

Introduction
Potassium (K+) was first isolated as an element in 1807 by Sir Humphrey Davy when he electrolyzed potash (plant ashes soaked in pots of water), for which its name is derived. Despite the organic origin of its isolation, however, K+’s role in biology was not elucidated until the 20th century. In this article, we discuss the basic science underlying the effects of both hypo- and hyperkalemia on cardiac excitability and arrhythmias. As the major intracellular cation, K+ is concentrated 30–40 fold over its extracellular concentration by the activity of Na+-K+ ATPase in the plasma membrane, which hydrolyzes ATP to pump three Na+ ions out of the cell in exchange for 2 extracellular K+ ions into the cell, generating an outward current in the process. Because most cells express time-independent K+ ion channels in their plasma membrane, the high selective permeability to K+ over other ions generates a negative resting membrane potential (Em) approaching the K+ equilibrium potential (EK) as determined by the Nernst equation (-95 mV for extracellular and intracellular [K+] of 4.0 and 140 mmol/l, respectively). In excitable tissues such as the heart, the negative resting Em stabilizes working atrial and ventricular myocytes during diastole, preventing spontaneous action potentials (AP’s) from causing premature extrasystoles. For this reason, serum [K+] is closely regulated physiologically, with normal values ranging from 3.5 to 5.0 mmol/l. Outside of this range, lower and higher values of serum [K+] have electrophysiological effects that commonly promote cardiac arrhythmias, not solely because of direct effects of K+, but also because the cellular balances of K+, Na+ and Ca2+ are interlinked through Na+-K+ ATPase and Na+-Ca2+ exchange (Fig. 1A). Thus, hypo- and hyperkalemia directly impact Na+ and Ca2+ as well as K+ balances. In this article, we discuss how these relationships promote cardiac arrhythmias. First, we review how hypokalemia not only reduces repolarization reserve by suppressing K+ conductances, but also significantly inhibits Na+-K+ ATPase activity, causing intracellular Na+ and Ca2+ accumulation. Increased intracellular Ca2+ loading activates Ca2+-calmodulin kinase (CaMK) signaling to further reduce repolarization reserve by inducing late Na+ and Ca2+ currents. This positive feedback loop promotes early afterdepolarization (EAD)-mediated arrhythmias, especially when K+ channel-blocking drugs are present, as well as delayed afterdepolarization (DAD)-mediated arrhythmias and automaticity. We then turn to hyperkalemia, both systemic and interstitial. We discuss how resting membrane depolarization by systemic hyperkalemia promotes conduction block and reentry and may also promote phase 2 reentry. Finally, we describe how interstitial hyperkalemia, a prominent feature of acute myocardial ischemia, contributes to injury currents causing electrocardiographic ST segment elevation and TQ segment depression that promote arrhythmias, including phase 2 reentry similar to Brugada and other early repolarization syndromes. Differences between responses of conduction system tissues and atrial and ventricular myocardium to changes in extracellular [K+] are also noted.
Figure 1.
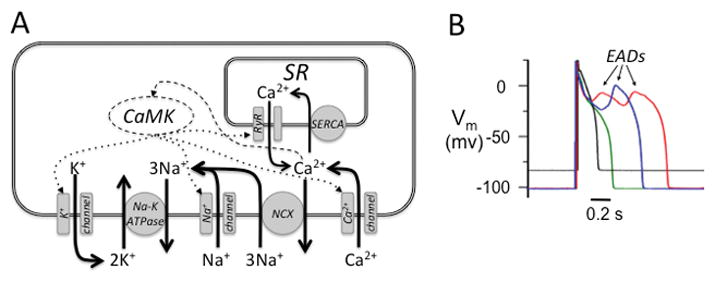
A. Interconnectedness of K+, Na+ and Ca2+ balances in the cardiac myocyte. Outward K+ loss through K+ channels (left) is recovered by the Na+-K+ ATPase removing 3 Na+ ions in exchange for 2 K+ ions. Some Na+ ions enter the cell via Na+ channels, but most via Na+-Ca2+ exchange (NCX) during diastole, which exchanges 1 Ca2+ ion for 3 Na+ ions. In the steady state, the Ca2+ removed by NCX balances the Ca2+ entering the cell via Ca2+ channels. Most Ca2+ in the cell recycles between the sarcoplasmic reticulum (SR) and cytoplasm, with uptake by sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) and release through ryanodine receptors (RyR). Cytoplasmic free Ca2+ activates CaMK, which regulates the properties of Na+, Ca2+ and K+ channels, and RyR in the SR (dotted arrows). B. Effects of hypokalemia on the AP. Superimposed AP recordings from an isolated rabbit ventricular myocyte with [K+]o=5.4 mmol/l (black trace) versus [K+]o= 2.7 mmol/l (red trace), showing hyperpolarized Em and EADs, the latter suppressed by the CaMK blocker KN-93 (green trace), but not by inactive KN-92 (blue trace). Modified from Pezhouman et al2 with permission.
