

Abstract
Begomoviruses (single-stranded DNA plant viruses) are responsible for serious agricultural threats. Begomovirus populations exhibit a high degree of within-host genetic variation and evolve as quickly as RNA viruses. Although the recombination-prone nature of begomoviruses has been extensively demonstrated, the relative contribution of recombination and mutation to the genetic variation of begomovirus populations has not been assessed. We estimated the genetic variability of begomovirus datasets from around the world. An uneven distribution of genetic variation across the length of the cp and rep genes due to recombination was evident from our analyses. To estimate the relative contributions of recombination and mutation to the genetic variability of begomoviruses, we mapped all substitutions over maximum likelihood trees and counted the number of substitutions on branches which were associated with recombination (ηr) and mutation (ημ). In addition, we also estimated the per generation relative rates of both evolutionary mechanisms (r/μ) to express how frequently begomovirus genomes are affected by recombination relative to mutation. We observed that the composition of genetic variation in all begomovirus datasets was dominated by mutation. Additionally, the low correlation between the estimates indicated that the relative contributions of recombination and mutation are not necessarily a function of their relative rates. Our results show that, although a considerable fraction of the genetic variation levels could be assigned to recombination, it was always lower than that due to mutation, indicating that the diversification of begomovirus populations is predominantly driven by mutational dynamics.
Keywords: evolution, geminivirus, genetic variability, phylogeny.
1. Introduction
Genetic variation allows the adaptation of populations to a changing environment. Many viral populations exist as complexes of closely related genomic variants as a result of high mutation rates, rapid replicative kinetics and large population sizes (Biebricher and Eigen 2006). The high mutation rates of RNA viruses are a consequence of their error-prone RNA-dependent RNA polymerases (RdRps) (Holland et al. 1982). It has been suggested that, relative to DNA viruses which replicate using proof-reading DNA-dependent DNA polymerases (DdDps), RNA viruses may explore their associated sequence spaces (all combinations of mutant sequences) more thoroughly and more rapidly (Eigen et al. 1988; Worobey and Holmes 1999). Consequently, it is likely that populations of RNA viruses possess greater adaptive capacities than those of DNA viruses, a factor which strongly impacts the formulation of strategies to control viral diseases (Gerrish and Garcia-Lerma 2003).
Nevertheless, a number of studies have shown that single-stranded (ss) DNA viruses may evolve as quickly as RNA viruses (Drake 1991; Shackelton et al. 2005; Shackelton and Holmes 2006; Duffy et al. 2008). Single-stranded DNA plant viruses of the families Geminiviridae and Nanoviridae exhibit high levels of within-host genetic variation (Ge et al. 2007; van der Walt et al. 2008; Grigoras et al. 2010), and substitution rates inferred for begomoviruses (whitefly-transmitted geminiviruses) are similar to those of RNA viruses (Duffy and Holmes 2008, 2009). It has been proposed that these viruses may replicate using low-fidelity DNA polymerases, and/or that spontaneous biochemical reactions which act preferentially on single-stranded nucleic acids (deamination, oxidation and methylation of bases) might contribute to their genetic variability (Duffy et al. 2008; van der Walt et al. 2008; Duffy and Holmes 2009). Although mutational dynamics is a primary factor in the diversification of viral populations (Roossinck 1997; García-Arenal et al. 2003; Balol et al. 2010), it does not account for all the standing genetic variation, since other evolutionary mechanisms including recombination might contribute significantly (Martin et al. 2011).
Recombination impacts the evolution of several families of viruses (Bonnet et al. 2005; Heath et al. 2006; Varsani et al. 2006; Fan et al. 2007; Martin et al. 2011) and has been extensively documented for geminiviruses [(Briddon et al. 1996; Padidam et al. 1999; Pita et al. 2001; García-Andrés et al. 2007); reviewed by Lefeuvre and Moriones (2015)]. The devastating mastrevirus Maize streak virus (MSV) in Africa, and begomoviruses associated with important disease complexes in Europe, Asia and Africa (tomato yellow leaf curl, cotton leaf curl and cassava mosaic diseases, respectively) seem to have evolved largely by recombination (Sanz et al. 2000; Pita et al. 2001; García-Andrés et al. 2007; Varsani et al. 2008). Although the mechanics of recombination in ssDNA viruses remains unknown, it has been proposed that their high recombination frequency may be a result of a recombination-dependent replication mechanism (Jeske et al. 2001).
While the impact of recombination in geminivirus populations is evident, most studies have focused on detecting recombination without determining the relative contribution of recombination and mutation to their genetic variation (Zhou et al. 1997; Padidam et al. 1999; Berrie et al. 2001; Pita et al. 2001; Monci et al. 2002; Silva et al. 2014). By applying a novel phylogeny-based partitioning method of genetic variability (Lima et al. 2013) and standard genetic population analysis tools to datasets of begomoviruses from around the world (available in public databases), we were able to estimate the minimal relative contribution of recombination to begomovirus evolutionary dynamics. Our results indicate that mutations are the main source of variation for most begomovirus populations. We further show that the rate of recombination poorly predicts the contribution of recombination to genetic variability.
2. Materials and Methods
2.1 Sequence Datasets
Fifteen datasets comprised of full-length DNA-A (or DNA-A-like) sequences of begomoviruses (a total of 887 sequences from 15 viral species; Supplementary Table S1) were downloaded from the GenBank database using Taxonomy Browser (www.ncbi.nlm.nih.gov) on July 2012. Only begomovirus species for which at least 20 sequences were available in GenBank were used. All genome sequences were organized to begin at the nicking site in the invariant nonanucleotide at the origin of replication (5′-TAATATT//AC-3′).
2.2 Multiple Sequence Alignments and Phylogenetic Analysis
Multiple sequence alignments for two viral genes (cp and rep) and full-length genome nucleotide (nt) sequences were constructed using Muscle (Edgar 2004) and manually corrected in Mega 5.0 (Tamura et al. 2011). The cp and rep genes were chosen due to their essential roles in virus transmission and replication, respectively. In addition, their combined lengths exceed 70% of the DNA-A components. Maximum likelihood (ML) trees were searched in PAUP* v. 4 (Swofford 2003) using the tree-bisection-reconnection algorithm under the best fit nucleotide substitution model determined in Modeltest (Posada and Crandall 1998) by the Akaike Information Criterion (AIC) (Supplementary Table S2). The support for each individual branch was assessed from 2,000 nearest neighbor interchange bootstrap replications. Trees were visualized and edited using FigTree (tree.bio.ed.ac.uk/software/figtree).
2.3 Nucleotide and Haplotype Diversity Indexes
The average pairwise number of nucleotide differences per site [nucleotide diversity, π; (Nei 1987)] was calculated using a script written in Python programming language (available from the authors upon request). The statistical significance of the differences amongst the mean π obtained from different data sets was assessed by estimating their 95% bootstrap confidence intervals from 15,000 nonparametric simulations in R software (R Development Core Team 2007) using the Simpleboot statistical package (Peng 2008). A nonparametric bootstrap resampling was chosen as it makes no assumptions concerning the distribution or sample size of the data (Young 1994; Carpenter and Bithell 2000). In addition, it represented the most practical approach to assess the statistical significance of the large number of datasets analyzed in this study. The nucleotide diversity at synonymous and nonsynonymous sites (defined as those sites in which synonymous and nonsynonymous substitutions, respectively, occurred, independently of them being at the first, second or third codon positions) and haplotype diversity indexes were calculated in DnaSP v.5 (Rozas et al. 2003).
2.4 Recombination Analysis
Full-length genome sequences of begomoviruses were scanned for recombination using the RDP, Geneconv, Bootscan, Maximum Chi Square, Chimaera, Sister Scan and 3Seq methods implemented in RDP version 4.51 (Martin et al. 2011) (RDP project files are available from the authors upon request). Statistical significance was inferred by P values lower than a Bonferroni-corrected α = 0.05 cutoff. Only recombination events detected by at least four of the analysis methods available in the program were considered reliable.
2.5 Detection of Positive and Negative Selection at Amino Acid Sites
Negatively and positively selected sites in the cp and rep datasets were identified using three distinct methods: Single Likelihood Ancestor Counting (SLAC), Partitioning for Robust Inference of Selection (PARRIS) and Fast Unconstrained Bayesian AppRoximation (FUBAR) implemented in DataMonkey (www.datamonkey.org) (Kosakovsky-Pond and Frost 2005; Scheffler et al. 2006; Murrell et al. 2013). All methods were applied using nucleotide substitution models determined to be the most appropriate for each dataset in the Datamonkey webserver. To avoid misleading selection results, we searched for recombination breakpoints in each dataset using GARD. dN/dS ratios (ω) for the cp and rep genes from all begomovirus datasets were also estimated using the SLAC method based on the inferred GARD-corrected phylogenetic trees.
