput molecular abundance experimental data, providing free distribution and shared access to comprehensive datasets (1). These data include single and multiple channel microarray-based experiments measuring the abundance of mRNA, genomic DNA and protein molecules. Data generated by innovative applications of microarray technology are also accepted, e.g. chromatin immunoprecipitation (ChIP-chips) for identifying protein-binding DNA regions and tiling arrays for genome annotation. Data from non-array-based high-throughput functional genomics and proteomics technologies are also archived, including serial analysis of gene expression (SAGE), and mass spectrometry peptide profiling.
The initial aim of GEO—to function as a robust, versatile high-throughput data repository—has been accomplished. As of fall 2004, GEO holds over 30 000 submissions representing approximately half a billion individual molecular abundance measurements, for over 100 organisms, submitted by over 600 researchers. Typically, GEO records are accessed over 15 000 times each weekday by over 1000 unique users, and bulk FTP downloads average 30 000 per month. Although GEO represents a huge reservoir of gene expression data that is widely used by the scientific community, it was recognized that the full potential of the repository could only be achieved by making these data easy to search and analyze, even by individuals having little experience in the field, without the need of massive data downloads. This paper describes database developments and tools that enable effective exploration, query and visualization of hundreds of experiments and millions of gene expression profiles using user-friendly Web-based interfaces.
REPOSITORY ORGANIZATION AND DATA FLOW
The principle architecture of the GEO database remains as described previously (1). Briefly, data submitted to GEO are stored in a relational database partitioned into three upper-level entity types: Platform, Sample and Series. A Platform describes the list of elements (e.g. oligonucleotide probesets, cDNAs, SAGE tags, antibodies) being assayed or that may be detected and quantified in that experiment. A Sample references a Platform and describes the abundance measurement of each feature element for a single hybridization or experimental condition. A Series brings together related Samples that make up an experiment and may include tables of extracted summary sets of significant genes or analysis as defined by the submitter. Each individual entity is assigned a unique and stable accession number; the accession number prefix indicates whether the record is a GEO Platform (GPL), Sample (GSM), or Series (GSE).
Unlike metadata that are stored in designated fields within database tables, Platform and Sample data tables are not fully granulated, but are stored as text objects. This design allows GEO to remain adaptable and responsive to developing technology trends, as it permits optimal flexibility in the quantity and type of data stored. For example, Platform elements may be described by any number of auxiliary attributes, and Sample data tables may contain all classes of supplementary and supporting measurements and calculations. The data within these tables may be extracted for higher-level rendering, indexing, search and retrieval purposes. Recent enhancements to the database include addition of supplementary metadata fields intended to facilitate and encourage MIAME (Minimum Information About Microarray Experiment) compliant data submissions (2), and acceptance of raw data contributions for storage and retrieval, e.g. Affymetrix .cel files or cDNA array scanned images.
Submission and standards
GEO aims at a balance between a submission procedure that is user-friendly and not overly rigid, while still encouraging high-quality data and a high level of experimental annotation. An infrastructure is provided so that submitters can present their data in a MIAME-compliant fashion (2). Submissions are validated syntactically according to a limited set of criteria and are subject to basic curation, assuring that records contain meaningful information and are organized correctly. Data depositors retain editorial control and are responsible for the content and quality of their records as outlined in the open letter published recently by the Microarray Gene Expression Data (MGED) Society board (3). GEO obviously could not attempt to independently verify the validity, merit, quality or biological significance of submitted data.
Once submitters establish their own private GEO accounts, there are three ways in which data may be deposited with GEO:
Interactive web-based forms. For each Platform and Sample submission, a text tab-delimited data table file is uploaded and validated. Metadata fields are entered interactively through a series of Web forms. This process is straightforward and is most useful when submitting relatively few entries. Updates to individual records may also be performed using similar interactive Web forms.
Direct submission using Simple Omnibus Format in text or SOFT format. SOFT was designed for rapid batch submission of data, and files may be easily produced from common spreadsheet and database applications. A single SOFT file can hold both data and metadata for multiple Platforms, Samples and Series, and can be uploaded directly to the database. Batch updates may also be quickly and efficiently performed using SOFT format. Detailed information on SOFT format is available on the GEO Web site.