Hypokalemia
Hypokalemia is most commonly encountered clinically as a complication of diuretic therapy1 used to treat hypertension, heart failure, renal disease and other conditions. Its direct electrophysiological effects include resting membrane hyperpolarization, Na+-K+ ATPase inhibition and suppression of K+ channel conductances resulting in AP duration (APD) prolongation, reduced repolarization reserve, EAD, DADs and automaticity.
Arrhythmia mechanisms
Reduced repolarization reserve predisposes the heart to EADs (Fig. 1B) and EAD-mediated arrhythmias including Torsades de pointes (TdP) and polymorphic ventricular tachycardia (VT), which can degenerate to ventricular fibrillation (VF) causing sudden cardiac death (Fig. 22).
Figure 2.
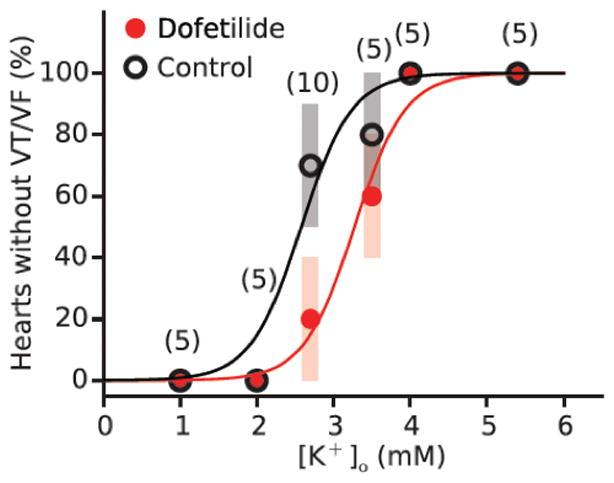
[K+]o dependence of hypokalemia-induced VT/VF in isolated rabbit hearts, without or with dofetilide. When [K+]o was lowered, the incidence of VT/VF within 90 minutes progressively increased to 100% at 2.0 and 1.0 mmol/l (black circles). Dofetilide (1 μmol/l) shifted the dose-response curve to the right. Reproduced with permission from Pezhouman et al.2
Reductions in repolarization reserve prolong APD in a heterogeneous manner, both because ion channel expression is heterogeneous throughout the atria and ventricles 3 and because EADs are intrinsically chaotic, occurring irregularly instead of reliably with every beat 4. Due to the gap junction coupling in cardiac tissue that prevents adjacent myocytes from exhibiting markedly different APD, the chaotic behavior causes some regions to exhibit EADs synchronously whereas other nearby regions do not. This dynamical process has been termed “regional chaos synchronization” and generates marked dispersion of repolarization since areas with long APD due to EADs are juxtaposed next to regions much shorter APD without EADs 4, 5 (Fig. 36). Moreover, if His-Purkinje fibers or regions of myocardium with EADs reach the threshold for triggered activity, the resulting extrasystoles can propagate into recovered regions without EADs, but may block when propagating into other regions with subthreshold EADs, thereby initiating reentry (Fig. 3, black arrows). Under these conditions, reentry can have a special property called biexcitability 7, in which two types of unstable rotors coexist. Incomplete repolarization allows slow meandering rotors to propagate using the L-type Ca2+ current (manifesting as TdP or polymorphic VT), whereas full repolarization allows fast rotors to propagate using the Na+ current (manifesting as polymorphic VT or VF). This arrhythmia mechanism is called mixed focal-reentrant fibrillation (as opposed the multiple wavelet or mother rotor fibrillation) since the unstable rotors often self-terminate but new rotors are then initiated by ongoing EAD-mediated triggered activity arising from His-Purkinje tissue and/or ventricular myocardium 4, 5.
Figure 3.
