


Abstract
Staphylococcus aureus displays a clonal population structure in which horizontal gene transfer between different lineages is extremely rare. This is due, in part, to the presence of a Type I DNA restriction–modification (RM) system given the generic name of Sau1, which maintains different patterns of methylation on specific target sequences on the genomes of different lineages. We have determined the target sequences recognized by the Sau1 Type I RM systems present in a wide range of the most prevalent S. aureus lineages and assigned the sequences recognized to particular target recognition domains within the RM enzymes. We used a range of biochemical assays on purified enzymes and single molecule real-time sequencing on genomic DNA to determine these target sequences and their patterns of methylation. Knowledge of the main target sequences for Sau1 will facilitate the synthesis of new vectors for transformation of the most prevalent lineages of this ‘untransformable’ bacterium.
INTRODUCTION
Type I DNA restriction–modification (RM) systems are found in about half of the sequenced prokaryotic genomes (1–4). They present a formidable barrier to the invasion of the host cell by foreign DNA whether by transduction, transformation or conjugation and thus exercise control over horizontal gene transfer (HGT) (1,4–8). As an example of their effectiveness, less than 1 in 104 or 105 phage infections can successfully avoid the classical EcoKI Type I RM system of Escherichia coli K12. In some circumstances, such as when anti-restriction systems are absent (9), when there are multiple target sites on the phage (10) or when RM expression is raised (11), the barrier due to this single RM system can be even greater. RM systems operate by methylating defined target sequences on the host genome and they maintain this methylation pattern through each round of DNA replication (modification). Foreign DNA entering the cell often contains the same target sequence but in an unmethylated state. These unmethylated target sequences are targeted for endonucleolytic cleavage by the RM system (restriction). The Type I RM system comprises three hsd (host specificity for DNA) genes, hsdR, hsdM and hsdS for restriction, modification and target sequence specificity respectively. The gene products form an R2M2S1 complex in which HsdS (or S) recognizes the target sequence, HsdM (or M) recognizes the methylation status of the target and methylates hemimethylated targets while HsdR (or R) cleaves the DNA containing unmethylated targets after a complex reaction involving adenosine triphosphate (ATP) hydrolysis and DNA translocation (12). An M2S1 complex can act solely as a methyltransferase (MTase) (13). Type I RM enzymes almost always recognize and methylate adenine nucleotides in their target sequences to form N6-methyl adenine (6mA) although a few forming N4-methyl cytosine (m4C) are now known (3,14). In addition to the protection offered by Type I, II and III RM systems, Type IV restriction systems can attack foreign DNA containing methylated sequences not found in the host (15).
The presence of multiple RM systems in a single host can increase the barrier to HGT still further. For instance, Staphylococcus aureus often contains two related Type I RM systems making its transformation extremely inefficient and hindering the genetic analysis of this organism (16–19). These genomes contain two hsdM and two hsdS and share a single hsdR, although some S. aureus strains have different numbers of hsdM and hsdS (Figure 1A). The presence of only a single hsdR is not a problem as it can interact with each hsdMhsdS pair. It has long been known that S. aureus displays a clonal population structure (20) in which HGT between different clonal complexes is exceedingly rare. Multilocus sequence typing, microarray analysis and whole genome sequencing divides lineages of S. aureus and close relatives into the clonal complexes (CC) (20–23), each of which carries a different range of mobile genetic elements and antibiotic resistance genes on the genome (24–27). Each CC can be further subdivided into sequence types (ST) (22). Waldron and Lindsay (16) first realized that each CC of S. aureus contained a unique pair of Type I RM systems. A Type IV restriction system, SauUSI, was also identified later and recognized as a methyl-dependent restriction enzyme which would prevent the uptake of foreign DNA containing C5-methyl cytosine (5mC) (28,29). Thus most genetic manipulation of S. aureus is confined to strain RN4220, which has a defective Type I RM system due to a premature stop codon in hsdR. Furthermore, to avoid the Type IV system, DNA needs to be prepared from an E. coli strain, such as E. coli ER2796, lacking the Dcm 5mC MTase (30).
Figure 1.

Staphylococcus aureus genomes showing the genes and the typical organization of target recognition domains (TRDs) in the HsdS DNA sequence specificity subunit. (A) Strain ED133 (CC133) has two hsdS; strain 11819–97 (CC80) has three hsdS (CC80) and strain JKD6159 (CC93) contains an extra Type I RM system from a different Type I RM family. From top to bottom: ED133, 11819–97, JKD6159. hsdR (red), hsdM (blue), hsdS (yellow). (B) The structural organization of the HsdS specificity subunit. The conserved regions (cr) are common to all S subunits within a family. The two TRDs (TRD1 and TRD2) define the target sequences recognized by the RM enzyme and can be swapped between S subunits of the same family to generate new specificities.
The Type I RM systems in different strains of S. aureus were given the informal name of Sau1 by Waldron and Lindsay (16) and it is clear from not only a comparison of the sequences of genes and proteins but also from the ability to use subunits from one strain to complement subunits from other strains (31) that the term Sau1 describes a classical ‘family’ of Type I RM systems. Type I RM families, Type IA to Type IE, were originally defined in E. coli and Salmonella enterica by DNA hybridization, antibody cross reactivity and subunit complementation (32,33), although now it is more usual to use the high levels of sequence identity (over 90%) in HsdM and HsdR to define a family in silico. Although the name Sau1 for this family of Type I RM systems in S. aureus is an informal one not following the usual conventions (34), we retain it as it is established in the literature. However, it is important to note that some strains of S. aureus show additional Type I RM systems, which show limited amino acid sequence identity to the HsdR, HsdM and HsdS of Sau1 (Figure 1A). For instance, Monk et al. (35) identified an active Type I RM system, SauJKDIII, in S. aureus JKD6159 which showed low sequence identity to members of the Sau1 family. This is clearly a member of a new and different Type I RM family whose subunits will be unable to interact with the Sau1 HsdM and HsdR (D. T. F. Dryden, J. A. Lindsay and M. T. G. Holden, in preparation).
The Sau1 Type I RM systems are so effective because they show great variability in the target sequences recognized thus preventing HGT between CC but allowing HGT between strains within a CC (31,35,36). This variability in target sequences is due to the modular construction of the Type I RM systems (Figure 1B). The S subunit contains two target recognition domains (TRDs) each of which recognizes one half of a bipartite target, for example the first Type I RM system in CC1, given the generic name CC1-1, recognizes CCAYNNNNNTTAA (adenine methylation sites are underlined) (35,36). Swapping TRDs between S subunits generates new targets, for example the second Type I RM enzyme in CC1, termed CC1-2, couples the first TRD of CC1-1 with a different second TRD to recognize CCAYNNNNNNTGT. This swapping is easy because the DNA for S subunits contain conserved sequences bounding each TRD. Most S. aureus strains have two copies of hsdS, two of hsdM and one of hsdR. Thus, there are often four TRDs in each CC, which define the restriction barrier against HGT. Some Type I RM enzymes have half-size HsdS incorporating only a single TRD. It has been shown that these products are often able to dimerize and recognize symmetric target sequences (37–39). We have been able to recapitulate these results on ‘half-HsdS’ enzymes by manipulating the CC398-1 S. aureus system (E. K. M. Bower and D. T. F. Dryden, unpublished results).
Previously we have identified the target sequences recognized by several common community-associated, hospital-associated and livestock-associated MRSA clonal complexes (31,36) and recently several more have been identified (3,35,40). Monk et al. (35) and Jones et al. (40) have used this information to prepare DNA methylated by the MTase M2S1 component enzymes to aid the transformation of S. aureus strains that are usually resistant to transformation.
The identification of further targets recognized by the S subunits of Sau1 Type I RM systems would in principle allow more CC to be transformed for genetic analysis. In addition, further understanding of the structural requirements for TRDs to recognize different specific DNA sequences is of intense interest as the Type I RM systems are very widespread in bacteria and archaea (1,4) and exert a considerable pressure on HGT and the evolution of prokaryotes. For instance, the use of multiple TRDs being exchanged between strains has been observed in Helicobacter (41), Mycoplasma (42,43), Streptococci (44,45) and Bacteroides (46).
Here we identify many further TRDs and their targets using both biochemical and PacBio single-molecule real-time (SMRT) sequencing methods to define the barriers to HGT in a wide range of S. aureus CC of global importance.