Submitters may FTP files in valid MAGE_ML (4) format to GEO.
Records may remain private for several months, typically pending journal publication. Manuscript reviewers may gain confidential access to data prior to publication using read-only passwords.
DataSets and profiles
It was evident early-on that retrieval of data by means of accession number alone, or browsing by categories, would be insufficient to allow effective data mining and essential linkage between expression data and other sequence information and publication resources. High-throughput molecular abundance data are inherently more complex than other data types, such as sequence or bibliographic records; the strong association between measured entities and the biological and statistical context in which they were extracted must be considered; GEO stores a wide assortment of high-throughput experimental data processed by multiple means and analyzed by various methods. To address these issues, an additional level of curation was introduced where submitted samples are assembled into biologically meaningful and statistically comparable GEO DataSets (GDS). GDS records provide a coherent synopsis about an experiment, and serve as the basis for downstream data mining and display tools.
Samples within a GDS refer to the same Platform, that is, a common set of elements are assayed. Calculations are computed on the ‘value’ column extracted from original Sample data tables. These value measurements are calculated in an equivalent manner for each Sample within a GDS, i.e. considerations such as background processing and normalization are consistent across the GDS. Samples within DataSets are further grouped and classified into subsets according to the experimental variables under examination in the study, for instance ‘tissue’ or ‘strain’.
The Sample-centric tabular data under the control of the GDS upper level object then undergoes a final re-factoring into a gene-oriented view, and the results are indexed into a query engine and retrieval system, and display suites. The NCBI Entrez (5) database system is used as grounds for the query engine and retrieval system; two databases are defined:
GEO DataSets stores all experimental metadata, providing an ‘experiment-centric’ perspective of GEO data. The query interface is accessible from the GEO homepage or directly at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=gds.
GEO Profiles stores individual gene expression profiles, providing a ‘gene-centric’ perspective of GEO data. The query interface is accessible from the GEO homepage or directly at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=geo.
Thus, each DataSet entity defines a single experiment in GEO DataSets, and each DataSet parents a multitude of profile entities in GEO Profiles (Figure 1).
Figure 1.

Schematic diagram of the relationships between GEO Platform, Sample, DataSet and Profiles. For each gene on a Platform (e.g. Gene A), multiple Sample measurement values are generated (Sample1–Sample3). Related Samples make up a DataSet, from which multiple, individual gene profile entities are generated.
RETRIEVAL, QUERY AND ANALYSIS
Basic retrieval
There are several ways and formats in which GEO data may be retrieved:
Individual Platform, Sample, Series and GDS records can be accessed directly on the Web via their GEO accession number. Related records are intra-linked on the GEO site, such that one may conveniently navigate to associated Platform, Sample, Series and GDS records.
GDS records may be browsed by title, type, Platform or organism at http://www.ncbi.nlm.nih.gov/geo/gds/gds_browse.cgi. User-submitted records may also be browsed by category or submitter.
All user-submitted records, GDS value matrices with annotation, and raw data are available for bulk download via FTP. User-submitted records are grouped as compressed Series and Platform ‘family’ files, which incorporate all related accessions. Equivalent files are available for individual download from each record on the Web.
Query and mining
Effective searches may be accomplished by querying Entrez GEO DataSets and/or Entrez GEO Profiles. As with other NCBI Entrez databases (5), both trivial and sophisticated query and mining is achieved using Boolean phrases that may be restricted to, or combined with, a number of supported attribute fields.
Experiments of interest may be located by searching GEO DataSets for attributes such as experimental variable information, technology type, author, organism or any text keywords from the GDS description or original submitter-supplied Sample and Series records. For example, all dual channel nucleotide microarray experimental DataSets exploring metastasis in humans can be located using the query ‘dual channel[Experiment Type] AND metastasis AND human[Organism]’. Retrievals display the DataSet title, a brief experiment description, taxonomy, experimental variable types and links to the parent Platform, reference Series record and the complete GDS record. Once a relevant DataSet has been identified, users may go on to further explore that experiment either by taking advantage of the various supplementary tools on the GDS record page (Figure 2C) or by restricting subsequent GEO Profiles searches to that DataSet.