2.6 Branch Length Frequencies in ML Phylogenetic Trees
The branch length information was extracted from cp and rep ML phylogenetic tree files using a custom written PERL script, BLExtractor. The branch length frequencies for each ML tree were calculated using a second customized PERL script, BLFrequency (both PERL scripts are available from the authors upon request). The statistical significance of the differences among the lengths of branches associated with recombination and mutation was also assessed by estimating their 95% bootstrap confidence intervals from 15,000 nonparametric simulations in R software (R Development Core Team 2007) using the Simpleboot statistical package (Peng 2008).
2.7 Relative Contribution of Recombination and Mutation to the Standing Genetic Variability of Begomovirus Populations
The relative contribution of recombination and mutation (ηr/ημ) to the standing genetic variability of the cp and rep genes was determined using a phylogeny-based partitioning method as previously described (Lima et al. 2013). To avoid overestimating the role of recombination due to putative sampling biases (e.g., situations in which several isolates descended from the same recombinant ancestor are sampled from a small area), nucleotide polymorphisms were computed from unique recombination events instead of ‘individual’ events. As an alternative approach, the population-scaled recombination rate (ρ = 2Ner) and Watterson’s infinite-sites estimator of the population-scaled mutation rate (θ = 2Neμ) were determined using the programs PAIRWISE and CONVERT, respectively, available in the LDhat package (McVean et al. 2002). A minimum minor allele frequency (MAF) cutoff of 0.01 was employed for all species datasets, except for those harboring large sample sizes (EACMV, N = 156 and TYLCV, N = 222) where a MAF of 0.05 was adjusted. In addition, for all species datasets analyzed in this study, a gene-conversion model was fitted. Precomputed likelihood lookup tables for per site mutation rates of 0.001 and 0.01, a maximum 2Ner of 100 and a 101-point grid were used for the ρ estimation of begomovirus datasets. The per-generation relative rates of recombination and mutation (r/μ) in the history of the samples was estimated as the ratio between the population-scaled recombination (ρ) and mutation (θ) rates. The Pearson correlation between the estimates from both approaches (relative contribution vs. relative rates) was assessed using the R package (R Development Core Team 2007).
3. Results
3.1 Genetic Variability in Begomovirus Datasets
In order to rigorously compare the standing genetic variability of the 15 begomovirus data sets, we applied a novel approach in which 95% bootstrap confidence intervals were estimated for the differences between nucleotide diversities (Fig. 1). This approach proved useful since it made no assumptions concerning the distribution or sample size of the data. In fact, although the sample sizes in most begomovirus datasets were similar (between 34 and 53 sequences; Supplementary Table S3), we were able to compare the genetic variability even between data sets with discrepant sample sizes (e.g., AgEV and ToYLCCNV, N = 21 and 26, respectively, and EACMV and TYLCV, N = 156 and 222, respectively) (full species names are provided in Table 1). Interestingly, the average pairwise number of nucleotide differences for the AgEV dataset (π = 0.04172) was statistically very similar to that from other datasets harboring larger sample sizes, for example, CLCuGV (N = 39, π = 0.04290) and TYLCV (N = 222, π = 0.04049) (Fig. 1 and Supplementary Table S3). Conversely, the genetic variation estimated for the EACMV and TYLCV datasets (π = 0.05672 and 0.04049; respectively) were lower than those from datasets harboring smaller sample sizes, for example, BhYVMV (N = 40, π = 0.06502) (Fig. 1 and Supplementary Table S3). The genetic variation estimated for the ToYLCCNV dataset (the second smallest dataset) was the highest amongst all begomovirus data sets analyzed in this study (π = 0.10414) (Fig. 1 and Supplementary Table S3). The haplotype diversity indexes calculated for all 15 begomovirus datasets were similar and close to 1, indicating that most isolates were unique within each dataset (Supplementary Table S3).
Figure 1.

Statistical significance of the differences amongst the average pairwise number of nucleotide differences per site (nucleotide diversity, π) calculated for full-length genomes of begomoviruses. Ninety-five percent bootstrap confidence intervals (CIs) for the difference between π values were estimated from 15,000 nonparametric simulations in R software (R Development Core Team 2007) using the Simpleboot statistical package (Peng 2008). Confidence intervals which include the value ‘zero’ denote no statistically significant difference between the means.
Table 1.
Mean dN/dS values estimated for the cp and rep genes of begomovirus datasets retrieved from the GenBank database.
| Species dataset | Acronym | cp | rep |
|---|---|---|---|
| African cassava mosaic virus | ACMV | 0.216 | 0.218 |
| Ageratum enation virus | AgEV | 0.068 | 0.291 |
| Bhendi yellow vein India virus | BhYVIV | 0.164 | 0.274 |
| Bhendi yellow vein mosaic virus | BhYVMV | 0.133 | 0.228 |
| Cotton leaf curl Burewala virus | CLCuBuV | 0.684 | 0.615 |
| Cotton leaf curl Gezira virus | CLCuGV | 0.059 | 0.178 |
| Cotton leaf curl Multan virus | CLCuMV | 0.124 | 0.312 |
| East African cassava mosaic virus | EACMV | 0.136 | 0.191 |
| Mungbean yellow mosaic India virus | MYMIV | 0.141 | 0.209 |
| Sweet potato leaf curl virus | SPLCV | 0.073 | 0.186 |
| Tomato leaf curl new Delhi virus | ToLCNDV | 0.093 | 0.172 |
| Tomato leaf curl Taiwan virus | ToLCTV | 0.144 | 0.229 |
| Tomato yellow leaf curl Thailand virus | TYLCTV | 0.102 | 0.260 |
| Tomato yellow leaf curl China virus | ToYLCCNV | 0.103 | 0.236 |
| Tomato yellow leaf curl virus | TYLCV | 0.198 | 0.270 |
3.2 Genetic Variation across Begomovirus Genomes/Genes Is Not Evenly Distributed
We also calculated nucleotide diversities for the cp and rep datasets, to assess the individual contribution of these genes to the standing genetic variation in the full-length DNA-A (or DNA-A-like sequences) (Fig. 2). Statistically significant differences between the diversities of the cp and rep genes were observed in 14 out of 15 datasets. The rep gene was more diverse than the cp gene in 11 out of the 15 datasets (ACMV, AgEV, BhYVIV, BhYVMV, CLCuBuV, CLCuMV, SPLCV, ToLCNDV, ToLCTV, TYLCTV and TYLCV; Fig. 2). In contrast, the CLCuGV, EACMV and MYMIV cp genes were more diverse than their cognate rep genes (Fig. 2). Although the differences between the nucleotide diversity indexes calculated for the cp and rep genes of the ACMV, AgEV, CLCuBuV and MYMIV datasets were statistically significant, they were very low, suggesting an approximately even distribution of the genetic variability. The ToYLCCNV dataset was the only one for which the difference between the nucleotide diversity indexes calculated for the two genes was not statistically significant.
Figure 2.
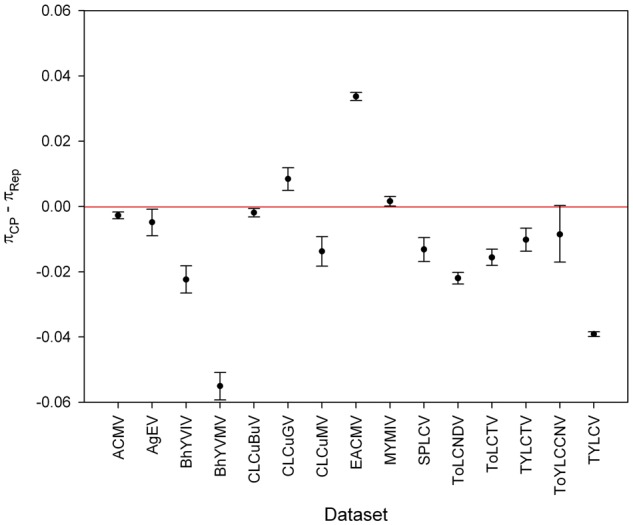
Statistical significance of the differences amongst the average pairwise number of nucleotide differences per site (nucleotide diversity, π) calculated for the cp and rep genes of begomoviruses. Ninety-five percent bootstrap confidence intervals (CIs) for the difference between π values (πcp–πrep) were estimated from 15,000 nonparametric simulations in R software (R Development Core Team 2007) using the Simpleboot statistical package (Peng 2008). Confidence intervals which include the value ‘zero’ denote no statistically significant difference between the means.