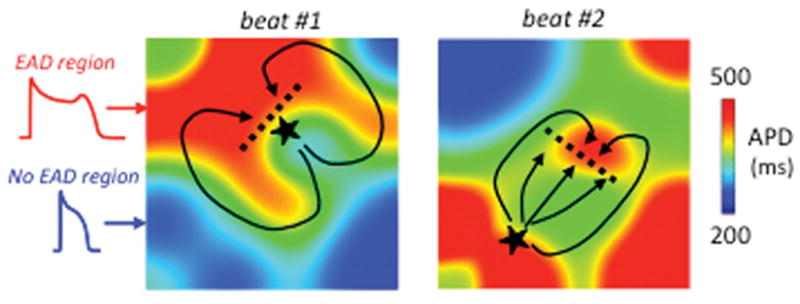
Regional chaos synchronization of EADs in tissue. In simulated paced homogeneous cardiac tissue, electrotonic coupling causes regional chaos synchronization to generate EAD islands (red regions), separated by regions without EADs (blue), whose position and size vary from beat-to-beat. Beat #1 illustrates a scenario in which a triggered PVC (★) arising from an EAD island blocks superiorly (dashed line) but conducts inferiorly (solid line), subsequently reentering the blocked region to induce reentry. Beat #2 illustrates a scenario in which the triggered PVC arising from an EAD island encounters another EAD island, resulting in conduction block (dashed line) and reentry (solid lines). Adapted from Weiss et al. 6 with permission.
Even moderate hypokalemia (2.5–3.0 mmol/l) can be highly arrhythmogenic in normal hearts. In isolated rabbit and rat hearts, we found that modestly reducing [K+]o to 2.7 mmol/l resulted in spontaneous EADs, polymorphic VT and VF in approximately over 50% of hearts studied, whereas severe hypokalemia (2.0 mmol/l) caused VF in 100% 2 (Fig. 2).
Molecular factors underlying hypokalemia-induced arrhythmias
The reduction in repolarization reserve by hypokalemia has classically been attributed to direct suppression of K+ channel conductances, but recent evidence indicates that indirect effects of hypokalemia leading to activation of late Na+ and Ca2+ currents play a key role as well 2. Together, these two factors are synergistic in reducing repolarization reserve sufficiently to induce EADs and EAD-mediated arrhythmias including TdP, polymorphic VT and VF.
Suppression of K+ channel conductances by hypokalemia
Many K+ channels, including the inward rectifier K+ current IK1, the rapid component of the delayed rectifier K+ current IKr and the transient outward current Ito, exhibit a strong allosteric dependence on extracellular K+ concentration [K+]o 8–12. In inward rectifier K+ channels (e.g. Kir2.1 encoded by the KCNJ2 gene), outward current through the channel is regulated by voltage-dependent block of the pore by cytoplasmic Mg2+ and polyamines that bind to the negative charges in the pore’s cytoplasmic vestibule and prevent passage of K+ ions. Extracellular K+ ions entering the pore from the outside electrostatically destabilize and “knock off” these blocking cations from their binding sites, restoring outward K+ flow 13. Thus, even though hypokalemia hyperpolarizes EK, increasing the driving force for outward K+ flow (Em - EK), the increased stability of blocking cations in the pore decreases the conductance more powerfully, resulting in decreased outward K+ current.
The mechanism by which extracellular K+ acutely regulates the conductance of voltage-dependent K+ channels, such as IKr (Kv11.1 encoded by KCHN2) and Ito (Kv1.4, Kv4.2, Kv4.3 encoded by KCNA4, KCND2, KCND3) is different. Hypokalemia speeds rapid inactivation of IKr 10 and slows reactivation kinetics of Ito 11, reducing outward repolarizing current even with moderate hypokalemia 12. Hypokalemia also downregulates IKr expression within hours 14. Thus, despite increasing the driving force for K+ efflux, hypokalemia reduces the number of conducting K+ channels during repolarization. His-Purkinje fibers are particularly susceptible to EADs, DADs and automaticity because of their lower resting K+ conductance (less IK1) compared to ventricular myocardium 15.
Na+-K+ ATPase inhibition by hypokalemia
The rate at which Na+-K+ ATPase transports ions depends both on the affinities of the extracellular and intracellular binding sites for Na+/K+ and Em, since the transport cycle moves one net positive charge outward. For the predominant α1 isoform of Na+-K+ ATPase in heart16, 17, the external K+ binding site is half-maximally saturated at [K+]o = 1.9 mmol/l, operating at half-maximal pumping rate at this concentration. Reducing [K+]o from 4.5 to 2.7 mmol/l decreases ion pumping rate by about 20% 18 (and by >50% for the α2 isoform that is half-maximally saturated at 2.9 mmol/l 16). In addition, hypokalemia hyperpolarizes Em, from -82 to -100 mV in isolated rabbit ventricular myocytes when [K+]o was reduced from 4.5 to 2.7 mmol/l (Fig 1B). Because Na+-K+ ATPase generates a net outward current and is inhibited by hyperpolarization, the combined effect reduced K+ binding and hyperpolarization is predicted to reduce Na+-K+ ATPase ion pumping rate by 43% 18. This agrees quantitatively with experimental measurements in isolated rat ventricular myocytes 19 reporting an approximately 50% suppression of Na+-K+ ATPase current when [K+]o was reduced from 5.4 to 2.7 mmol/l. The consequence was a slow rise in intracellular [Na+] that inhibited the ability of the Na+-Ca2+ exchanger to remove Ca2+ from the cell resulting in spontaneous diastolic Ca2+ waves.