MATERIALS AND METHODS
Nomenclature for expression plasmids encoding new MTases
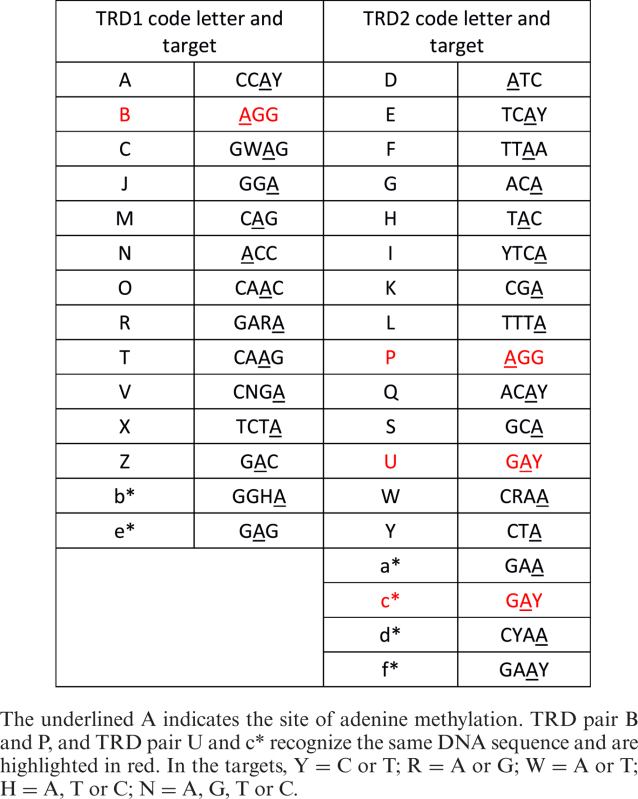
As each Type I S subunit contains two TRDs and we propose to determine the targets recognized by each TRD, we have given each TRD a single letter code, Table 1, and refer to the plasmids as pSauTRD1-TRD2, e.g. pSauBI expresses an S subunit containing TRD B and TRD I and the M subunit. If the TRD combination is the same as that found in a known clonal complex, then that CC is also given in brackets. The MTase would be called M.SauBI in this example and the S subunit S.SauBI and is from CC22. All sequences are given in the Supplementary Data.
Table 1. TRD targets shown from 5΄ to 3΄.
|
Preparation of M.SauBI (CC22-1), M.SauCD (CC30-1), M.SauJK (CC30-2) and M.SauCL (CC45-1)
These four MTases were prepared as EGFP-His tag fusions as described in Roberts et al. (31). pSauBI-EGFP (CC22-1, genomic DNA from MRSA5906), pSauCD-EGFP (CC30-1, genomic DNA from MRSA252), pSauJK-EGFP (CC30-2, genomic DNA from MRSA252) and pSauCL-EGFP (CC45-1, genomic DNA from strain 70642) were all constructed by the polymerase chain reaction (PCR) with their hsdS fused to DNA encoding EGFP and a His-tag, with the following locus-specific oligonucleotides priming from the 3΄ end of the genes encoding the S subunits:
CC22-1 BI BS 5΄GATCGAATTCCGGATCCAATAAACATCTTTTGTAAAAACAC3΄
CC30-1 CD BS 5΄GATCGAATTCCGGATCCTAAGAACATCTTTTGTAAAAAGG3΄
CC30-2 JK BS 5΄GATCGAATTCCGGATCCTATAAAAATTTTTTGAAGTAATCCTTG3΄
CC45-1 CL R167K BS 5΄GATCGAATTCCGGATCCAATAAACATCGATTTAAGTAAGGC3΄
The sequence for CC45-1 introduced a single mutation R167K in the first TRD in the S subunit but since this change is found in other S. aureus isolates containing this TRD, the change is presumed to be completely neutral.
A new vector for MTase expression: pJF118his
Although we had not experienced problems in examining the fusion proteins of S subunits and EGFP in biochemical work, we decided to construct a vector encoding hsdS with only a C-terminal His-tag. Vector pJF118his was made by PCR of the plasmid encoding the MTase CC5-1-EGFP constructed in Roberts et al. (31) with these two oligonucleotides:
pJFMShisTS 5΄AGCTTCGAGAGGATCCCATCATCATCATCATCATTAAGAATTCAGCTTGGCTGTTTTGGCGG3΄ and pJFMSEGFPhisBS 5΄GAGTGAATCCCCGGGGATCCGTCGACC3΄.
The resulting PCR product was cut with BamHI and unimolecular religation gave pJF118his into which the hsdMS operon could be ligated as BamHI fragments and from which all subsequent MTase clones were descended.
Construction of an MTase plasmid to allow TRD swaps: pSaudeltaXmaI
A PCR-based strategy was devised to allow free pairwise assortment of desired TRDs in HsdS. Many, but not all of the HsdS subunits, including that encoded by the Type I system in CC398 (36), have a predicted proline-glycine sequence near the N-terminus. This dipeptide can be encoded by CCCGGG, which would be a target site for SmaI or XmaI. Oligonucleotides were designed which would introduce this motif in the N-terminus (a replacement with no amino acid changes) and at the C terminus (an insertion of two amino acids) of the S subunit of the CC398 system (36), by a two stage PCR fusion. Thus, primary PCR products were generated by reactions primed by: PromoterJF 5΄GCTTCTGGCGTCAGGCAGCC3΄ with 398SmaIOligoBS 5΄CCCATTCGCCTTCAAACCCGGGGAATCTCAACTCTGGCAC3΄ and 398SmaIOligoTS 5΄GTGCCAGAGTTGAGATTCCCCGGGTTTGAAGGCGAATGGG3΄ with 398SmaIBamHI 5΄GATCGATCGGATCCCCCGGGAATAAACATCTTTTGAAGTAATGAC3΄.
The purified PCR products were then fused in a secondary PCR reaction primed by PromoterJF with 398SmaIBamHI. The product was then cut with BamHI, and ligated into the BamHI site of pJF118his as pSauNE-XmaI. This mutated form of the CC398-1 MTase, could assemble the complete restriction enzyme that proved to be active in endonucleolytic cleavage (36). This indicated that insertion of a proline and glycine toward the C-terminus did not affect the function of the enzyme. Subsequently, on reanalyzing the DNA sequence, a single PCR mutation was discovered within the XmaI fragment. This caused a mutation A50S but this clearly did not affect the specificity or function of the S subunit in our assays. Digestion of pSauNE-XmaI with XmaI followed by intramolecular religation of the vector fragment generates pSaudeltaXmaI, into which any pairwise combination of TRDS with XmaI cohesive ends may be inserted.
Construction of MTases M.SauNI, M.SauND, M.SauNK, M.SauNL, M.SauBE, M.SauJE and M.SauCE (ST425-1) containing hybrid S subunits
The DNA for each TRD of these S subunits was fused to the DNA for the reciprocal TRD of S.SauNE (CC398-1). This was achieved by creating primary PCRs with a short area of homology, which then allowed base pairing of single strands of each PCR, in a secondary PCR. For example, S.SauBE TRD B was generated from an appropriate plasmid template by PCR with oligonucleotides, TRD1FOR398SmaIOligoTS 5΄GTGCCAGAGTTGAGATTCCCCGGGTTTGAAGGCGAATGGG3΄ paired with TRD1nearuniversal 5΄GTTCTTCTAATTCAATTTGT3΄. TRD E was similarly generated by PCR from plasmid template with oligonucleotides TRD2nearuniversal 5΄ACAAATTGAATTAGAAGAAC3΄ and 398SmaIBamHI 5΄GATCGATCGGATCCCCCGGGAATAAACATCTTTTGAAGTAATGAC3΄. The final insert was then generated by PCR with the two gel-purified primary oligonucleotides and TRD1FOR398SmaIOligoTS 5΄GTGCCAGAGTTGAGATTCCCCGGGTTTGAAGGCGAATGGG3΄ and 398SmaIBamHI 5΄GATCGATCGGATCCCCCGGGAATAAACATCTTTTGAAGTAATGAC3΄. S.SauCL was the only subunit for which we could not use the central universal oligonucleotides for PCR and required specific substitutes: TRDLFOR/CC45-1 5΄ACAAATTGAATTAGAAGAACAAAAACTTGAATTACTTCAACAACAG3΄ and TRDC/CC45-1 5΄GTTCTTCTAATTCAATTTGTCGATCGAGTTTGCTGAAGAAG3΄. Each C-terminus is unique and where TRD2 was not TRD E, a specific oligonucleotide was employed: TRDIREV/CC22-1c-termsmaI 5΄GATCGATCGGATCCCCCGGGAATAAACATCTTTTGTAAAAACAC3΄, TRDDREV/CC30-1c-termsmaI 5΄GATCGATCGGATCCCCCGGGTAAGAACATCTTTTGTAAAAAGGATTG3΄, TRDKREV/CC30-2c-termsmaI 5΄GATCGATCGGATCCCCCGGGTATAAAAATTTTTTGAAGTAATCCTTG3΄ and TRDLREV/CC45-1c-termsmaI 5΄GATCGATCGGATCCCCCGGGAATAAACATCGATTTAAGTAAGGC3΄. Each pure secondary PCR product was cut with XmaI and ligated into the XmaI site of pSaudeltaXmaI.