The uneven distribution of genetic variation between these two genes prompted us to examine intragenic genetic variation. We divided each gene into three regions: 5′-terminal, central and 3′-terminal. Our analysis yielded statistical support for increased levels of genetic variation at the central portion and 3′-terminal regions of the cp gene of most begomovirus datasets, with fewer examples of increased diversity at the 5′-terminal region (Fig. 3). Similar levels of genetic variation were observed between the 5′-terminal and the central or 3′-terminal regions of the cp gene of CLCuMV, ToLCNDV and TYLCTV (Fig. 3). We also observed an uneven distribution of genetic variation levels across the length of the rep gene in most datasets (Fig. 3). Statistically significantly increased levels of genetic variation were readily observed at the 5′-terminal region of the rep gene and/or at the central region in most begomovirus datasets. The single exception was TYLCTV, whose differences amongst the nucleotide diversities calculated in its three regions were not statistically significant (Fig. 3). In contrast to the similar nucleotide diversity calculated for both ToYLCCNV genes (Fig. 2), both genes had a significantly uneven distribution of intragenic variation (Fig. 3).
Figure 3.

Statistical significance of the differences amongst the average pairwise number of nucleotide differences per site (nucleotide diversity, π) calculated for the 5′-terminal, central and 3′-terminal regions of the cp and rep genes of begomoviruses. The whole sequences of the cp and rep genes were partitioned into three segments of identical lengths and the π values were calculated using an algorithm written in Python programming language. Ninety-five percent bootstrap confidence intervals (CIs) for the difference between π values were estimated from 15,000 nonparametric simulations in R software (R Development Core Team 2007) using the Simpleboot statistical package (Peng 2008). Confidence intervals which include the value ‘zero’ denote no statistically significant difference between the means.
3.3 Synonymous Site Variation in the Cp and Rep Genes
We assessed the putative role of positive and negative selection in shaping the uneven distribution of genetic variation levels across the cp and rep genes. The dN/dS ratio (ω) estimated for each gene in all begomovirus datasets were <1, signifying predominant negative selection (Table 1). However, there was wide variation amongst genes/datasets (from 0.059 to 0.684 for the CLCuGV and CLCuBuV cp datasets, respectively), indicating distinct selective constraints in different datasets. The cp gene was under stronger negative selection in most begomovirus data sets. The single exception was the CLCuBuV dataset, in which the ω value for the cp was similar to that of the rep gene (0.684 and 0.615, respectively). Both genes in CLCuBUV have relatively high ω values, which suggests that this virus is evolving under more relaxed negative selection.
Very few sites showed evidence of positive selection, in both genes. SLAC detected positive selection on one site in each BhYVMV and ToLCNDV cp gene (codon positions 146 and 2, respectively; Supplementary Table S4). Positive selection was also detected on one site in the rep gene of MYMIV and SPLCV (codon positions 181, 112, respectively), and on two sites in ToLCNDV rep (codon positions 40 and 352) (Supplementary Table S4). All sites under positive selection detected by SLAC were also detected by FUBAR, however, the latter detected additional sites under positive selection in both genes. No positively selected sites were detected by PARRIS in any dataset. As a general perception, the vast majority of sites in both genes showed evidence of negative selection. As expected, SLAC and FUBAR detected a larger number of sites with statistical evidence of negative selection from most variable datasets, (e.g., BhYVIV, BhYVMV, SPLCV, ToLCNDV and ToYLCCNV, with 42.6%, 38.3%, 39.5%, 56.6% and 57.4% of the sites under negative selection in the cp gene and 29.9%, 41.3%, 45.0%, 51.1% and 39.4% in the rep gene; Supplementary Table S4). Taken together, the number and distribution of sites under positive selection detected by SLAC and/or FUBAR do not consistently explain the discrepant degrees of genetic variability across the cp and rep genes.
We then analyzed the distribution of the genetic variation levels at synonymous and nonsynonymous sites across the length of both genes. While there were few sites showing nonsynonymous variation in the two genes, regardless of regions (5′-terminal, central and 3′-terminal), variation at synonymous sites explained the uneven intragenic distribution of genetic variation (Fig. 4). These results indicate that the most variable regions in the cp and rep genes are the consequence of higher local synonymous site variation. In addition, we concluded that adaptive selection does not make a detectable contribution to the high genetic variation levels in the cp gene central/3′-terminal and rep gene 5′-terminal/central regions.
Figure 4.

Average pairwise number of nucleotide differences per site (nucleotide diversity, π) at synonymous (a, c) and nonsynonymous (b, d) sites calculated for three distinct regions (5′-terminal, central and 3′-terminal) of the cp (a, b) and rep (c, d) genes of begomoviruses using DnaSP v.5.
3.4 The Mosaic Structure of Begomovirus Genomes
Well-supported recombination events (detected by at least four methods in RDP) were detected in 14 of the 15 datasets (Supplementary Table S5), which was expected considering the notorious recombination-prone nature of begomovirus genomes. No reliable recombination events were detected in the ACMV dataset, while 26 unique events were detected in the BhYVMV dataset. A high number of recombination events was also detected in the SPLCV and BhYVIV datasets (15 and 16 unique events, respectively). As expected, there was a low correlation between the number of unique recombination events and the standing genetic variation estimated for the full-length DNA-A (r = 0.327280), underscoring the dependence of the effect of each recombination event on the genetic distance between the parental viruses. For example, five unique recombination events were detected in both the highly genetically diverse ToYLCCNV (the most variable begomovirus dataset; π = 0.10414, N = 26; Supplementary Table S3) and the much less diverse AgEV (π = 0.04172, N = 21).
A nonrandom distribution of recombination breakpoints was observed across begomovirus genomes/genes, with most events showing at least one breakpoint within the rep gene (especially at the 5′/central regions) and less frequently in the cp gene (at the central/3′ regions; Supplementary Table S5). Interestingly, breakpoint distribution was associated with an uneven distribution of the genetic variation levels across viral genomes/genes. Together, these results indicate that the uneven distribution of genetic variation levels across genomes/genes is a consequence of high synonymous site variation due to recombination. However, the opposite is not necessarily true, that is, the even distribution of variability levels across genomes does not indicate the absence of recombination events. For instance, both ToYLCCNV genes were equally diverse (Fig. 2), but four out of the five unique recombination events detected involved the rep gene, and only one involved the cp gene (Supplementary Table S5).
3.5 Long Branches in the Begomovirus Phylogeny Are Associated with Recombination Events
Isolates sharing well-supported recombination events tended to form clades in maximum likelihood (ML) phylogenetic trees based on both the cp or rep genes. In addition, these clades were frequently connected to others by long branches, whose associated nucleotide polymorphisms delimited by the breakpoints were inherited en masse during the ancestral recombination event. For example, the longest branch in the EACMV cp tree (representing 0.2604 substitutions/site) was clearly associated with a well-supported recombination event shared by 53 isolates [Fig. 5a andSupplementary Table S5 (event 1) and Supplementary Figure S1h]. Long branches were also associated with recombination events involving the rep gene. In the EACMV rep tree, the longest branch (0.0477 subs/site) leading to a clade composed of eight isolates was associated with a recombination event with a P value of 8.02 × 10 − 9 [Fig. 5b andSupplementary Table S5 (event 3) and Supplementary Figure S1w].
Figure 5.

Midpoint-rooted maximum likelihood trees based on the EACMV cp (a) and rep (b) gene nucleotide sequences. Nodes to the right of branches with bootstrap support ≥90% are indicated by filled circles, and those with support between 70% and 89% by empty circles. The unique recombination events detected within the cp (event 1) and rep sequences (events 2 and 3) are shown as diagrams close to the branches where the substitutions due to each recombination event were mapped. Branches in red, blue and green colors were assigned to events 1, 2 and 3, respectively.