Intracellular Ca2+ overload and CaMK activation
Hypokalemia prolongs APD by reducing outward current through both K+ channels and Na+-K+ ATPase. The prolonged APD results in increased Ca2+ influx through Ca2+ channels. At the same time, intracellular Ca2+ removal via NCX is compromised by the elevated intracellular [Na+] from Na+-K+ ATPase inhibition. Together, these factors cause an increase in cytoplasmic [Ca2+] 19 sufficient to activate CaMK, as documented directly in rabbit hearts exposed to 2.7 mmol/l [K+]o 18 (Fig. 4). When activated, CaMK phosphorylates a variety of protein targets, including Na+ channels, L-type Ca2+ channels and RyRs 20 (Fig. 1). Na+ channel phosphorylation by CaMKII increases late Na+ current which further decreases repolarization reserve and also further exacerbates intracellular Na+ loading 21. L-type Ca2+ channel phosphorylation by CaMKII both increases current amplitude and slows inactivation 20, increasing the Ca2+ window current that plays a critical role in EAD generation 22–24. RyR phosphorylation by CaMKII increases RyR leakiness, further elevating diastolic [Ca2+] and promoting Ca2+ waves and DADs20. At the same time hyperkalemia reduces the outward current hump of inward rectifier K+ channels and shifts its peak to a more negative voltage25, requiring less opposing inward current to depolarize Em. This effect, combined with enhancement of inward Na+-Ca2+ exchange current by hypokalemia-induced hyperpolarization of Em, lowers the threshold for DADs to cause triggered activity (for detailed explanation see 25). CaMK has other targets as well 20, whose role in the genesis of EADs, DADs and automaticity is less clear, but may also be important. Thus, the effect of CaMK activation during hypokalemia is to create the positive feedback scenario illustrated in Fig. 4 that further reduces repolarization reserve and exacerbates intracellular Ca2+ overload, culminating in the appearance of afterdepolarization-mediated arrhythmias. Purkinje fibers are particularly sensitive due to their already low IK1 density and reduced repolarization reserve compared to ventricular myocardium15. In our rabbit heart experiments, the role of CaMK activation was critical, since blocking CaMK with KN-93 completely prevented hypokalemia-induced VT/VF in both myocytes (Fig 1B) and isolated hearts 2, as did the selective late Na+ current blocker GS-967.
Figure 4.
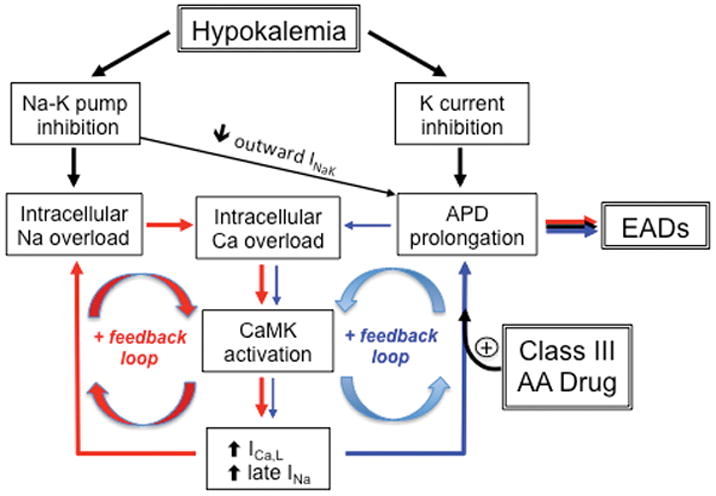
Hypokalemia-induced positive feedback loops (red and blue arrows) promoting intracellular Na+ and Ca overload, CaMK activation and EADs during hypokalemia. The potentiation of the blue positive feedback loop by Class III antiarrhythmic (AA) drugs is also shown. INaK=Na+-K+ ATPase outward current. Reproduced with permission from Pezhouman et al 2.