Construction of further MTases with further combinations of TRDs using synthetic genes
Additional hsdS sequences were obtained as synthetic genes from GeneArt (ThermoFisher Scientific) with sequences optimized for expression in E. coli (Supplementary Data). All the first TRDs begin with 5΄CCCGGGTTTGAAGGCGAATGGGAG3΄, except that for CC80-2 which begins with 5΄CCCGGGTTTGAAGGCGAATATTCT3΄. All the first TRDs end with 5΄CAAATTGAATTAGAAGAACAGAAG3΄. All the second TRDs begin with 3΄CAAATTGAATTAGAAGAACAGAAG5΄ and have a universal reverse oligonucleotide, Trd2unirev 5΄GATCGATCGGATCCCCCGGG3΄. These conserved sequences were used to create oligonucleotides to prime PCR reactions. Each pure secondary PCR product was cut with XmaI and ligated into the XmaI site of pSaudeltaXmaI. The orientation of the fragments was determined by PCR.
Expression and purification of MTases
These new MTases and the R subunit of CC5 were expressed in E. coli BL21(DE3) and purified via HisTrap chromatography, size exclusion chromatography, diethylaminoethyl anion exchange chromatography and, if necessary, Heparin HiTrap chromatography (GE Healthcare, Uppsala, Sweden) as described previously (31).
Nuclease and ATPase assays
Purified MTases were mixed with the CC5 R subunit and used in assays for ATP hydrolysis (ATPase) activity (coupled enzyme assay following a change in absorbance of NADH) and DNA cleavage activity (plasmid cutting assay with analysis via agarose gel electrophoresis) as previously described (31,36).
Preparation of genomic DNA for SMRT sequencing
The expression plasmids harboring the various MTases were used to transform a non-methylating (dam− dcm−) strain of E. coli ER2796 (30). Single colonies from the transformation plate of Lysogeny Broth (LB) agar medium supplemented with 10 μg/ml kanamycin, 10 μg/ml tetracycline as well as 100 μg/ml carbenicillin, which acted as a selection marker for the expression construct, were picked and used to inoculate 5 ml of LB containing the same cocktail of antibiotics. The cultures were incubated overnight with shaking at 37°C and 1 ml aliquots of the overnight culture were then pelleted by centrifugation (6000 g, 6 min, 4°C). The culture medium was carefully removed and the cell pellets stored at −20°C until required. Genomic DNA was prepared from each cell pellet using the Wizard Genomic DNA purification kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. The quality of the genomic DNA preparations was initially assessed by agarose gel electrophoresis and from the shape of the absorbance profile from 240 to 340 nm. Genomic DNA from S. aureus strains LGA251 (a kind gift from Mark Holmes) and NCTC13435 (a kind gift from Angela Kearns) was prepared by using the PurElut Bacterial Genomic Kit (EdgeBio, Gaithersburg, MD 20877, USA). The DNA library for SMRT sequencing was prepared and subsequently analyzed as described in Anton et al. (30).
Methylation of plasmids using M.EcoGII
M.EcoGII was kindly supplied by Dr Iain Murray (New England Biolabs) and used to modify plasmids E2, E5, E10, E11 and E12 previously described (31) and plasmid pCN36 (47). A total of 0.45 μg DNA was methylated using 2.0 U of M.EcoGII for 100 min at 37°C in a 50 μl volume. The reaction was in 1×NEB4 buffer (50 mM potassium acetate, 20 mM Tris acetate, 10 mM Mg acetate, 1 mM dithiothreitol, pH 7.9, 25°C) supplemented with 320 μM S-adenosyl-L-methionine (SAM). As a negative control, DNA was incubated in the same buffer without M.EcoGII. The DNA samples were then supplemented with ATP (20 μM) and additional SAM (160 μM) and then digested with a Type I enzyme (CC5-1, CC5-2, CC30-1, CC45-1 or the NY TRD hybrid) for 14 min at 37°C. As a control, methylated and unmethylated DNA was digested with EcoRI.
RESULTS AND DISCUSSION
Assigning TRDs to target sequences
Each TRD was given a one letter code (A to Z and a* to f*), Table 1. There were 14 TRD1 examples and 18 TRD2 examples in our survey and these are found in 17 different CC or ST groups. Table 1 lists the target specificity and site of methylation for each TRD in our survey. These data were obtained by pairing TRDs and determining the complete target for each TRD pair as described in the next section and in full in the Supplementary Data. Of interest are the TRD pairs B and P and U and c*. These pairs recognize the same DNA sequence namely AGG and GAY respectively. Amino acid sequence comparisons of B with P and U with c* are shown in Figure 2.
Figure 2.

Amino acid sequence and secondary structure alignment of two pairs of TRDs recognizing the same DNA target. The TRD sequences are highlighted in yellow. Consensus secondary structure shows ‘h’ for α helix and ‘e’ for β sheet. (A) TRDs B and P are examples of a first and a second TRD respectively recognizing 5΄-AGG-3΄. (B) TRDs U and c* are both examples of second TRDs with the same specificity, 5΄-GAY-3΄. The long predicted α helices at the start and the end of the sequences are the conserved helical spacer regions in the HsdS subunits while the sequence between these helices makes up the TRD.
TRD B and TRD P are virtually identical throughout the TRD region even though TRD B is the first TRD in the HsdS subunit and TRD P is the second TRD in the HsdS subunit, (Figure 2A). While the high level of sequence identity is expected for Type I systems in the same family, the high level of identity between TRDs found in the first or second position in the HsdS subunit is more unusual. However, such a situation has previously been observed in comparisons of the Type I systems in Salmonella blegdam and E. coli R124 (48).
In contrast, TRDs U and c* are both examples of the second TRD in the HsdS subunit recognizing 5΄-GAY-3΄ but the level of identity between them is much lower (∼36%) (Figure 2B). This level of identity between TRDs recognizing the same target is expected if the TRDs are from different Type I RM families so the low level of identity observed here is unusual. Despite this low level of sequence identity, the predicted secondary structure elements are the same as expected from the early work of Sturrock and Dryden (49). In fact, all of the TRDs in the Sau1 family of RM systems align well when secondary structure elements are taken into consideration (50) and they will have the same protein fold (Supplementary Data: PROMALS alignments). Therefore, it should in future be possible to predict the precise amino acid to nucleotide contacts involved in sequence recognition as was done for the Type IIG TRDs (51,52).
Determination of complete target sequences recognized by pairs of TRDs
Tables 2, 3 and 4 show the TRD combinations investigated in this work and those investigated previously by ourselves and others along with their combined target sequences, methylation specificity and the methods used to determine these parameters. The full experimental data are given in the Supplementary Data. Many of the TRDs were investigated in more than one MTase and in more than one assay thus our set of data represents a self-consistent set. DNA cleavage and ATP hydrolysis assays were performed on purified MTases mixed with purified R subunit while SMRT data were collected from E. coli genomic DNA isolated after the hosts were transformed with a plasmid expressing the MTase or directly from S. aureus genomic DNA. The adenines targeted for methylation were determined easily by SMRT sequencing but for systems not examined in this manner, it was assumed if there was a single adenine in the site recognized by the TRD that this was the target for methylation.