Single recombinant sequences also tended to be connected to the ML tree by long branches. For example, the longest branches in the BhYVMV and CLCuBuV cp trees were associated with recombination events detected in single sequences (Supplementary Figure S2d and e, respectively). Overall, the longest branches of 8 out of the 15 cp trees were associated with well-supported recombination events (Supplementary Figure S1b, d-h, j and m). A similar pattern was observed in 10 out of 15 rep trees, in which the longest branches were associated with well-supported recombination events (Supplementary Figure S1r-u, y-ad).
We assessed the statistical significance of the differences between mean lengths of branches associated with recombination and mutation in the ML phylogenetic trees (except for ACMV cp and rep trees, whose branches were all associated with mutation). We observed significant differences in 5 out of 14 cp trees (CLCuBuV, EACMV, ToLCNDV, TYLCTV and TYLCV; Fig. 6a) and in 8 out of 14 rep trees (BYVIV, BYVMV, CLCuBuV, CLCuGV, EACMV, SPLCV, TYLCTV and TYLCV; Fig. 6b). It is important to note that in most trees where the differences were not statistically significant, the longest branches were associated with well supported recombination events, however, short branches were also associated with recombination. As a consequence, the mean length of branches associated with recombination was comparatively lower. While there are several reasons why long branches can occur in phylogenetic trees, our results strongly support that recombination events are associated with long branches in ML trees based on the cp and rep genes of begomoviruses.
Figure 6.
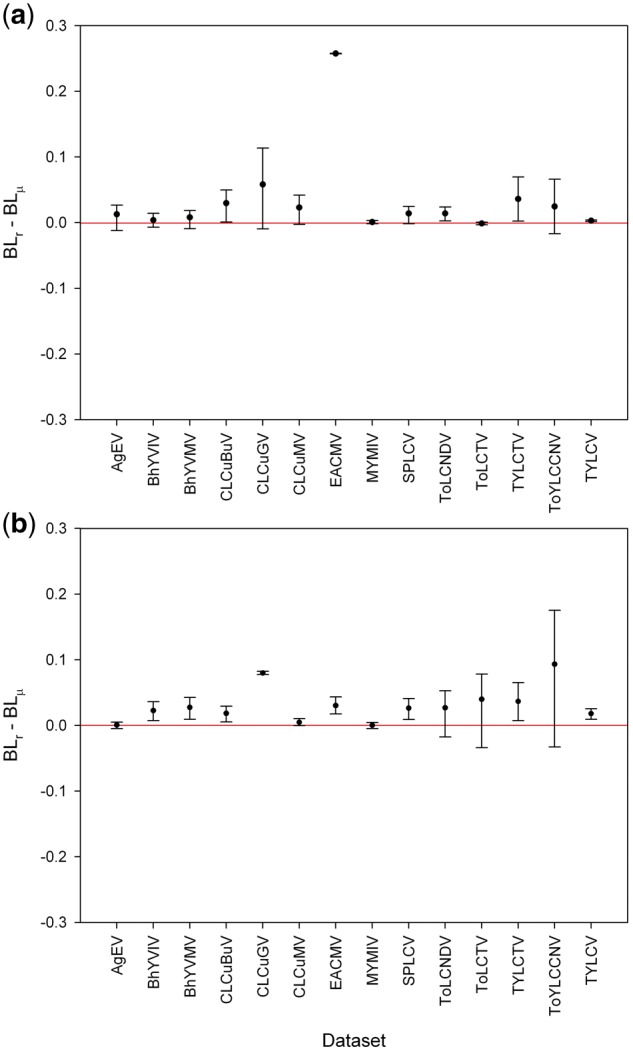
Statistical significance of the differences between branch lengths associated with recombination (BLr) and mutation (BLμ) in ML phylogenetic trees for the cp (a) and rep (b) genes. Ninety-five percent bootstrap confidence intervals (CIs) for the difference between branch lengths were estimated from 15,000 nonparametric simulations in R software (R Development Core Team 2007) using the Simpleboot statistical package (Peng 2008). Confidence intervals which include the value ‘zero’ denote no statistically significant difference between the means.
3.6 Relative Contribution of Mutation and Recombination to Begomovirus Variability
We mapped all substitutions over the ML trees constructed for the cp and rep genes and counted the number of substitutions on branches which were associated with recombination and the number which were associated with mutation, to estimate the relative contributions of these processes to the standing genetic variation in each begomovirus dataset. In addition, we also estimated the per generation relative rates of both evolutionary mechanisms (r/μ; equivalent to ρ/θ) as the ratio between the population-scaled recombination (ρ = 2Ner) and mutation rates (θ = 2Neμ), to express how frequently these sequences are targeted by recombination relative to mutation. In this latter approach, likelihood lookup tables for per site mutation rates of 0.001 and 0.01 were used, both yielding very similar estimates of the population-scaled recombination rates (Table 2 and Supplementary Table S6, respectively).
Table 2.
The relative contribution (ηr/ημ) and relative rates (ρ/θ) of recombination and mutation for the cp and rep genes of begomovirus datasets retrieved from the GenBank database.
| Dataset |
cp |
rep |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ηra | ημb | ηr/ημ | ρc | θwd | ρ/θwe | ηr | ημ | ηr/ημ | ρ | θw | ρ/θw | |
| ACMV | 0 | 312 | 0.000 | 5.101 | 35.994 | 0.142 | 0 | 694 | 0.000 | 5.101 | 48.147 | 0.106 |
| AgEV | 55 | 93 | 0.591 | 1.020 | 26.128 | 0.039 | 70 | 103 | 0.680 | 1.020 | 32.242 | 0.032 |
| BhYVIV | 105 | 503 | 0.209 | 12.242 | 52.560 | 0.233 | 360 | 527 | 0.683 | 4.081 | 81.912 | 0.050 |
| BhYVMV | 184 | 192 | 0.958 | 7.141 | 40.907 | 0.175 | 511 | 775 | 0.659 | 7.141 | 86.986 | 0.082 |
| CLCuBuV | 99 | 152 | 0.651 | 0.000 | 42.089 | 0.000 | 145 | 190 | 0.763 | 4.081 | 47.377 | 0.086 |
| CLCuGV | 98 | 138 | 0.710 | 1.020 | 29.092 | 0.035 | 56 | 317 | 0.177 | 3.061 | 40.446 | 0.076 |
| CLCuMV | 114 | 189 | 0.603 | 2.040 | 41.577 | 0.049 | 54 | 433 | 0.125 | 2.040 | 59.920 | 0.034 |
| EACMV | 104 | 541 | 0.192 | 1.020 | 15.825 | 0.064 | 61 | 1372 | 0.044 | 5.101 | 13.692 | 0.373 |
| MYMIV | 11 | 238 | 0.046 | 2.040 | 32.768 | 0.062 | 13 | 342 | 0.038 | 3.061 | 39.973 | 0.077 |
| SPLCV | 166 | 290 | 0.572 | 4.081 | 33.656 | 0.121 | 164 | 985 | 0.166 | 13.263 | 73.623 | 0.180 |
| ToLCNDV | 128 | 644 | 0.199 | 14.283 | 42.452 | 0.336 | 160 | 1136 | 0.141 | 11.222 | 77.347 | 0.145 |
| ToLCTV | 1 | 163 | 0.006 | 3.061 | 23.889 | 0.128 | 146 | 301 | 0.485 | 1.020 | 59.131 | 0.017 |
| TYLCTV | 46 | 132 | 0.348 | 2.040 | 29.867 | 0.068 | 37 | 309 | 0.120 | 2.040 | 49.779 | 0.041 |
| ToYLCCNV | 40 | 444 | 0.090 | 2.040 | 46.384 | 0.044 | 263 | 406 | 0.648 | 1.020 | 84.907 | 0.012 |
| TYLCV | 45 | 420 | 0.107 | 5.101 | 5.315 | 0.960 | 41 | 1040 | 0.039 | 2.040 | 29.320 | 0.070 |
ACMV, African cassava mosaic virus; AgEV, Ageratum enation virus; BhYVIV, Bhendi yellow vein India virus; BhYVMV, Bhendi yellow vein mosaic virus; CLCuBuV, Cotton leaf curl Burewala virus; CLCuGV, Cotton leaf curl Gezira virus; CLCuMV, Cotton leaf curl Multan virus; EACMV, East African cassava mosaic virus; MYMIV, Mungbean yellow mosaic India virus; SPLCV, Sweet potato leaf curl virus; ToLCNDV, Tomato leaf curl new Delhi virus; ToLCTV, Tomato leaf curl Taiwan virus; TYLCTV, Tomato yellow leaf curl Thailand virus; ToYLCCNV, Tomato yellow leaf curl China virus; TYLCV, Tomato yellow leaf curl virus.