Figure 2.
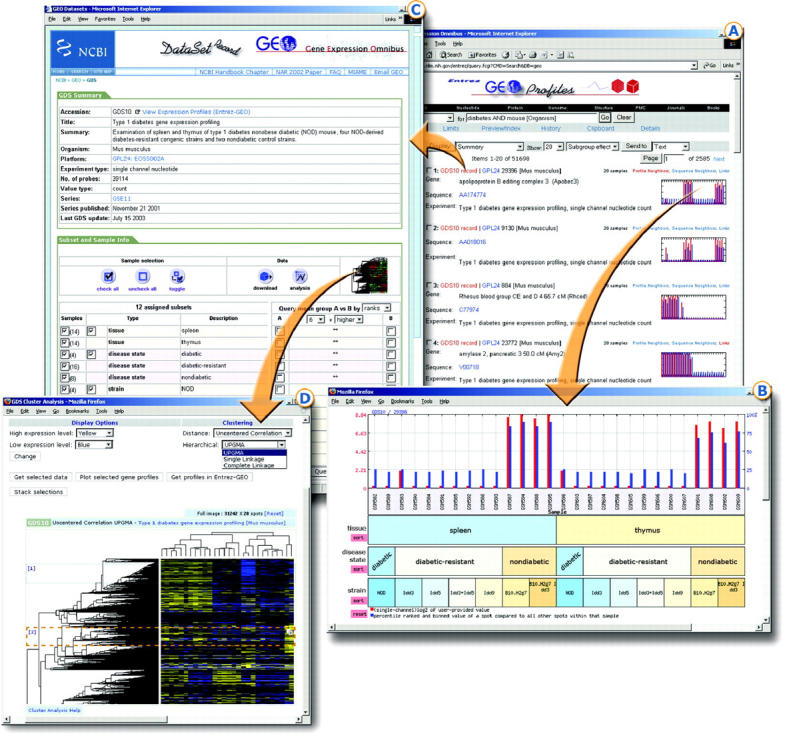
Selection of GEO web screenshots and how they link with each other. (A) GEO Profiles retrieval results; each entity includes sequence identifier and DataSet information, and a thumbnail profile image. Links to other Entrez databases or related profiles are provided above the thumbnail image. (B) Expanded profile chart depicts values (red bars) and rank (blue bars) information for one gene across each Sample in a GEO DataSet. Experimental subset groupings are reflected in labels at foot of chart. (C) DataSet record includes experiment summary information, DataSet subset classifications, and access to data mining features such as hierarchical cluster heat map and ‘Query subset A versus B’ tool. (D) DataSet hierarchical cluster heat map calculated by un-centered correlation coefficient/average linkage option. Regions of interest are selected using the red image cropper box, then either expanded to view Sample and gene annotation, downloaded, charted as line plots, or linked directly to corresponding Entrez GEO Profiles records.
The elemental unit in GEO Profiles is a gene, sequence or other reporter molecule, and its traced behavior along the measured conditions of the experiment, hence a ‘profile’. GEO Profiles are annotated in accordance with concurrent Entrez Gene and UniGene resources, and may be queried for attributes such as gene name, GenBank accession number, SAGE tag, GDS accession, DataSet description or profiles flagged as having significant effects with regards to specific experimental variables. For example, the query ‘Type 1 diabetes[GDS Text] AND apolipoprotein[Gene Description] NOT Homo sapiens[Organism]’ retrieves all apolipoprotein-related gene profiles in Type 1 diabetes-related datasets in organisms other than human. Query results display reporter annotation, brief experimental information, taxonomy and a bar-graph thumbnail image of the profile (Figure 2A). The thumbnail images are helpful for rapid batch profile scanning and comparison. A click on a thumbnail reveals the profile details (Figure 2B). Gene expression values extracted from original sample records are represented by red bars. Blue bars represent intra-sample percentile rank information, providing an indication of the relative expression level of that gene compared to all other genes on the array. Experimental structure is reflected in subgroup labels along the bottom of each chart allowing even complex experiments involving multiple and overlapping subset types to be clearly visualized. Standard GEO Profile retrievals are ordered according to subset effect flags by default, bringing potentially significant and interesting profiles to the fore. However, users may select alternative sorting schema based on mean value, deviation or outliers.