Hypokalemia and class III antiarrhythmic drugs
Hypokalemia is well known to potentiate the proarrhythmic effects of class III antiarrhythmic drugs 26, 27, especially in diseased hearts. This is not surprising since hypokalemia further reduces the conductance of K+ channels not already blocked by the drug. In addition, affinity of some Class III antiarrhythmic drugs is increased by hypokalemia28. Most experimental models in which EAD-mediated arrhythmias were induced by K+ channel blocking drugs in normal heart tissue have included hypokalemia (and often hypomagnesemia) as a co-factor. In our isolated rabbit heart preparation, the class III antiarrhythmic drug dofetilide, despite significantly prolonging APD, failed to induce EAD-mediated arrhythmias when extracellular [K+] was normal 2. Rather dofetilide sensitized the heart to hypokalemia, such that VT/VF occurred at a higher (but still abnormal) [K+]o when dofetilide was present (Fig. 2).
Other evidence also suggests that drug-induced suppression of K+ currents alone may not be sufficient to induce EAD-mediated arrhythmias. Anderson and colleagues29, 30 previously showed that CaMK activation by the K+ channel blocker clofilium played key role in EAD formation in isolated rabbit hearts. Moreover, CaMK inhibitors abolished clofilium-induced EADs. Recently, Yang et al31 reported that various class III antiarrhythmic drugs such as dofetilide contribute to EAD formation by activating the late Na+ current (INa) via phosphoinositide 3-kinase inhibition. Thus, the concomitant activation of pathologic late Na+ or Ca2+ currents by CaMK, phosphoinositide 3-kinase or other signaling pathways appears to be an essential co-requirement for eliciting EAD-mediated arrhythmias by either hypokalemia or class III drugs. These findings may provide insight into why failing hearts, in which CaMK activity is chronically elevated 20, are pre-sensitized to the pro-arrhythmic effects of Class III antiarrhythmic drugs even when serum [K+] is normal. Finally, targeting late INa to break the CaMK-facilitated positive feedback cycle promoting EAD formation (Fig. 4) may be a promising antiarrhythmic strategy.
Hyperkalemia
Hyperkalemia can be systemic or interstitial (confined to cardiac or other tissue as a result of acute global or regional ischemia). Both settings confer a high arrhythmia risk, whose mechanisms are discussed below.
Systemic hyperkalemia
Systemic hyperkalemia (mild 5.5–6.0, moderate 6.0–7.0, severe >7.0 mmol/l) is most commonly encountered clinically in chronic and acute renal failure, K+ supplementation for diuretic therapy, diuretic therapy with K+-sparing drugs such as spironolactone and triamterene, therapy with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers in cardiorenal disease, acute digitalis toxicity, massive hemolysis or muscle trauma, etc. Because one or more of these conditions are often present in chronic heart failure, they combine with the inherently increased arrhythmia susceptibility of diseased myocardium to further enhance risk.
Electrophysiological effects
The major cardiac electrophysiological effects of systemic hyperkalemia are depolarization of Em as EK becomes less negative (+18 mV change for a doubling of [K+]o from 4.0 to 8.0 mmol/l), APD shortening and altered conduction velocity (CV). APD shortening is due to the allosteric effect of [K+]o at increasing K+ channel conductances (despite a decreased driving force Em-EK) 9, 10, thereby creating excess repolarization reserve. APD shortening by hyperkalemia initially decreases the effective refractory period (ERP), but as hyperkalemia worsens, increased K+ channel conductances can induce post-repolarization refractoriness, such that the AP remains refractory for a period of time after full repolarization has occurred, prolonging the ERP. The effect of hyperkalemia on CV is biphasic, determined jointly by: 1) the voltage difference between Em and the Na+ channel activation threshold (around -55 mV); and 2) the effect of Em on the steady-state inactivation of Na+ channels, which increases with depolarization so that fewer Na+ channels are available to be activated during the AP upstroke. Up to approximately 8 mmol/l, the first factor dominates such that hyperkalemia-induced depolarization of Em accelerates CV. At higher [K+]o, however, the second factor of decreased Na+ channel availability becomes more important, causing CV slowing eventually to the point of propagation failure and inexcitability at [K+]o >14 mmol/l 32. Despite its biphasic effects on CV, however, hyperkalemia uniformly accentuates CV restitution, i.e. the dependence of CV on the previous diastolic interval. This is because time required for Na+ channels to recover from inactivation after repolarization prolongs as resting Em depolarizes, such that fewer Na+ channels are available to contribute to the AP upstroke at short diastolic intervals. Thus, although CV may be accelerated by hyperkalemia at normal heart rates, premature beats exhibit slowed CV due to incomplete Na+ channel recovery from inactivation. The arrhythmogenic consequences of these changes are described below.