Number of substitutions due to recombination over the ML phylogenetic tree.
Number of substitutions due to mutation over the ML phylogenetic tree.
Population-scaled recombination rate estimated using a likelihood lookup table for a per site mutation rate of 0.001.
Watterson’s infinite-sites estimator of the population-scaled mutation rate (θ).
Equivalent to r/μ.
All values of the ηr/ημ ratio were <1, however, there was a wide variation amongst genes/datasets. While all substitutions over the ACMV cp and rep trees were assigned to mutations, a considerable fraction of the substitutions in the CLCuMV, CLCuBuV and CLCuGV cp trees were assigned to recombination (relative contributions of 0.603, 0.651 and 0.710, respectively; Table 2). In contrast, the values of the ρ/θ ratio estimated for the cp gene of these latter datasets were comparatively low amongst all datasets analyzed in this study. Importantly, some specific recombination events accounted for a considerable individual contribution. Indeed, the number of substitutions assigned to individual recombination events was rather variable in each phylogenetic tree (data not shown), with those events that mapped to the longest branches being the main contributors to the total number of substitutions due to recombination (ηr). For example, a single recombination event (event 1, P value = 1.75 × 10 − 6; Supplementary Table S5) mapping to the longest branch of the CLCuBuV cp tree (Supplementary Figure S2e) accounted for ∼30% of all substitutions (72 out of 251). A similarly high contribution (∼30% of all substitutions) was observed for the CLCuBuV rep gene, in which recombination events 2, 5 and 6 (mapped to the three longest branches in the tree; Supplementary Figure S3e) accounted for 100 out of 335 substitutions. As a consequence, the relative contribution of recombination to the variability of CLCuBuV rep was high (0.763). The variable contribution of individual recombination events to the overall sequence diversity in a population means that the relative contribution of recombination is not necessarily a function of the relative rates of recombination and mutation. Moreover, even very low recombination rates (such as those estimated for CLCuD-causing begomoviruses) can provide a substantial amount of variation. More than the number of recombination events, the genetic distance between parental sequences strongly determines the contribution of recombination to the total variability. Consequently, we did not observe a significant correlation between the estimates obtained from both approaches (relative contribution vs. relative rates) for the cp and rep genes of begomoviruses (r = −0.286484 and −0.411914, respectively).
No recombination events were detected in the ACMV dataset, and therefore no substitutions were assigned to the evolutionary mechanism of recombination. Nevertheless, the ρ/θ ratios for the ACMV cp and rep genes (0.142 and 0.106, respectively; Table 2) indicate that they are considerably targeted by recombination relative to mutation. These results might be, in part, due to the conservative cut off used in our recombination analysis, since a number of unique recombination events were detected by less than four methods but were not considered in the relative contribution calculation.
Considerable values of ρ/θ were estimated for the cp gene of both okra-infecting begomoviruses (ρ/θ = 0.233 and 0.175 for BhYVIV and BhYVMV, respectively), ToLCNDV (ρ/θ = 0.336) and TYLCV, the latter having the greatest skew towards recombination (ρ/θ = 0.960). Although <1, relatively high values of the ρ/θ ratio were also observed for the rep gene of EACMV, SPLCV and ToLCNDV (ρ/θ = 0.373, 0.180 and 0.145, respectively). The relative contributions calculated for the rep gene of both okra-infecting begomoviruses were also comparatively high amongst the datasets analyzed. These results indicate distinct evolutionary dynamics of BhYVIV, BhYVMV, ToLCNDV and TYLCV, all of which are more recombination-prone (in addition to the well-known SPLCV). However, for both genes in most begomovirus datasets, mutation seems to occur more often and play the larger role in shaping the standing genetic variation.
4. Discussion
The high genetic variability of begomovirus populations has been primarily attributed to two factors: (1) their high rates of nucleotide substitution, which are similar to those of RNA viruses (Duffy and Holmes 2008, 2009) and (2) the frequent occurrence of recombination, which may significantly accelerate their evolution by maximizing the combinations of pre-existing nucleotide polymorphisms created by mutation (Padidam et al. 1999; Pita et al. 2001). Thus, mutation and recombination are often referred as the major contributors to the genetic variability of begomovirus populations. However, until recently (Lima et al. 2013), no attempt had been made to determine the individual contribution of each of these mechanisms. Lima et al. (Lima et al. 2013) analyzed only two data sets corresponding to bipartite, New World begomoviruses (Macroptilium yellow spot, MaYSV, and Tomato severe rugose virus, ToSRV). Here, we extend this analysis to an additional 15 data sets, representing both mono- and bipartite, New World and Old World begomoviruses.
Based on the analysis of about 900 full-length DNA-A sequences representing 15 species, we showed that the genetic variability across the DNA-A segments of 14 out of 15 species datasets was not evenly distributed, with the rep gene being more variable than its cognate cp gene in 11 of these 14 datasets (the exceptions being CLCuGV, EACMV and MYMIV, for which the cp gene was more variable). In addition, the central/3′-terminal regions of the cp gene and the 5′-terminal/central regions of the rep gene were often more variable than other regions of these genes in most datasets analyzed. We also observed that these increased levels of genetic variability in specific regions from both genes consisted of a high content of synonymous substitutions. Then, we applied a range of ML-based methods for detection of positive selection to verify the role of diversifying or positive selection in shaping the uneven levels of genetic variability across both genes. As has been shown for several viruses (García-Arenal et al. 2001), purifying selection seems to act on most sites in both genes and only a few sporadic cases of positively selected sites were detected. Thus, our results clearly exclude positive selection as responsible for the increased levels of genetic variability in specific regions of the cp and rep genes.
Our data sets are composed of full-length sequences of begomoviruses collected from around the world, and we cannot rule out the occurrence of population admixture and recent demographic events (contraction/expansion of populations) which might, respectively, affect the standing genetic variability and create confounding artifacts with negative selection. In fact, such factors might explain, at least in part, the discrepant levels of genetic variation observed amongst the begomovirus species data sets.
To rule out any others biases that might result in significant differences between the degrees of interspecific variability, we checked whether there was a positive correlation between the nucleotide diversity indexes (π) and either the sampling time period (STP) or the number of hosts from which the isolates were sampled in each data set. Data analysis indicated a lack of positive correlation between STP and the nucleotide diversity (π) calculated for full-length genome sequences. The 2-year STP determined for the highly variable BhYVIV data set (π = 0.06293) was particularly short amongst the data sets analyzed. A very similar STP (3 years) was determined for the second least variable data set from this study (TYLCTV, π = 0.02643). Moreover, sampling dates available from Genbank records for 19 out of 41 isolates from the SPLCV data set suggest a 15-year minimum STP. However, its degree of genetic variability (π = 0.07167) was very similar to that of the BhYVIV data set, whose STP spanned exactly 2 years. The STP does not seem to affect other parameters analyzed in this study, such as the number of unique recombination events detected by RDP. For example, although the STP determined for the SPLCV and BhYVIV data sets were considerably discrepant (15 and 2 years, respectively), both showed similar numbers of unique recombination events (16 and 15 events, respectively). On the other hand, the EACMV data set showed an 8-year minimum STP and only three unique recombination events. We calculated the coefficients of linear correlation (Pearson’s r) between the number of hosts from which the sequences from each begomovirus data set were obtained and (1) the total number of substitutions on the phylogeny likely due to recombination and mutation (ηr e ημ, respectively), (2) the population-scaled recombination and mutation rates (ρ and θ, respectively), and (3) the nucleotide diversity (π) obtained for both cp and rep genes (data not shown). Most coefficients indicated no significant correlation (P values > 0.05) between the number of hosts and the other variables listed above.
On the other hand, it is important to note that the key issue addressed in this study refers to the uneven distribution of the genetic variation across begomovirus genomes/genes. In this context, it is unlikely that the above-mentioned factors are responsible for the nonrandom distribution of polymorphisms in equivalent genomic regions of all begomovirus data sets. Similarly, the occurrence of a recent population expansion (whose effect in its essence would involve solely the mechanism of mutation) does not explain the high content of synonymous substitutions detected at specific regions of the cp and rep genes in most begomovirus species. The pattern of variation across begomovirus genomes/genes suggests the action of an additional mechanism able to affect the genetic variability in a nonrandom manner.