Table 2. The Sau1 RM systems with published recognition sequences.
| Strain name and genome reference | Clonal Complex or Sequence Type | S subunit name in REBASE | Recognition sequence | TRDs assigned | Suggested generic name | Experimental method | Reference for target specificity and method |
|---|---|---|---|---|---|---|---|
| MW2 (53) | CC1 | S.SauMW2I | CCAY-5-TTAA | AF | CC1-1 | g, s, a | g (31) |
| S.SauMW2II | CCAY-6-TGT | AG | CC1-2 (CC8-2) | g, s, a | a (36) | ||
| N315 (54) | CC5 | S.SauN315II | CCAY-6-GTA | AH | CC5-2 | g, s, a | s (CC8-1 and CC8-2 in strain NRS384 are from ref. 35) |
| S.SauN315I | AGG-5-GAT | BD | CC5-1 (CC8-1) | g, s, a | |||
| MRSA252 (55) | CC30 | S.SauMRSII | GWAG-5-GAT | CD | CC30-1 | g, s | s (35) g, s (this work) |
| S.SauMRSI | GGA-7-TCG | JK | CC30-2 | s | s (35) s (this work) | ||
| JKD6159 (56) | CC93 | S.SauJKDIII | GAAG-5-TAC or complement | Not a Sau1 system | CC93-3 | s | s (35) |
| S.SauJKDII | GGHA-7-TCG | b*K | CC93-2 | s | Note the ambiguity in assigning CC93-1 and CC93-3 is clarified with strains ED133 and 32320 and from Table 3. | ||
| S.SauJKDI | CAG-6-TTC | Ma* | CC93-1 | s | |||
| ED133 (57) | CC133 | S.Sau133ORF451P | CAG-5-RTGA | ME | CC133-1 | g | g (36) |
| S.Sau133ORF1794P | GGA-7-TTRG | Jd* | CC133-2 | s | s (this work) | ||
| 32320 (58) | CC133 | S.Sau32320ORFAP | CAG-5-RTGA | ME | CC133-1 | g | g (36) |
| S0385 (59) | CC398 | S.SauSTORF499P | ACC-5-RTGA | NE | CC398-1 | g, s | g (36) s (this work) |
Target sites are shown from 5΄ to 3΄ with the length of the non-specific spacer shown as a number. Underlined A or T indicates the site of adenine methylation on the top or bottom strands respectively. The experimental methods used are indicated as g = target obtained by DNA cleavage with a purified enzyme, s = target obtained by SMRT sequencing of E. coli ER2796 genomic DNA, a = target obtained by ATPase assay with a purified enzyme. Full details are given in the Supplementary Data. S.Sau133ORF1794P is characterized in this work but is included here as it is part of the RM system found in strain ED133. SauMRSI and SauMRSII characterized by Monk et al. and S.SauSTORF499P characterized by Chen et al. are also further characterized in this work.
Table 3. The ‘artificial’ Sau1 systems containing novel pairings of TRDs.
| ‘Artificial’ Sau1 RM systems. | |||||
|---|---|---|---|---|---|
| Recognition sequence | TRDs assigned | Experimental method | Recognition sequence | TRDs assigned | Experimental method |
| AGG-5-RTGA | BE | a | ACC-6-TTC | Na* | s |
| GGA-6-RTGA | JE | g, s | ACC-6-RTC | Nc* | s |
| ACC-6-TGAR | NI | g | ACC-6-TTRG | Nd* | g, s |
| ACC-6-TCG | NK | g | GARA-6-RTGA | RE | s |
| ACC-6-TAAA | NL | g | CAAG-5-RTGA | TE | s |
| ACC-5-CCT | NP | s | CNGA-6-RTGA | VE | s |
| ACC-5-RTGT | NQ | g, s | TCTA-6-RTGA | XE | g, s |
| ACC-6-TGC | NS | s | GAC-5-RTGA | ZE | a |
| ACC-5-RTC | NU | g, s | GAC-6-TGC | ZS | a |
| ACC-6-TTYG | NW | g, s | GGHA-6-RTGA | b*E | s |
| ACC-6-TAG | NY | g, s | GAG-6-RTGA | e*E | g, s |
Target sites are shown from 5΄ to 3΄ with the length of the non-specific spacer shown as a number. Underlined A or T indicates the site of adenine methylation on the top or bottom strands respectively. The experimental methods used are indicated as g = target obtained by DNA cleavage with a purified enzyme, s = target obtained by SMRT sequencing of E. coli ER2796 genomic DNA, a = target obtained by ATPase assay with a purified enzyme. Full details are given in the Supplementary Data.
Table 4. The Sau1 RM systems investigated in this project.
| Strain name and genome reference | Clonal Complex or Sequence Type | S subunit name in REBASE | Recognition sequence | TRDs assigned | Suggested generic name | Experimental method |
|---|---|---|---|---|---|---|
| CO1791 (58) | CC97 | S.SauC01791ORFAP | CCAY-6-RTC | Ac* | CC97-1 | s |
| HO5096 (60) | CC22 | S.Sau5096I | AGG-6-TGAR | BI | CC22-1 | g, s |
| LGA251 (61) | ST425 | S.Sau251I | GWAG-5-RTGA | CE | ST425-1 | g, s* |
| S.Sau251ORF16900P | GAG-?-RTTC | e*f* | ST425-2 | Not expressed, no signature with s*. | ||
| S.Sau251II | GAAG-5-TAC or complement | Not a Sau1 system | Same as CC93-3 | s* | ||
| Isolate 3 (19) | CC51 | S.SauL3ORFAP | GGA-6-CCT | JP | CC51-1 | s |
| Isolate 3067 (19) | CC45 | S.Sau347I | GWAG-6-TAAA | CL | CC45-1 | g |
| Isolate 3150 (19) | CC15 | S.SauL315ORFAP | CAAC-5-RTGA | OE | CC15-1 | s |
| SA40 (62) | CC59 | S.SauSA40ORF370P | GGA-6-RTGT | JQ | CC59-1 | a |
| CN1 (63) | CC72 | S.SauCN1ORF415P | GARA-6-RTGT | RQ | CC72-1 | a |
| S.SauCN1ORF1757P | GGA-7-TGC | JS | CC72-2 | a | ||
| MSHR1132 (64) | CC75 | S.Sau1132ORF3780P | CAAG-5-RTC | TU | CC75-1 | g |
| S.Sau1132ORF16570P | CNGA-7-TTYG | VW | CC75-2 | s | ||
| NCTC13435 NCBI Biosample identifier: | ST80 | S.Sau13435ORF394P | TCTA-?-TAG | XY | ST80-1 | Not expressed, no signature with s or s*. |
| SAMEA2479566 | S.Sau13435ORF1751P | GAC-6-TTYG | ZW | ST80-2 | a, s* | |
| S.Sau13435ORF2165P | TCTA-6-RTTC | Xf* | ST80-3 | s, s* | ||
| 32326 (58) | CC873 | S.Sau32326ORFAP | GAG-6-GAT | e*D | CC873-1 | a |
Target sites are shown from 5΄ to 3΄ with the length of the non-specific spacer shown as a number. Underlined A or T indicates the site of adenine methylation on the top or bottom strands respectively. TRD pair e*f* in strain LGA251 was not cloned in E. coli while TRD pair XY was cloned. However, no target modification was observed using SMRT on genomic DNA from either E. coli or S. aureus for these TRD pairs. If the genes are translated, their target is inferred from other TRDs in this table although the spacer length remains undefined. The experimental methods used are indicated as g = target obtained by DNA cleavage with a purified enzyme, s = target obtained by SMRT sequencing of E. coli ER2796 genomic DNA, s* = target obtained by SMRT sequencing of S. aureus genomic DNA, a = target obtained by ATPase assay with a purified enzyme. Full details are given in the Supplementary Data.
Table 2 contains systems from a range of CC investigated previously as well as several examined in this study. It is important to note that in our work those systems containing M.SauMRSII plus S. SauMRSII, M.Sau133ORF1794P plus S.Sau133ORF1794P and M.SauMRSI plus S.SauMRSI are paired with the HsdR (SauN315ORF189P) from the N315 strain of CC5 in DNA cleavage and ATPase assays. Those shown in Tables 3 and 4 are studied as HsdS paired with the HsdM (M.SauSTORF499P) from strain S0385 of CC398 and the HsdR (SauN315ORF189P) from the N315 strain of CC5 (if used in DNA cleavage or ATPase assays). Therefore, these HsdS are not examined in the context of their natural genome, but since they are all from the Sau1 family of Type I RM systems and the HsdM and HsdR of these RM systems are essentially identical in all of the strains, it is reasonable to assume that the target specificities identified are those that would be recognized in their natural host.