Arrhythmia mechanisms
As hyperkalemia worsens, the electrocardiogram first demonstrates peaked T waves resulting from global APD shortening causing more synchronous repolarization across the ventricular wall. Subsequently, the P wave broadens and decreases in amplitude, eventually disappearing, and the QRS widens due to CV slowing. Severe hyperkalemia ([K+]o > 7.0 mmol/l) can lead to heart block, asystole and VT/VF. In humans, the precise level of hyperkalemia producing (or not producing) these changes varies considerably. For example, in trained athletes, short duration maximal exercise to exhaustion increased serum [K+] to an average of 8.2 mmol/l at peak exercise, which recovered with a half-time of 25 sec post exercise without apparent arrhythmias (perhaps protected by the high catecholamine state stimulating Na+-K+ ATPase activity33), although electrocardiograms were not recorded 34. Yet in a hospitalized patient, a serum [K+] of 8 mM is considered a dire medical emergency. It is not uncommon for systemic hyperkalemia to cause the P wave to disappear and the QRS to widen at serum [K+] between 7–8 mmol/l, i.e. below the threshold at which hyperkalemia causes CV slowing in animal models. Although visible P waves may disappear completely, conduction from the sinus node to ventricle may persist (sinoventricular conduction), taken as circumstantial evidence favoring specialized hyperkalemia-resistant atrial internodal tracts connecting the sinoatrial node to the atrioventricular node 35. Pacemaking by the sinus node is less sensitive to hyperkalemia because of its low resting K+ conductance, as reflected in its partially depolarized resting potential (-50 to -60 mV) and slow response (Ca2+ current-dependent) AP 36. However, the conduction through the atrioventricular node is often impaired 37, which has been attributed to potentiation of adenosine-sensitive inward rectifier K+ channels (Kir3 family encoded by KCNJ3) by hyperkalemia 38 and may also apply to the sinus node. Compared to nodal tissue, His-Purkinje tissue has higher resting K+ conductance and its secondary pacemaking capability (driven by the hyperpolarization-activated nonselective cation current If encoded by the HCN gene family) is suppressed by hyperkalemia, such that infranodal escape pacemakers become unreliable after heart block develops, which, together with propagation failure, can cause frank asystole.
Several mechanisms predispose hyperkalemic hearts to reentrant tachyarrhythmias. Decreased Na+ channel availability after premature extrasystoles can result in localized conduction block initiating reentry. As heart rate increases, the accentuation of CV restitution by hyperkalemia, combined with post-repolarization refractoriness, may predispose the heart to spatially discordant APD alternans, the classic mechanism causing localized conduction block and initiation of reentrant VT/VF during rapid pacing 39, 40. To our knowledge, however, this mechanism has not been demonstrated experimentally for hyperkalemia. By increasing repolarization reserve, hyperkalemia also potentiates all-or-none early repolarization of the AP, potentially predisposing the heart to phase 2 reentry, as in Brugada, Short QT and Early Repolarization (J-wave) Syndromes 41. It is interesting that phase 2 reentry was originally described in hearts exposed to simulated global ischemia in which hyperkalemia (6 mmol/l) was included as a co-factor together with hypoxia and acidosis 42 (Fig. 5). However, whether hyperkalemia alone is capable of inducing phase 2 reentry and initiating VF has not, to our knowledge, been systematically investigated, even though hyperkalemia is classified as an acquired form of Short QT Syndrome 43. It is intriguing to speculate that the effectiveness of Ca2+ at “stabilizing the heart” during systemic hyperkalemia may be related in part to its enhancement of the L-type Ca2+ current, thereby suppressing all-or-none repolarization of the AP that is critical for the development of phase 2 reentry. Sympathetic enhancement of the L-type Ca2+ current during exercise might similarly protect the heart from phase 2 reentry-mediated arrhythmias during exercise-induced hyperkalemia. Further experimental studies are needed to better clarify the specific mechanisms of tachyarrhythmia initiation by systemic hyperkalemia. In simulated ventricles, once reentry has been initiated, hyperkalemia tends to stabilize spiral/scroll wave reentry by flattening APD restitution slope 44. The shorter APD increases the dominant frequency until CV slowing becomes prominent. The latter setting tends to promote the sinusoidal appearance of VT on the electrocardiogram in severe hyperkalemia.