Recombination events were detected in 14 out of 15 species datasets, and the uneven distribution of recombination breakpoints readily resembled that of levels of genetic variability in both genes, that is, regions with increased levels of genetic variability were often targeted by a number of recombination breakpoints. Together, these results implicated recombination, and not diversifying selection, as responsible for shaping the levels of genetic variability across begomovirus genomes. Nevertheless, the standing genetic variability in all begomovirus populations was dominated by mutation, since all ηr/ημ and ρ/θ ratios were <1, that is, for both genes of begomovirus datasets point mutations were more frequent than recombination events.
Over the last years, various studies have shown that recombination occurs at high frequencies in begomovirus populations (Padidam et al. 1999; Pita et al. 2001; Martin et al. 2011). A nonrandom location of recombination breakpoints is a conserved feature amongst ssDNA viruses which use a rolling-circle mechanism for replicating their genomes (Lefeuvre et al. 2007a,b; Prasanna and Rai 2007; Martin et al. 2011). For example, most recombination events detected in our analyses had at least one breakpoint in the origin of replication and in the 5′-terminal/central regions of the rep gene, known hotspots of begomovirus recombination. It has been shown that recombinants that exchange whole domains have less disrupted intragenome interaction networks and are favored by selection (Martin et al. 2011).
In agreement, the functional modular structure of the geminivirus coat protein (CP) might provide important clues to explain the uneven distribution of genetic variation and the exchangeability via recombination of the 5′-terminal, central and 3′-terminal regions of the cp gene. In the 5′-terminal region are encoded a nuclear localization signal (NLS) (Unseld et al. 2001) and the ss/dsDNA binding domain (Liu et al. 1997). In addition, the N-terminal 3D structure of the CP includes an α-helix, which is involved in the maintenance of the geminate particle architecture, and two out of eight β-strands (βB e βC) that compose the β-barrel structure (Zhang et al. 2001; Bottcher et al. 2004). The structural and functional roles played by this region could explain, at least in part, its strong conservation at the nucleotide level and, consequently, a lower propensity to be disrupted by recombination (in other words, a strong purifying selection may act against recombination when breakpoints are located at the 5′-terminal region of the cp gene). The central region of the cp gene encodes for a second NLS, a nuclear export signal (NES) and the cell wall targeting motif (CW). Functionally, the central portion of the CP is involved in insect transmission (Hohnle et al. 2001) and multimerization during particle assembly (Hallan and Gafni 2001). Its 3D structure involves three β-strands (βD, βE and βF) with a long loop connecting the βE and βF strands (Zhang et al. 2001; Bottcher et al. 2004). Although this region plays important structural and functional roles, it was more flexible to variation at the nucleotide level than the 5′-terminal region. Finally, the C-terminal of the CP protein includes a third NLS (Unseld et al. 2001) and, structurally, three β-strands (βG, βH and βI). It seems that the lack of a structural role for this region makes it even more flexible to variation and possibly more susceptible to being disrupted by recombination (i.e., a more relaxed purifying selection acts against recombinantion when breakpoints are located in this region).
On the other hand, the increased variability in the 5′-terminal and central regions of the rep gene contrast with the multifunctional nature of the Rep protein. The 5′-terminal and central regions encode the DNA binding, cleavage/ligation and oligomerization domains of the protein (Heyraud-Nitschke et al. 1995; Horvath et al. 1998). In addition, there are also binding sites for viral (replication enhancer protein, REn) (Settlage et al. 2005) and host factors (retinoblastoma-related protein [RBR], proliferating cell nuclear antigen [PCNA], GRIK, small ubiquitin-related modifier [SUMO]-conjugating enzyme [SCE1] and ATP) (Ach et al. 1997; Gutierrez 2000; Castillo et al. 2004). Although the 3′-terminal region seems to encode few functional domains (only the helicase domain and an ATP binding site have been identified in C-terminal of the Rep protein), it was less tolerant to variation and disruption by recombination.
The intergenic (common) region was also targeted by a number of recombination events. The precise features of this region that either make it mechanistically recombination-prone, or enable the preservation of its function following recombination, are unknown. However, its role as both the origin and termination point of both complementary and virion strand replication (through rolling-circle replication and/or recombination-dependent replication) could provide the explanation (Lefeuvre et al. 2009).
Estimates of recombination rates are scarce for plant viruses. Experimental recombination rates determined for a dsDNA (Cauliflower mosaic virus, CaMV; family Caulimoviridae, genus Caulimovirus) and a ssRNA plant virus (Tobacco etch virus, TEV; family Potyviridae, genus Potyvirus) were of the order of 10 − 5, very similar to their predicted mutation rates (Froissart et al. 2005; Tromas et al. 2014). However, the statistically detectable recombination rate appears to be lower than the mutation rate in begomoviruses. Even cases in which our results indicated a highly recombination-prone nature, that is, showed high population-scaled recombination rates (e.g., BhYVIV cp, SPLCV rep and both genes of ToLCNDV), the mutational dynamics was still the dominant force shaping the standing genetic variability. These three viruses may have evolutionary dynamics distinct from most other begomoviruses, with recombination playing a more prominent role. But once again, although recombination may occur frequently, the mechanism of mutation was responsible for the largest fraction of nucleotide polymorphisms in these species datasets (nr/nµ <1).
Notably, our results indicate that even low recombination rates could provide a significant amount of genetic variability. This effect was more evident in less variable datasets (e.g., CLCuBuV, CLCuGV, CLCuMV and TYLCTV) in which few recombination events (frequently mapped onto long branches) were responsible for up to 30% of the substitutions. It is important to note that our relative contribution calculations were based on recombination events detected by RDP, which is able to capture only a small fraction of recombination events that occurred in the history of the samples. Ancient recombination events shared by all members of the current species or those involving closely related parental sequences are poorly detected by the methods contained in the RDP package. In fact, the performance of all of the different methods employed in recombination detection is significantly affected by the levels of divergence and recombination in the datasets (most of them show improved performance under increased levels of genetic variation and/or when recombination has occurred frequently) (Posada and Crandall 2001; Posada 2002). Therefore, our calculations most likely represent the minimal contribution of recombination to the standing genetic variation. In other words, the relative contribution is potentially greater than that estimated from our analyses. Obviously, the contribution of recombination events involving very similar parental sequences is expected to be low (in terms of number of substitutions), but is not possible to speculate about the contribution of ancient recombination events or other types of undetectable events. While we used a conservative cut off to detect events, it is unlikely that a more liberal cut off would lead to dramatically higher ηr/ημ. Using a cut off of two methods instead of four did not result in many additional recombination events in most begomoviruses datasets, and these additional events did not provide a considerable increase in the ηr calculations (data not shown).
Our results demonstrate the utility of a method to calculate the relative contribution of recombination and mutation to the standing genetic variability, since this is not a function of their relative frequencies. Obviously, the discrepancies observed between the estimates using each approach could be, at least in part, due to the ability of the different methods in detecting recombination events. Indeed, the seven methods used in RDP and the coalescent-based method employed by LDhat could recover different recombination scenarios. While estimates of the population scaled-recombination rate (ρ) typically capture the combined effects of all recombination events (providing a more accurate picture of recombination), RDP identifies and individually characterizes the easily detectable ones (Brown et al. 2001; Posada and Crandall 2001; Posada 2002). The variable contribution of individual recombination events corroborates the lack of correlation between recombination rates and the proportion of standing genetic variability due to recombination (even under similar performances of recombination detection).
The phylogeny-based partitioning method was shown to be useful in quantifying the effect of each well-supported recombination event. It is reasonable to assume that recombination events involving more divergent parental viruses could provide more genetic variation to the population, causing even longer branches on resulting phylogenetic trees. Recombination between divergent parental sequences might play an important role in the diversification of viruses, and might not even be captured when looking at the diversity of one species. However, the level of divergence between parental viruses seems to be constrained by the mandatory maintenance of intragenome interaction networks (Martin et al. 2005). Experimental evidence from chimaeric MSV genomes indicates that viral fitness is reduced with increasing divergence of exchanged sequences. Moreover, the modularity of the exchanged sequences is correlated with the complexity of intragenome interaction networks, indicating that some portions of the genome could be more tolerant than others to recombination events involving more divergent parental viruses (Martin et al. 2005).
In summary, our findings indicate that the relative evolutionary value of recombination and mutation cannot be explained solely by their relative frequencies, and that the rapid evolution of begomovirus populations is primarily a consequence of their rapid mutational dynamics. Therefore, in spite of the recombination-prone nature of begomovirus genomes, this evolutionary mechanism does not seem to be the main contributor to their high levels of genetic variation.