Identifying the complete target recognized by a member of the Sau1 Type I RM family when both TRDs have unknown targets is difficult and ambiguous as either orientation may be correct. Hence, we combined TRDs with unknown targets with TRD E or TRD N to make a protein recognizing a hybrid sequence in which one half of the target was already known (Table 3). A variety of methods were used to determine the target associated with each hybrid including DNA cleavage and ATP hydrolysis assays when the hybrid enzyme could be expressed and purified from E. coli and SMRT sequencing when the expression and purification levels were low, for example, the SauJK enzyme corresponding to the second Type I RM enzyme in CC30 did not express in E. coli despite its expression in S. aureus by Monk et al. (35). The ambiguity in assignment of targets in CC93 in Monk et al. (35) is resolved because the TRDs M and b* occur in more than one HsdS in our survey.
The DNA sequences for further pairs of TRDs found in a wide range of CC and ST groups were then inserted after the hsdM of CC398-1 in our expression vector and examined to ascertain the spacer sequence in the natural system (Table 4).
Genomic DNA from S. aureus strains NCTC13435 and LGA251 was prepared and examined using SMRT sequencing as these strains contain two TRD pairs, XY and e*f* respectively, which we could not express in E. coli. While SMRT signatures for the other Type I HsdS in these strains were very clear (Supplementary Data) and in agreement with our results from E. coli (Table 4) and those of Monk et al. (35), these TRD pairs still showed no methylation activity even in their normal host. Thus, these TRDs pairs are not active.
Analysis of spacer sequence length in S. aureus Type I RM systems
It is apparent that the number of base pairs separating the adenines targeted for methylation and the number of base pairs in the non-specific spacer between the sequences recognized by the TRDs is not constant, with the former varying between 7 and 9 bp and the latter varying between 5 and 7 bp. This variation makes it very difficult to predict a Type I RM recognition sequence if one knows only the targets recognized by the two TRDs as the length of the spacer in the target is not recognized in any obvious manner by the TRDs. An example of this is the CC80-1 enzyme (Table 4) containing TRDs X and Y of known specificity. Since the enzyme did not methylate DNA in vivo for the SMRT analysis, the spacer and hence the complete target for CC80-1 remain unknown until the enzyme is purified and analyzed biochemically. While it has been observed that insertions of multiples of four amino acids into the alpha helical spacers separating the TRDs can increase the length of the spacer in the target sequence in a predictable manner (65–67), it is clear from the structure of HsdS subunits (Figure 1B) that the junction between the TRDs and the alpha helical spacers in the conserved region is going to be of crucial importance for determining the fine details of the length of the spacer in the target sequence as was found for some Type IIB RM enzymes which contain a subunit equivalent to HsdS (68). Perhaps even single amino acid insertions or deletions will serve to rotate the TRD with respect to the rest of the subunit and thereby change the length of the spacer. Further progress in understanding the correlation between amino acid sequence and the length of the target spacer would be greatly aided by an accurate atomic structure of a Type I enzyme with DNA as the current models (12,13) lack sufficient resolution to be informative on this point.
Linking TRDs pairs to further clonal complexes and sequence types
After determining the recognition sequences for all of the TRDs in Table 1 by creating artificial hybrids (Table 3) we also found that some of these TRD combinations do actually occur in natural systems as given in Table 5 (and Supplementary Data) (69). As sequence databases expand, more and more of the possible TRD combinations based on the TRDs in Table 1 will be found. As mentioned above, although the sequences recognized by the TRDs are known, the length of the non-specific spacer separating them is unknown so that the complete target cannot be specified accurately without experimentation.
Table 5. Further TRD pairs found in sequenced strains of Staphylococcus aureus.
| TRD pair | Example strain | Clonal complex or sequence type of example strain | REBASE name |
|---|---|---|---|
| AD | FDAARGOS_159 | ST5 | S.Sau159ORF12345P |
| AL | K12S0375 | ST692 | S.Sau375ORFDP |
| AU | Staphylococcus schweitzeri FSA084 | S.SauFSA084ORF355P | |
| AW | FDA209P | ST464 | S.Sau209ORF1697P |
| BG | MRSN8611 | ST8 | S.Sau8611ORF11430P |
| BH | PLAC6019 | ST5 | S.Sau6019ORF851P |
| BU | SA-083 | ST101 | S.Sau083ORF9680P |
| BY | Staphylococcus argenteus M260-MSHR | S.SarM260ORF2316P | |
| Bf* | SA-083 | ST101 | S.Sau083ORF1720P |
| JE | Tager 104 | ST49 | S.Sau104ORF1102P |
| JL | W56227 | ST45 | S.Sau56227ORF970P |
| JW | CIG290 | ST45 | S.SauCIG290ORF2408P |
| JW | APS211 | ST45 | S.SauAPS211ORF9230P |
| MW | FSA037 | ST1872 | S.SauFSA037ORF2487P |
| NQ | KPL1845 | ST96 | S.Sau1845ORF2596P |
| Of* | USA300-TCH959 | ST1159 | S.SauTCH959ORF2844P |
| Rf* | Tager 104 | ST49 | S.Sau104ORF2433P |
| TY | M21126 | ST2250 | S.Sau21126ORF1065P |
| XF | 21334 | ST109, CC9 | S.Sau21334ORF1353P |
| XF | RKI4 | ST27 | S.SauRKI4ORF1905P |
| XW | 103564 | ST80-PVL carrier | S.Sau103564ORF678P |
| ZY | D139 | ST145 | S.SauD139ORF2470P |
| b*W | ST20130941 | CC15 | S.Sau941ORF4310P |
| e*f* | SA-120 | ST425 | S.Sau120ORF4875P |
Every pair of TRD1 with TRD2 in Table 1 was used in a BLASTP sequence search to identify HsdS subunit sequences in publicly accessible databases. Examples of strains containing these TRD pairs are shown. ST and CC are from the PATRIC database (69) or derived using www.cbs.dtu.dk/services/MLST (73). Some TRD pairs are present in many strains while others are rare.
Further TRDs in S. aureus Type I RM systems
Searching the publicly available sequences in the NCBI database with individual TRD sequences revealed that some of those given in Table 1 can be found paired up with further novel TRDs. We have found four new TRDs shown in Table 6 in S. aureus strains 21343 and KPL1845. Strain 21343 contains ‘NOVEL 1’ paired with TRD K and the TRD pair NQ described in Table 3. Strain KPL1845 also contains the TRD pair NQ and two further systems comprised of ‘NOVEL 2’ paired with ‘NOVEL 3΄ and ‘NOVEL 4’ paired with TRD f*. Undoubtedly further TRDs will be found as sequencing continues.
Table 6. New TRD pairs associated with pairs shown in Tables 2, 3 and 4.