Selected GEO Profile entities possess intra-database links. ‘Profile neighbors’ connects genes that show a similar profile shape within a DataSet, as calculated by Pearson correlation coefficients. ‘Sequence neighbors’ retrieves related profiles based on nucleotide sequence similarity by BLAST (6) across all DataSets, and ‘Homologs’ retrieves profiles of genes belonging to the same HomoloGene group. Sequence and profile neighbor retrievals are weighted by presumed relevance, and are subject to cutoffs so as to limit the number of links that can be managed.
Entrez GEO DataSets and GEO Profiles are fully integrated with each other, as well as with other NCBI Entrez databases (7). Where possible, links are provided to GenBank, PubMed, Gene, UniGene, OMIM, Homologene, SNP, Taxonomy, SAGEMap and MapViewer. These links are reciprocal, meaning they can be traced back to GEO from any of the above resources, and facilitate seamless navigation and cross-referencing between databases.
Supplementary features
In addition to the Entrez query system, several supporting tools and features are provided to assist with enhanced mining and visualization of data:
Cluster heat maps. Pre-computed sample and gene hierarchical cluster heat maps are provided for most DataSets (Figure 2). Users are given the option to view clusters calculated using a variety of distance metrics (Euclidean distance, Pearson correlation or un-centered correlation coefficient) and clustering methods (single linkage, complete linkage or average linkage). Multiple cluster portions of interest may be selected, expanded, downloaded, charted as line plots or linked directly to Entrez GEO-Profile records.
Query subset A versus B. This feature identifies gene expression profiles of interest by calculating average rank or value differences between experimental subsets within a DataSet. For example, a user can specify that he wants to locate genes displaying 10-fold higher expression values in time point ‘A’ compared to time point ‘B’, and he will be directed to profiles matching those criteria.
Subset effects. Profiles are flagged if they display significant differences in expression values or ranks between subsets. This feature retrieves all profiles, either DataSet-specific or across all DataSets, that are flagged as having significant profiles with respect to a specific experimental variable, e.g. ‘age’ or ‘strain’.
Value distribution. Box and whisker plots for each Sample within a DataSet are presented, allowing an overview of the distribution of values across a DataSet.
GEO BLAST. This interface allows users to search for GEO Profiles of interest based on nucleotide sequence similarity using BLAST. The GEO BLAST database contains all GenBank sequences represented in GEO DataSets. Furthermore, standard BLAST output as performed using NCBI's BLAST interface, displays ‘E’ icon links where appropriate, linking directly to GEO Profiles expression data.
CONCLUSION
GEO represents a large compendium of gene expression data, addressing a wide range of biological issues across many organisms. The database already contains approximately half a billion measurements, and continues to grow at an average rate of >20 million per month. While very valuable, these data are not immediately interpretable or human readable in the raw form. To address this issue, database applications have been developed to facilitate complex data mining by providing query capabilities and concise displays that allow human scanning and data reduction. Tools are provided to help identify and categorize gene and sample relationships. Additional context is provided through comprehensive integration with sequence information, mapping and bibliographic resources.
As an open repository, the data in GEO have typically been analyzed and studied, and in most cases, the results published in journals. Nonetheless, pooling disparate data into one location and organizing them to be analyzable and cross-comparable using common interfaces adds a valuable analytic layer not attainable when considering individual experiments. Mining GEO data can provide clues as to the function of uncharacterized genes and genetic networks by examining spatial and temporal expression patterns (8–10), and co-regulation with well-characterized markers. Cross-comparison of independently generated but experimentally similar datasets can corroborate interesting gene expression trends that may be overlooked in one experiment alone (11). The GEO database and tools may also substantiate laboratory findings, or suggest supportive or negating evidence for research proposals and hypotheses (12). Reanalysis and reinterpretation of GEO data can provide valuable insights into other fields (13,14). Such opportunities for discovery will only increase as the database continues to grow in size and diversity.