Supplementary data
Supplementary data are available at Virus Evolution online.
Supplementary Material
Acknowledgements
The authors wish to thank Darren P. Martin for critical review of the article. This work was funded by CNPq grant 483607/2013-4 and FAPEMIG grant APQ-02037-13 to F.M.Z. S.D. was supported by the NSF (DEB 1026095). A.T.M.L. was the recipient of a CNPq doctoral fellowship.
Data Availabitity
All data retrieved, generated and presented in this article is available either in the supplementary material or from the authors upon request.
Conflict of interest: None declared.
References
- Ach R. A. et al. (1997) ‘RRB1 and RRB2 Encode Maize Retinoblastoma-Related Proteins that Interact with a Plant D-Type Cyclin and Geminivirus Replication Protein’, Molecular and Cellular Biol, 17: 5077–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balol G. B. et al. (2010) ‘Sources of Genetic Variation in Plant Virus Populations’, Journal of Pure and Applied Microbiology, 4: 803–8 [Google Scholar]
- Berrie L. C., Rybicki E. P., Rey M. E. C. (2001) ‘Complete Nucleotide Sequence and Host Range of South African cassava Mosaic Virus: Further Evidence for Recombination Amongst Begomoviruses’, Journal of General Virology, 82: 53–8 [DOI] [PubMed] [Google Scholar]
- Biebricher C. K., Eigen M. (2006) ‘What is a Quasispecies?’, Current Topics in Microbiology and Immunology, 299: 1–31 [DOI] [PubMed] [Google Scholar]
- Bonnet J. et al. (2005) ‘Role of Recombination in the Evolution of Natural Populations of Cucumber Mosaic Virus, A Tripartite RNA Plant Virus’, Virology, 332: 359–68 [DOI] [PubMed] [Google Scholar]
- Bottcher B. et al. (2004) ‘Geminate Structures of African Cassava Mosaic Virus’, Journal of Virology, 78: 6758–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briddon R. W. et al. (1996) ‘Analysis of the Nucleotide Sequence of the Treehopper-Transmitted Geminivirus, Tomato Pseudo-Curly Top Virus, Suggests a Recombinant Origin’, Virology, 219: 387–94 [DOI] [PubMed] [Google Scholar]
- Brown C. J. et al. (2001) ‘The Power to Detect Recombination Using the Coalescent’, Molecular Biology and Evolution, 18: 1421–4 [DOI] [PubMed] [Google Scholar]
- Carpenter J., Bithell J. (2000) ‘Bootstrap Confidence Intervals: When, Which, What? A Practical Guide for Medical Statisticians’, Statistical Medicine, 19: 1141–64 [DOI] [PubMed] [Google Scholar]
- Castillo A. G. et al. (2004) ‘Interaction Between a Geminivirus Replication Protein and the Plant Sumoylation System’, Journal of Virology, 78: 2758–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davino S. et al. (2009) ‘Two New Natural Begomovirus Recombinants Associated with the Tomato Yellow Leaf Curl Disease Co-Exist with Parental Viruses in Tomato Epidemics in Italy’, Virus Research, 143: 15–23 [DOI] [PubMed] [Google Scholar]
- Drake J. W. (1991) ‘A Constant Rate of Spontaneous Mutation in DNA-Based Microbes’, Proceedings of the National Academy of Sciences, USA, 88: 7160–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., Holmes E. C. (2008) ‘Phylogenetic Evidence for Rapid Rates of Molecular Evolution in the Single-Stranded DNA Begomovirus Tomato Yellow Leaf Curl Virus’, Journal of Virology, 82: 957–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., Holmes E. C. (2009) ‘Validation of High Rates of Nucleotide Substitution in Geminiviruses: Phylogenetic Evidence from East African Cassava Mosaic Viruses’, Journal of General Virology, 90: 1539–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., Shackelton L. A., Holmes E. C. (2008) ‘Rates of Evolutionary Change in Viruses: Patterns and Determinants’, Nature Reviews Genetics, 9: 267–76 [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2004) ‘MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity’, BMC Bioinformatics, 5: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M., Winkler-Oswatitsch R., Dress A. (1988) ‘Statistical Geometry in Sequence Space: A Method of Quantitative Comparative Sequence Analysis’, Proceedings of the National Academy of Sciences, USA, 85: 5913–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Negroni M., Robertson D. L. (2007) ‘The Distribution of HIV-1 Recombination Breakpoints’, Infection, Genetics and Evolution, 7: 717–23 [DOI] [PubMed] [Google Scholar]
- Froissart R. et al. (2005) ‘Recombination Every Day: Abundant Recombination in a Virus During a Single Multi-Cellular Host Infection’, PLoS Biology, 3: e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Andrés S. et al. (2006) ‘Begomovirus Genetic Diversity in the Native Plant Reservoir Solanum nigrum: Evidence for the Presence of a New Virus Species of Recombinant Nature’, Virology, 350: 433–42 [DOI] [PubMed] [Google Scholar]
- García-Andrés S. et al. (2007) ‘Founder Effect, Plant Host, and Recombination Shape the Emergent Population of Begomoviruses That Cause the Tomato Yellow Leaf Curl Disease in the Mediterranean Basin’, Virology, 359: 302–12 [DOI] [PubMed] [Google Scholar]
- García-Arenal F., Fraile A., Malpica J. M. (2001) ‘Variability and Genetic Structure of Plant Virus Populations’, Annual Review of Phytopathology, 39: 157–86 [DOI] [PubMed] [Google Scholar]
- García-Arenal F., Fraile A., Malpica J. M. (2003) ‘Variation and Evolution of Plant Virus Populations’, International Microbiology, 6: 225–32 [DOI] [PubMed] [Google Scholar]
- Ge L. M. et al. (2007) ‘Genetic Structure and Population Variability of Tomato Yellow Leaf Curl China Virus’, Journal of Virology, 81: 5902–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish P. J., Garcia-Lerma J. G. (2003) ‘Mutation Rate and The Efficacy of Antimicrobial Drug Treatment’, Lancet Infectious Diseases, 3: 28–32 [DOI] [PubMed] [Google Scholar]
- Grigoras I. et al. (2010) ‘High Variability and Rapid Evolution of a Nanovirus’, Journal of Virology, 84: 9105–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez C. (2000) ‘DNA Replication and Cell Cycle in Plants: Learning from Geminiviruses’, EMBO Journal, 19: 792–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallan V., Gafni Y. (2001) ‘Tomato Yellow Leaf Curl Virus (TYLCV) Capsid Protein (CP) Subunit Interactions: Implications for Viral Assembly’, Archives of Virology, 146: 1765–73 [DOI] [PubMed] [Google Scholar]
- Heath L. et al. (2006) ‘Recombination Patterns in Aphthoviruses Mirror Those Found in Other Picornaviruses’, Journal of Virology, 80: 11827–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyraud-Nitschke F. et al. (1995) ‘Determination of the Origin Cleavage and Joining Domain of Geminivirus Rep Proteins’, Nucleic Acids Research, 23: 910–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohnle M. et al. (2001) ‘Exchange of Three Amino Acids in the Coat Protein Results in Efficient Whitefly Transmission of a Nontransmissible Abutilon Mosaic Virus Isolate’, Virology, 290: 164–71 [DOI] [PubMed] [Google Scholar]
- Holland J. et al. (1982) ‘Rapid Evolution of RNA Genomes’, Science, 215: 1577–85 [DOI] [PubMed] [Google Scholar]
- Horvath G. V. et al. (1998) ‘Prediction of Functional Regions of the Maize Streak Virus Replication-Associated Proteins by Protein-Protein Interaction Analysis’, Plant Molecular Biology, 38: 699–712 [DOI] [PubMed] [Google Scholar]
- Jeske H., Lutgemeier M., Preiss W. (2001) ‘DNA Forms Indicate Rolling Circle and Recombination-Dependent Replication of Abutilon Mosaic Virus’, EMBO Journal, 20: 6158–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky-Pond S. L., Frost S. D. W. (2005) ‘Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection’, Molecular Biology and Evolution, 22: 1208–22 [DOI] [PubMed] [Google Scholar]
- Lefeuvre P. et al. (2007a) ‘Avoidance of Protein Fold Disruption in Natural Virus Recombinants’, PLoS Pathogens, 3: e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefeuvre P. et al. (2007b) ‘Begomovirus ‘Melting Pot’ in the South-West Indian Ocean Islands: Molecular Diversity and Evolution Through Recombination’, Journal of General Virology, 88: 3458–68 [DOI] [PubMed] [Google Scholar]