Figure 5.
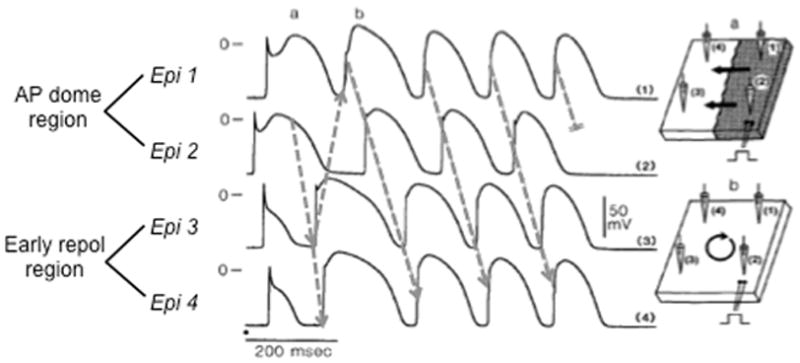
Phase 2 reentry during simulated ischemia in canine epicardium. Traces show AP recordings from four sites (Epi 1–4) in a canine epicardial sheet exposed to simulated ischemia ([K+]o=6 mmol/l, hypoxia, pH=6.8). Sites 1 and 2 exhibit normal APs with accentuated AP domes, whereas sites 3 and 4 show early repolarization. Arrows show re-excitation of site 3 by the AP dome at site 2, inducing phase 2 reentry that self-terminates after 4 beats. Adapted from Lukas & Antzelevitch 42.
Interstitial hyperkalemia
Interstitial hyperkalemia refers to elevated interstitial [K+] in tissue with normal serum [K+] in the circulation. Interstitial hyperkalemia is a prominent feature of acute myocardial ischemia due to global or regional cessation of coronary blood. During acute myocardial ischemia, intracellular Na+ ([Na+]i) accumulates due to a net imbalance involving both increased Na+ influx and decreased Na+-K+ ATPase reserve caused by the fall in tissue ATP levels, although the relative contributions are still debated. When [Na+]i increases, however, a counter charge movement (i.e. concomitant efflux of a positively-charged ion or influx of a negatively-charged ion) is also required, since a 1 mmol/l increase in positive charge from Na+ ions would depolarize membrane voltage by >30 volts if uncompensated. As the most ubiquitous intracellular cation with a high membrane permeability, K+ serves the bulk of this charge-compensating role 45. Without washout by coronary blood flow, the net K+ efflux causes interstitial [K+]o to accumulate rapidly, typically reaching 10–15 mmol/l in the first 10 minutes of ischemia 46, 47 (Fig. 648). Interstitial hyperkalemia during acute ischemia has all of the electrophysiological actions described for systemic hyperkalemia, complicated by additional changes related to hypoxia, acidosis and other ischemic components 44, 48. In particular, APD shortening is accelerated by activation of sarcolemmal ATP-sensitive K (KATP) channels due the ischemic fall in the cytoplasmic ATP/ADP ratio. The activation of sarcolemmal KATP channels, however, does not itself accelerate the net K+ loss during ischemia 45. This is because the APD shortening induced by the increase in K+ conductance is offset by the decrease in driving force Em-EK for K+ efflux over the cardiac cycle due to the prolongation of diastole (Fig. 6A). APD shortening is also accelerated by intracellular acidosis during ischemia, which suppresses both L-type Ca2+ and Na+ currents. Other ischemic components, such as catecholamines, adenosine, fatty acid metabolites, lysophospholipids, etc. also contribute to complex, temporally evolving electrophysiological changes 49. The net result is more rapid APD shortening, onset of post-repolarization refractoriness, CV slowing and conduction block, typically leading to inexcitability within 15–25 min (Fig 6B).
Figure 6.