| Subspecies 21343 Bioproject accession: PRJNA53699 |
| > S.Sau21343ORF2597P TRD NOVEL 1 + TRD K |
| MSNTQKKNVPELRFPGFEGEWEEKKLGEVATFAKGKLGAKKDVSQNGVPVILYGELYTKYGAIVSKIFSKTDIPENKLKMAKKNDVLIPSSGETAIDIATASCIYLNKGVAVGGDINILTPQKQDGRFISLSING INKNELSKYAQGKTVVHLYNNDIKNLKIAFPSEFEEQVRIGNFFSKLDRQIELEEQKLELLQQQKKGYMQKIFSQELRFKDENGNDYPKWEEKKIEDIASQVYGGGTPNTKIKEFWNGDIPWIQSSDVKVNDLIL QQCNKFISKNSIELSSAKLIPANSIAIVTRVGVGKLCLVEFDYATSQDFLSLSSLKYDKLYSLYSLLYTMKKISANLQGTSIKGITKKELLDSIIKIPHNLEEQQKIGDLFYKIDKYISFNKCKIEILKSLKQGLLKKMFI |
| Species KPL1845. Bioproject accession: PRJNA169473 |
| > S.Sau1845ORF1619P TRD NOVEL 2 + NOVEL 3 |
| MTEQINTPELRFPEFKNEWSYDLVSDVVTNKSKKFDPKKEEAKKDIELDSIEQNTGRLLDTYISNDFTSQKNKFNKGNVLYSKLRPYLNKYYYATIDGVCSSEIWVLNTLNKDVLANKFLYYFIQTNRFSSVTN KSAGSKMPRADWELVKNIRLYKGSIEEQEKIGYFFSKLDRQIELEEKKLELLEQQKKGYMQKIFAQELRFKDENGNDYPDWVTKKLGDIGKVAMNKRIYKNETTENGEIPFYKIGNFGKNADTFITREKFDEYK EKYPYPNVGDILISASGSIGRTIEYTGEDAYYQDSNIVWLNHNDEVINKYLKYFYKIVKWSGIEGTTIKRLYNKNILNTKIELPTVEEQYKMANFLSKLDKIIDIQIEKIELLKQRKQGLLQKMFV |
| > S.Sau1845ORF2199P TRD NOVEL 4 + TRD f* |
| MSNTQKKNVPELRFPEFEGEWKDVKFVSIFQEVSNKTSDLAKYPLFSLTVEKGITPKTERYKRDFLVKKSDNFKIVEPRDIVYNPMNVTLGAIDLSKYNYDIALSGYYHVMKIINSFNPDFISNFLKTEKMIIHYK KIATGSLMEKQRVHFSEFKNIIKKFPTNKEQQKIGDFFSKLDRQIELQVQKLELLQQQKKGYMQKIFSQELRFKDENGEDYPDWKEKKLGDITEQSMYGIGASATRFDSKNIYIRITDIDEKSRKLNYQNLTTPDE LNNKYKLKRNDILFARTGASTGKSYIHKEEKDIYNYYFAGFLIKFEIDEQNNPLFIYQFTLTSKFNKWVKVMSVRSGQPGINSEEYAKLPLVLPNKLEQQKIAEFLDRFDQQIELEKQKIEILQQQKKGLLQSMFI |
The new TRDs of unknown specificity are termed NOVEL 1, NOVEL 2, NOVEL 3 and NOVEL 4. TRD NOVEL 3 is a second TRD while the others are first TRDs in the HsdS amino acid sequence. Subspecies 21343 and species KPL1845 also contain S.SauNQ (S.Sau21343ORF1169P and S.Sau1845ORF2596P respectively).
Improving transformation of S. aureus by avoiding targets recognized by the Sau1 Type I RM family
A general method of preparing DNA suitable for transformation of S. aureus which can overcome the RM barrier should be possible. Several DNA MTases belonging to Type II RM systems have been found which have extremely short target recognition sites, namely Hin1523, Nma1821 and Hia5 (70) and EcoGII recognizing and methylating adenine in the targets 5΄-A-3΄, 5΄-AB-3΄ or 5΄-BA-3΄. The methylation performed by these enzymes should protect any DNA molecule from the RM enzymes described here (or indeed any RM barrier relying upon adenine methylation). Thus, DNA methylated in vitro with these unusual MTases could be used in subsequent transformation experiments even when major RM barriers are present.
We used the M.EcoGII adenine MTase (a kind gift from Iain Murray, New England Biolabs) to modify all adenines in several plasmids in vitro. The plasmids were from our collection of plasmids used to determine the target sequences of the S. aureus Type I enzymes and have been previously described (31). These plasmids were then mixed with various purified S. aureus Type I restriction enzymes or, as a control, the EcoRI restriction enzyme. After one hour of methylation by M.EcoGII, the plasmids were completely resistant to digestion by EcoRI and by the S. aureus restriction enzymes (Figure 3). Furthermore, the shuttle vector pCN36 (47) was also protected from digestion by these same enzymes (data not shown). Subsequent experiments using the methylated pCN36 to transform S. aureus were unfortunately entirely unsuccessful (J. A. Lindsay, unpublished results using strains HO5096 (CC22), JE2 (CC8) and RN4220 (CC8, hsdR−). The reason for the failure of transformation with the highly-methylated pCN36 when it should be resistant to all Sau1 RM systems is not clear. This result may imply a further unrecognized barrier to transformation of S. aureus or some aspect of the physical properties of highly methylated DNA. Nevertheless, the method using MTases with very short target recognition sequences may be of use for transformation of other bacterial species.
Figure 3.

General protection from endonuclease activity using M.EcoGII MTase to methylate all adenines. Plasmid without M.EcoGII treatment is digested (− lanes) but plasmid with M.EcoGII treatment is protected from digestion (+ lanes). Panel (A) uses Sau347I (CC45-1, TRDs C and L) restriction enzyme against plasmids E2, E5 and E10 described in (31). Panel (B) uses SauNY (TRDs N and Y) against plasmids E10, E11 and E12 described in (31). Panel (C) uses three different enzymes, SauN315I (CC5-1, TRDs B and D), SauN315II (CC5-2, TRDs A and H) and SauMRSII (CC30-1, TRDs C and D), against plasmid E10. In each panel EcoRI restriction enzyme was used as a control and markers (M) are in kb.
CONCLUSIONS
In conclusion, we have determined the target recognition sequences of a considerable number of TRDs and HsdS specificity subunits of the Type I RM systems in S. aureus. This was achieved using a combination of gene synthesis, endonuclease activity, ATP hydrolysis activity and single molecule real-time genome sequencing. The systems analyzed cover a large proportion of the known sequence types and clonal complexes of S. aureus and delineate more clearly the barrier to HGT within the S. aureus population.
The data obtained here will allow the construction of new E. coli strains for preparing methylated shuttle vectors (35) and MTase reagents for in vitro methylation of DNA (40) to assist transformation of further S. aureus strains. However, these approaches are time consuming and it is worth noting that the common shuttle vector used for transformation of S. aureus, pCN36 (47), contains a target site for almost every TRD pair investigated in this paper. This means that pCN36 is inevitably a poor vector for transformation of S. aureus. The construction of new shuttle vectors completely lacking Sau1 targets via DNA synthesis, coupled with careful analysis of the fragments to be ligated into the vector so that they also lack targets, may be an effective way forward to improve transformation of S. aureus now that so many target specificities have been determined. Obviously, the avoidance of the sequence AN6-9T, although difficult to achieve without altering protein coding sequences in a vector, would be a general method to negate the effect of the Type I RM systems in S. aureus and other prokaryotes.
Lastly, the determination of so many recognition sequences of Type I RM systems in different lineages of S. aureus, in effect a ‘Rosetta Stone’, means that now the population structure of S. aureus can be investigated from an epigenetic/evolutionary perspective (4) as performed previously with, for example, Helicobacter pylori (71) and Streptococcus pneumoniae (72).
Supplementary Material
ACKNOWLEDGEMENTS
D.T.F.D. thanks the Institute of Advanced Study, Durham University for providing a fellowship from January to April 2016 and an excellent environment for writing this paper. We thank Dr Iain Murray, New England Biolabs for supplying M.EcoGII, Mark Holmes for donating strain LGA251 and Angela Kearns for donating strain NCTC13435.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Biotechnology and Biological Sciences Research Council (BBSRC) [BB/K005804/1 to D.T.F.D. ]; Wellcome Trust [GR080463MA, 090288/Z/09/ZA to D.T.F.D., J.A.L.]; Institute of Advanced Study, Durham University Fellowship (to D.T.F.D.). Funding for open access charge: Wellcome Trust; BBSRC.
Conflict of interest statement. None declared.