- Lefeuvre P. et al. (2009) ‘Widely Conserved Recombination Patterns Among Single-Stranded DNA Viruses’, Journal of Virology, 83: 2697–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefeuvre P., Moriones E. (2015) ‘Recombination as a Motor of Host Switches and Virus Emergence: Geminiviruses as Case Studies’, Current Opinion in Virology, 10: 14–9 [DOI] [PubMed] [Google Scholar]
- Lima A. T. M. et al. (2013) ‘Synonymous Site Variation Due to Recombination Explains Higher Genetic Variability in Begomovirus Populations Infecting Non-Cultivated Hosts’, Journal of General Virology, 94: 418–31 [DOI] [PubMed] [Google Scholar]
- Liu H. T., Boulton M. I., Davies J. W. (1997) ‘Maize Streak Virus Coat Protein Binds Single- and Double-Stranded DNA In Vitro’, Journal of General Virology, 78: 1265–70 [DOI] [PubMed] [Google Scholar]
- Lozano G. et al. (2009) ‘Novel Begomovirus Species of Recombinant Nature in Sweet Potato (Ipomoea batatas) and Ipomoea indica: Taxonomic and Phylogenetic Implications’, Journal of General Virology, 90: 2550–62 [DOI] [PubMed] [Google Scholar]
- Martin D. P. et al. (2005) ‘The Evolutionary Value of Recombination is Constrained by Genome Modularity’, PLoS Genetics, 1: e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P. et al. (2011) ‘Complex Recombination Patterns Arising During Geminivirus Coinfections Preserve and Demarcate Biologically Important Intra-Genome Interaction Networks’, PLoS Pathogens, 7: e1002203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P. et al. (2011) ‘Recombination in Eukaryotic Single Stranded DNA Viruses’, Viruses, 3: 1699–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G., Awadalla P., Fearnhead P. (2002) ‘A Coalescent-Based Method for Detecting and Estimating Recombination from Gene Sequences’, Genetics, 160: 1231–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monci F. et al. (2002) ‘A Natural Recombinant Between the Geminiviruses Tomato Yellow Leaf Curl Sardinia Virus and Tomato Yellow Leaf Curl Virus Exhibits a Novel Pathogenic Phenotype and is Becoming Prevalent in Spanish Populations’, Virology, 303: 317–26 [DOI] [PubMed] [Google Scholar]
- Murrell B. et al. (2013) ‘FUBAR: A Fast, Unconstrained Bayesian Approximation for Inferring Selection’, Molecular Biology and Evolution, 30: 1196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas-Castillo J. et al. (2000) ‘Natural Recombination Between Tomato Yellow Leaf Curl Virus-Is and Tomato Leaf Curl Virus’, Journal of General Virology, 81: 2797–801 [DOI] [PubMed] [Google Scholar]
- Nei M. (1987) Molecular Evolutionary Genetics. New York: Columbia University Press. [Google Scholar]
- Padidam M., Sawyer S., Fauquet C. M. (1999) ‘Possible Emergence of New Geminiviruses by Frequent Recombination’, Virology, 265: 218–24 [DOI] [PubMed] [Google Scholar]
- Peng R. D. (2008) simpleboot: Simple Bootstrap Routines Available at: https://CRAN.R-project.org/package=simpleboot
- Pita J. S. et al. (2001) ‘Recombination, Pseudorecombination and Synergism of Geminiviruses are Determinant Keys to the Epidemic of Severe Cassava Mosaic Disease in Uganda’, Journal of General Virology, 82: 655–65 [DOI] [PubMed] [Google Scholar]
- Posada D. (2002) ‘Evaluation of Methods for Detecting Recombination from DNA Sequences: Empirical Data’, Molecular Biology and Evolution, 19: 708–17 [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K. A. (1998) ‘MODELTEST: Testing the Model of DNA Substitution’, Bioinformatics, 14: 817–8 [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K. A. (2001) ‘Evaluation of Methods for Detecting Recombination from DNA Sequences: Computer Simulations’, Proceedings of the National Academy of Sciences, USA, 98: 13757–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna H. C., Rai M. (2007) ‘Detection and Frequency of Recombination in Tomato-Infecting Begomoviruses of South and Southeast Asia’, Virology Journal, 4: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. (2007) R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Roossinck M. J. (1997) ‘Mechanisms of Plant Virus Evolution’, Annual Review of Phytopathology, 35: 191–209 [DOI] [PubMed] [Google Scholar]
- Rozas J. et al. (2003) ‘DnaSP: DNA Polymorphism Analyses by the Coalescent and Other Methods’, Bioinformatics, 19: 2496–7 [DOI] [PubMed] [Google Scholar]
- Sanz A. I. et al. (2000) ‘Multiple Infection, Recombination and Genome Relationships Among Begomovirus Isolates Found in Cotton and Other Plants in Pakistan’, Journal of General Virology, 81: 1839–49 [DOI] [PubMed] [Google Scholar]
- Saunders K., Bedford I. D., Stanley J. (2001) ‘Pathogenicity of a Natural Recombinant Associated with Ageratum Yellow Vein Disease: Implications for Geminivirus Evolution and Disease Aetiology’, Virology, 282: 38–47 [DOI] [PubMed] [Google Scholar]
- Scheffler K., Martin D. P., Seoighe C. (2006) ‘Robust Inference of Positive Selection from Recombining Coding Sequences’, Bioinformatics, 22: 2493–9 [DOI] [PubMed] [Google Scholar]
- Settlage S. B., See R. G., Hanley-Bowdoin L. (2005) ‘Geminivirus C3 Protein: Replication Enhancement and Protein Interactions’, Journal of Virology, 79: 9885–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton L. A. et al. (2005) ‘High Rate of Viral Evolution Associated with the Emergence of Carnivore Parvovirus’, Proceedings of the National Academy of Sciences, USA, 102: 379–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton L. A., Holmes E. C. (2006) ‘Phylogenetic Evidence for the Rapid Evolution of Human B19 Erythrovirus’, Journal of Virology, 80: 3666–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva F. N. et al. (2014) ‘Recombination and Pseudorecombination Driving the Evolution of the Begomoviruses Tomato Severe Rugose Virus (ToSRV) and Tomato Rugose Mosaic Virus (ToRMV): Two Recombinant DNA-A Components Sharing the Same DNA-B’, Virology Journal, 11: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D. L. (2003) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Tamura K. et al. (2011) ‘MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods’, Molecular Biology and Evolution, 28: 2731–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromas N. et al. (2014) ‘Estimation of the In Vivo Recombination Rate for a Plant RNA Virus’, Journal of General Virology, 95: 724–32 [DOI] [PubMed] [Google Scholar]
- Unseld S. et al. (2001) ‘Subcellular Targeting of the Coat Protein of African Cassava Mosaic Geminivirus’, Virology, 286: 373–83 [DOI] [PubMed] [Google Scholar]
- van der Walt E. et al. (2008) ‘Experimental Observations of Rapid Maize Streak Virus Evolution Reveal a Strand-Specific Nucleotide Substitution Bias’, Virology Journal, 5: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varsani A. et al. (2006) ‘Evidence of Ancient Papillomavirus Recombination’, Journal of General Virology, 87: 2527–31 [DOI] [PubMed] [Google Scholar]
- Varsani A. et al. (2008) ‘Recombination, Decreased Host Specificity and Increased Mobility May Have Driven the Emergence of Maize Streak Virus as an Agricultural Pathogen’, Journal of General Virology, 89: 2063–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey M., Holmes E. C. (1999) ‘Evolutionary Aspects of Recombination in RNA Viruses’, Journal of General Virology, 80: 2535–45 [DOI] [PubMed] [Google Scholar]
- Young G. A. (1994) ‘Bootstrap: More Than a Stab in the Dark? ’, Statistical Science, 9: 14 [Google Scholar]
- Zhang W. et al. (2001) ‘Structure of the Maize Streak Virus Geminate Particle’, Virology, 279: 471–7 [DOI] [PubMed] [Google Scholar]
- Zhou X. et al. (1997) ‘Evidence that DNA-A of a Geminivirus Associated with Severe Cassava Mosaic Disease in Uganda has Arisen by Interspecific Recombination’, Journal of General Virology, 78: 2101–11 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.