Future plans for GEO are continued development of submission and retrieval formats, further integration with NCBI resources, and enhancements to data visualization and mining features. The features described herein are mostly relevant to gene expression studies; separate tools and graphical representations specific to other data types, such as proteomic technologies and comparative genomic hybridization, are also planned.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the Entrez/PubMed development team for ongoing support. Michael Domrachev implemented the first GEO database and provided support while moving to the MIAME enhanced schema. We thank Sergey Kurdin for web page designs and JS code, Todd Groesbeck for generation of manuscript figures, and Jim Ostell and David Lipman for advising on this project and review of this manuscript.
REFERENCES
- 1.Edgar R., Domrachev,M. and Lash,A.E. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res., 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brazma A., Hingamp,P., Quackenbush,J., Sherlock,G., Spellman,P., Stoeckert,C., Aach,J., Ansorge,W., Ball,C.A., Causton,H.C. et al. (2001) Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nature Genet., 29, 365–371. [DOI] [PubMed] [Google Scholar]
- 3.Ball C., Brazma,A., Causton,H., Chervitz,S., Edgar,R., Hingamp,P., Matese,J.C., Parkinson,H., Quackenbush,J., Ringwald,M. et al. (2004) Microarray Data Standards: An Open Letter. PLoS Biol., 2, 23–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spellman P.T., Miller,M., Stewart,J., Troup,C., Sarkans,U., Chervitz,S., Bernhart,D., Sherlock,G., Ball,C., Lepage,M. et al. (2002) Design and implementation of microarray gene expression markup language (MAGE-ML). Genome Biol., 3, RESEARCH0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuler G.D., Epstein,J.A., Ohkawa,H. and Kans,J.A. (1996) Entrez: molecular biology database and retrieval system. Methods Enzymol., 266, 141–162. [DOI] [PubMed] [Google Scholar]
- 6.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 7.Wheeler D.L., Church,D.M., Edgar,R., Federhen,S., Helmberg,W., Madden,T.L., Pontius,J.U., Schuler,G.D., Schriml,L.M., Sequeira,E. et al. (2004) Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res., 32, D35–D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tasheva E.S., Ke,A. and Conrad,G.W. (2004) Analysis of the expression of chondroadherin in mouse ocular and non-ocular tissues. Mol. Vis., 10, 544–554. [PubMed] [Google Scholar]
- 9.Oliver B. (2003) Fast males. Heredity, 91, 535–536. [DOI] [PubMed] [Google Scholar]
- 10.Gomez-Merino F.C., Brearley,C.A., Ornatowska,M., Abdel-Haliem,M.E., Zanor,M.I. and Mueller-Roeber,B. (2004) AtDGK2, a novel diacylglycerol kinase from Arabidopsis thaliana, phosphorylates 1-stearoyl-2-arachidonoyl-sn-glycerol and 1,2-dioleoyl-sn-glycerol and exhibits cold-inducible gene expression. J. Biol. Chem., 279, 8230–8241. [DOI] [PubMed] [Google Scholar]
- 11.Lee H.K., Hsu,A.K., Sajdak,J., Qin,J. and Pavlidis,P. (2004) Coexpression analysis of human genes across many microarray data sets. Genome Res., 14, 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puffenberger E.G., Hu-Lince,D., Parod,J.M., Craig,D.W., Dobrin,S.E., Conway,A.R., Donarum,E.A., Strauss,K.A., Dunckley,T., Cardenas,J.F. et al. (2004) Mapping of sudden infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification of TSPYL loss of function. Proc. Natl Acad. Sci. USA, 101, 11689–11694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reverter A., McWilliam,S.M., Barris,W. and Dalrymple,B.P. (2004) A rapid method for computationally inferring transcriptome coverage and microarray sensitivity. Bioinformatics, doi:10.1093/bioinformatics/bth472. [DOI] [PubMed] [Google Scholar]
- 14.Cheadle C., Cho-Chung,Y.S., Becker,K.G. and Vawter,M.P. (2003) Application of z-score transformation to Affymetrix data. Appl. Bioinformatics, 2, 209–217. [PubMed] [Google Scholar]