REFERENCES
- 1. Oliveira P.H., Touchon M., Rocha E.P.. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014; 42:10618–10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE - a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015; 43:D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blow M.J., Clark T.A., Daum C.G., Deutschbauer A.M., Fomenkov A., Fries R., Froula J., Kang D.D., Malmstrom R.R., Morgan R.D. et al. . The epigenomic landscape of prokaryotes. PLoS Genet. 2016; 12:e1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oliveira P.H., Touchon M., Rocha E.P.. Regulation of genetic flux between bacteria by restriction-modification systems. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:5658–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loenen W.A.M., Dryden D.T.F., Raleigh E.A., Wilson G.G., Murray N.E.. Highlights of the DNA cutters: a short history of the restriction enzymes. Nucleic Acids Res. 2014; 42:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Loenen W.A.M., Dryden D.T.F., Raleigh E.A., Wilson G.G.. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014; 42:20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pingoud A., Wilson G.G., Wende W.. Type II restriction endonucleases–a historical perspective and more. Nucleic Acids Res. 2014; 42:7489–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rao D.N., Dryden D.T.F., Bheemanaik S.. Type III restriction-modification enzymes: a historical perspective. Nucleic Acids Res. 2014; 42:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. King G., Murray N.E.. Restriction alleviation and modification enhancement by the Rac prophage of Escherichia coli K-12. Mol. Microbiol. 1995; 16:769–777. [DOI] [PubMed] [Google Scholar]
- 10. Murray N.E., Batten P.L., Murray K.. Restriction of bacteriophage lambda by Escherichia coli K. J. Mol. Biol. 1973; 81:395–407. [DOI] [PubMed] [Google Scholar]
- 11. Webb J.L., King G., Ternent D., Titheradge A.J.B., Murray N.E.. Restriction by EcoKI is enhanced by co-operative interactions between target sequences and is dependent on DEAD box motifs. EMBO J. 1996; 15:2003–2009. [PMC free article] [PubMed] [Google Scholar]
- 12. Kennaway C.K., Taylor J.E., Song C.F., Potrzebowski W., Nicholson W., White J.H., Swiderska A., Obarska-Kosinska A., Callow P. et al. . Structure and operation of the DNA-translocating Type I DNA restriction enzymes. Genes Dev. 2012; 26:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kennaway C.K., Obarska-Kosinska A., White J.H., Tuszynska I., Cooper L.P., Bujnicki J.M., Trinick J., Dryden D.T.F.. The structure of M.EcoKI Type I DNA methyltransferase with a DNA mimic antirestriction protein. Nucleic Acids Res. 2009; 37:762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morgan R.D., Luyten Y.A., Johnson S.A., Clough E.M., Clark T.A., Roberts R.J.. Novel m4C modification in type I restriction-modification systems. Nucleic Acids Res. 2016; 44:9413–9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loenen W.A.M., Raleigh E.A.. The other face of restriction: modification-dependent enzymes. Nucleic Acids Res. 2014; 42:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Waldron D.E., Lindsay J.A.. Sau1: a novel lineage-specific Type I Restriction-Modification system that blocks horizontal gene transfer into Staphylococcus aureus, and between S. aureus isolates of different lineages. J. Bacteriol. 2006; 188:5578–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lindsay J.A. Genomic variation and evolution of Staphylococcus aureus. Intl J. Med. Microbiol. 2010; 300:98–103. [DOI] [PubMed] [Google Scholar]
- 18. Monk I.R., Shah I.M., Xu M., Tan M.W., Foster T.J.. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. Mbio. 2012; 3, doi:10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lindsay J.A. Staphylococcus aureus genomics and the impact of horizontal gene transfer. Intl. J. Med. Microbiol. 2014; 304:103–109. [DOI] [PubMed] [Google Scholar]
- 20. Feil E.J., Cooper J.E., Grundmann H., Robinson D.A., Enright M.C., Berendt T., Peacock S.J., Smith J.M., Murphy M., Spratt B.G. et al. . How clonal is Staphylococcus aureus?. J. Bacteriol. 2003; 185:3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sung J.M., Lloyd D.H., Lindsay J.A.. Staphylococcus aureus host specificity: comparative genomics of human versus animal isolates by multi-strain microarray. Microbiol. 2008; 154:1949–1959. [DOI] [PubMed] [Google Scholar]
- 22. Monecke S., Coombs G., Shore A.C., Coleman D.C., Akpaka P., Borg M., Chow H., Ip M., Jatzwauk L., Jonas D. et al. . A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One. 2011; 6:e17936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Méric G., Miragaia M., de Been M., Yahara K., Pascoe B., Mageiros L., Mikhail J., Harris L.G., Wilkinson T.S., Rolo J. et al. . Ecological Overlap and Horizontal Gene Transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol. Evol. 2015; 7:1313–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCarthy A.J., Lindsay J.A.. Genetic variation in Staphylococcus aureus surface and immune evasion genes is lineage associated: implications for vaccine design and host-pathogen interactions. BMC Microbiol. 2010; 10:173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCarthy A.J., Lindsay J.A.. The distribution of plasmids that carry virulence and resistance genes in Staphylococcus aureus is lineage associated. BMC Microbiol. 2012; 12:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCarthy A.J., Witney A.A., Lindsay J.A.. Staphylococcus aureus temperate bacteriophage: carriage and horizontal gene transfer (HGT) is lineage associated. Front. Cell. Infect. Microbiol. 2012; 2, doi:10.3389/fcimb.2012.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCarthy A.J., Loeffler A., Witney A.A., Gould K.A., Lloyd D.H., Lindsay J.A.. Extensive horizontal gene transfer during Staphylococcus aureus co-colonization in vivo. Genome Biol. Evol. 2014; 6:2697–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Corvaglia A.R., François P., Hernandez D., Perron K., Linder P., Schrenzel J.. A Type III-like restriction endonuclease functions as a major barrier to horizontal gene transfer in clinical Staphylococcus aureus strains. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:11954–11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu S.Y., Corvaglia A.R., Chan S.H., Zheng Y., Linder P.. A type IV modification-dependent restriction enzyme SauUSI from Staphylococcus aureus subsp. aureus USA300. Nucleic Acids Res. 2011; 39:5597–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anton B.P., Mongodin E.F., Agrawal S., Fomenkov A., Byrd D.R., Roberts R.J., Raleigh E.A.. Complete genome sequence of ER2796, a DNA methyltransferase-deficient strain of Escherichia coli K-12. PLoS One. 2015; 10:e0127446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roberts G.A., Houston P.J., White J.H., Chen K., Stephanou A.S., Cooper L.P., Dryden D.T.F., Lindsay J.A.. Impact of target site distribution for Type I restriction enzymes on the evolution of methicillin-resistant Staphylococcus aureus (MRSA) populations. Nucleic Acids Res. 2013; 41:7472–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barcus V.A., Titheradge A.J.B., Murray N.E.. The diversity of alleles at the hsd locus in natural populations of Escherichia coli. Genetics. 1995; 140:1187–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Titheradge A.J.B., King J., Ryu J., Murray N.E.. Families of restriction enzymes: an analysis prompted by molecular and genetic data for type ID restriction and modification systems. Nucleic Acids Res. 2001; 29:4195–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev S.Kh., Dryden D.T.F. et al. . A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003; 31:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Monk I.R., Tree J.J., Howden B.P., Stinear T.P., Foster T.J.. Complete bypass of restriction systems for major Staphylococcus aureus lineages. Mbio. 2015; 6:e00308–e00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen K., Stephanou A.S., Roberts G.A., White J.H., Cooper L.P., Houston P.J., Lindsay J.A., Dryden D.T.F.. The Type I restriction enzymes as barriers to horizontal gene transfer: determination of the DNA target sequences recognised by livestock-associated methicillin-resistant Staphylococcus aureus clonal complexes 133/ST771 and 398. Adv. Exp. Med. Biol. 2016; 915:81–97. [DOI] [PubMed] [Google Scholar]
- 37. Abadjieva A., Patel J., Webb M., Zinkevich V., Firman K.. A deletion mutant of the type IC restriction endonuclease EcoR1241 expressing a novel DNA specificity. Nucleic Acids Res. 1993; 21:4435–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meister J., MacWilliams M., Hübner P., Jütte H., Skrzypek E., Piekarowicz A., Bickle T.A.. Macroevolution by transposition: drastic modification of DNA recognition by a type I restriction enzyme following Tn5 transposition. EMBO J. 1993; 12:4585–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. MacWilliams M.P., Bickle T.A.. Generation of new DNA binding specificity by truncation of the type IC EcoDXXI hsdS gene. EMBO J. 1996; 15:4775–4783. [PMC free article] [PubMed] [Google Scholar]
- 40. Jones M.J., Donegan N.P., Mikheyeva I.V., Cheung A.L.. Improving transformation of Staphylococcus aureus belonging to the CC1, CC5 and CC8 clonal complexes. PLoS One. 2015; 10:e0119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Furuta Y., Kawai M., Uchiyama I., Kobayashi I.. Domain movement within a gene: a novel evolutionary mechanism for protein diversification. PLoS One. 2011; 6:e18819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dybvig K., Sitaraman R., French C.T.. A family of phase-variable restriction enzymes with differing specificities generated by high-frequency gene rearrangements. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:13923–13928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiao L., Ptacek T., Osborne J.D., Crabb D.M., Simmons W.L., Lefkowitz E.J., Waites K.B., Atkinson T.P., Dybvig K.. Comparative genome analysis of Mycoplasma pneumoniae. BMC Genomics. 2015; 16, doi:10.1186/s12864-015-1801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li J., Li J.W., Feng Z., Wang J., An H., Liu Y., Wang Y., Wang K., Zhang X., Miao Z. et al. . Epigenetic switch driven by DNA inversions dictates phase variation in Streptococcus pneumoniae. PLoS Pathog. 2016; 12:e1005762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Willemse N., Schultsz C.. Distribution of Type I restriction–modification systems in Streptococcus suis: an outlook. Pathogens. 2016; 5, doi:10.3390/pathogens5040062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cerdeño-Tárraga A.M., Patrick S., Crossman L.C., Blakely G., Abratt V., Lennard N., Poxton I., Duerden B., Harris B., Quail M.A. et al. . Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science. 2005; 307:1463–1465. [DOI] [PubMed] [Google Scholar]
- 47. Charpentier E., Anton A.I., Barry P., Alfonso B., Fang Y., Novick R.P.. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 70:6076–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thorpe P.H., Ternent D., Murray N.E.. The specificity of sty SKI, a type I restriction enzyme, implies a structure with rotational symmetry. Nucleic Acids Res. 1997; 25:1694–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sturrock S.S., Dryden D.T.F.. A prediction of the amino acids and structures involved in DNA recognition by type I DNA restriction and modification enzymes. Nucleic Acids Res. 1997; 25:3408–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pei J., Kim B.H., Tang M., Grishin N.V.. PROMALS web server for accurate multiple protein sequence alignments. Nucleic Acids Res. 2007; 35:W649–W652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morgan R.D., Luyten Y.A.. Rational engineering of type II restriction endonuclease DNA binding and cleavage specificity. Nucleic Acids Res. 2009; 37:5222–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Callahan S.J., Luyten Y.A., Gupta Y.K., Wilson G.G., Roberts R.J., Morgan R.D., Aggarwal A.K.. Structure of Type IIL restriction-modification enzyme MmeI in complex with DNA has implications for engineering new specificities. PLoS Biol. 2016; 14:e1002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baba T., Takeuchi F., Kuroda M., Yuzawa H., Aoki K., Oguchi A., Nagai Y., Iwama N., Asano K., Naimi T. et al. . Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002; 359:1819–1827. [DOI] [PubMed] [Google Scholar]
- 54. Kuroda M., Ohta T., Uchiyama I., Baba T., Yuzawa H., Kobayashi I., Cui L., Oguchi A., Aoki K., Nagai Y. et al. . Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet. 2001; 357:1225–1240. [DOI] [PubMed] [Google Scholar]
- 55. Holden M.T., Feil E.J., Lindsay J.A., Peacock S.J., Day N.P., Enright M.C., Foster T.J., Moore C.E., Hurst L., Atkin R. et al. . Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:9786–9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chua K., Seemann T., Harrison P.F., Davies J.K., Coutts S.J., Chen H., Haring V., Moore R., Howden B.P., Stinear T.P.. Complete genome sequence of Staphylococcus aureus strain JKD6159, a unique Australian clone of ST93-IV community methicillin-resistant Staphylococcus aureus. J. Bacteriol. 2010; 192:5556–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guinane C.M., Ben Zakour N.L., Tormo-Mas M.A., Weinert L.A., Lowder B.V., Cartwright R.A., Smyth D.S., Smyth C.J., Lindsay J.A., Gould K.A. et al. . Evolutionary genomics of Staphylococcus aureus reveals insights into the origin and molecular basis of ruminant host adaptation. Genome Biol. Evol. 2010; 2:454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sung J.M., Lloyd D.H., Lindsay J.A.. Staphylococcus aureus host specificity: comparative genomics of human versus animal isolates by multi-strain microarray. Microbiology. 2008; 154:1949–1959. [DOI] [PubMed] [Google Scholar]
- 59. Schijffelen M.J., Boel C.H., van Strijp J.A., Fluit A.C.. Whole genome analysis of a livestock-associated methicillin-resistant Staphylococcus aureus ST398 isolate from a case of human endocarditis. BMC Genomics. 2010; 11:376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Holden M.T., Hsu L.Y., Kurt K., Weinert L.A., Mather A.E., Harris S.R., Strommenger B., Layer F., Witte W., de Lencastre H. et al. . A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 2013; 23:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Garcia-Alvarez L., Holden M.T., Lindsay H., Webb C.R., Brown D.F., Curran M.D., Walpole E., Brooks K., Pickard D.J., Teale C. et al. . Meticillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect. Dis. 2011; 11:595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen C.J., Unger C., Hoffmann W., Lindsay J.A., Huang Y.C., Götz F.. Characterization and comparison of 2 distinct epidemic community-associated methicillin-resistant Staphylococcus aureus clones of ST59 lineage. PLoS One. 2013; 8:e63210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Y., Chatterjee S.S., Porcella S.F., Yu Y.S., Otto M.. Complete genome sequence of a Pantón-Valentine leukocidin-negative community-associated methicillin-resistant Staphylococcus aureus strain of sequence type 72 from Korea. PLoS One. 2013; 8:e72803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Holt D.C., Holden M.T., Tong S.Y., Castillo-Ramirez S., Clarke L., Quail M.A., Currie B.J., Parkhill J., Bentley S.D., Feil E.J. et al. . A very early-branching Staphylococcus aureus lineage lacking the carotenoid pigment staphyloxanthin. Genome Biol. Evol. 2011; 3:881–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Price C., Lingner J., Bickle T.A., Firman K., Glover S.W.. Basis for changes in DNA recognition by the EcoR124 and EcoR124/3 type I DNA restriction and modification enzymes. J. Mol. Biol. 1989; 205:115–125. [DOI] [PubMed] [Google Scholar]
- 66. Gubler M., Braguglia D., Meyer J., Piekarowicz A., Bickle T.A.. Recombination of constant and variable modules alters DNA sequence recognition by type IC restriction-modification enzymes. EMBO J. 1992; 11:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Adamczyk-Popławska M., Kondrzycka A., Urbanek K., Piekarowicz A.. Tetra-amino-acid tandem repeats are involved in HsdS complementation in type IC restriction-modification systems. Microbiology. 2003; 149:3311–3319. [DOI] [PubMed] [Google Scholar]
- 68. Jurenaite-Urbanaviciene S., Serksnaite J., Kriukiene E., Giedriene J., Venclovas C., Lubys A.. Generation of DNA cleavage specificities of type II restriction endonucleases by reassortment of target recognition domains. Proc. Natl. Acad. Sci. U.S.A.. 2007; 104:10358–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wattam A.R., Abraham D., Dalay O., Disz T.L., Driscoll T., Gabbard J.L., Gillespie J.J., Gough R., Hix D., Kenyon R. et al. . PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014; 42:D581–D591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Drozdz M., Piekarowicz A., Bujnicki J.M., Radlinska M.. Novel non-specific DNA adenine methyltransferases. Nucleic Acids Res. 2012; 40:2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kojima K.K., Furuta Y., Yahara K., Fukuyo M., Shiwa Y., Nishiumi S., Yoshida M., Azuma T., Yoshikawa H., Kobayashi I.. Population evolution of Helicobacter pylori through diversification in DNA methylation and interstrain sequence homogenization. Mol. Biol. Evol. 2016; 33:2848–2859. [DOI] [PubMed] [Google Scholar]
- 72. Croucher N.J., Coupland P.G., Stevenson A.E., Callendrello A., Bentley S.D., Hanage W.P.. Diversification of bacterial genome content through distinct mechanisms over different timescales. Nat. Commun. 2014; 5, doi:10.1038/ncomms6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Larsen M.V., Cosentino S., Rasmusen S., Hasman H., Marvig R.L., Jelsbak L., Sicheritz-Ponten T., Ussery D.W., Aarestrup F.M., Lund O.. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012; 50:1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.