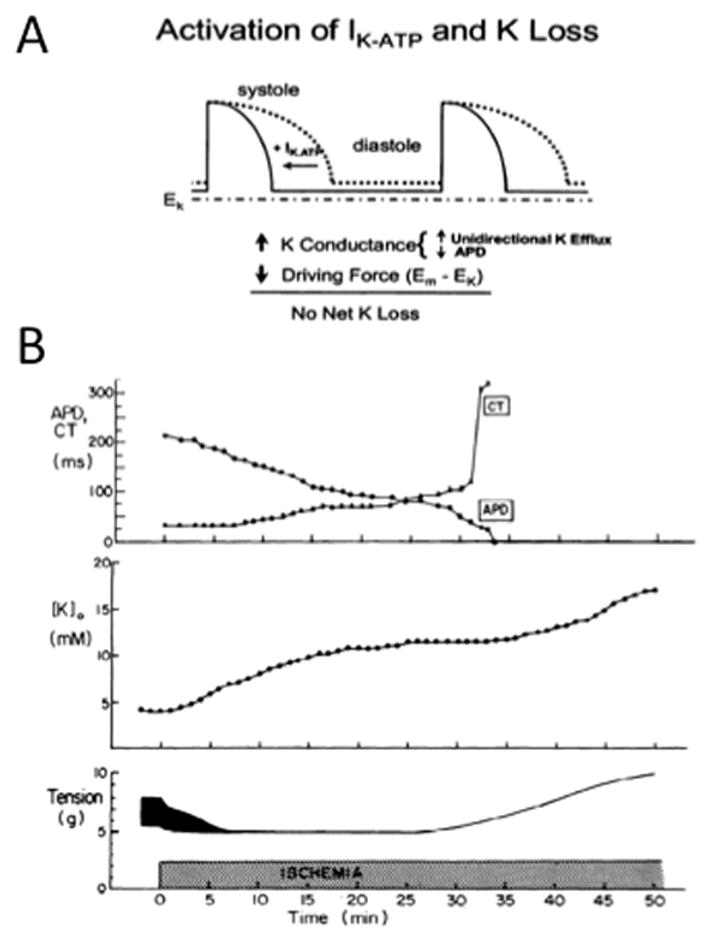
A. Schematic illustrating how APD shortening due to ATP-sensitive K+ current (IKATP) activation offsets net cellular K+ loss by decreasing the average driving force Em-EK for K+ efflux over the cardiac cycle. B. ADP shortening, conduction time (CT) delay, tension development and interstitial [K+]o accumulation versus time during acute global ischemia in rabbit ventricle. Reproduced from Weiss & Shine 48 with permission.
Acute global ischemia (or hypoxia with maintained coronary blood flow to wash out ischemic metabolites), however, is less arrhythmogenic than acute regional ischemia following coronary artery occlusion. The link between ischemic [K+]o accumulation and arrhythmias following coronary occlusion was first delineated by Harris et al 50 (Fig. 7). Subsequent studies with K+-selective electrodes documented that [K+]o increased to 10–15 mmol/l in the central ischemic zone within the first 10 min of coronary occlusion, creating a steep [K+]o gradient between ischemic and adjacent non-ischemic tissue 46. As [K+]o increases, the subepicardial tissue layer, with its greater repolarization reserve due to higher expression of the transient outward current Ito, becomes inexcitable prior to the subendocardial tissue layer 51. After 10–15 mins, [K+]o reaches a plateau followed by a secondary rise after 20–30 min associated with contracture and irreversible injury upon reperfusion 47. This sequence accounts for both the electrocardiographic changes and early arrhythmia phases following coronary occlusion, as follows.
Figure 7.
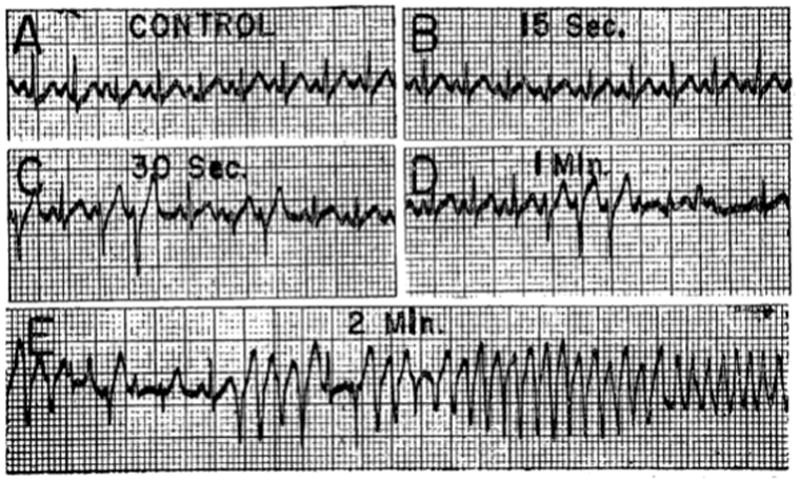
Ventricular arrhythmias following injection of KCl (2.8 mg/kg) into the left anterior descending coronary artery of a dog. From Harris & Rojas 50 with permission.
Electrocardiographic changes
When APD shortens in the ischemic region due to [K+]o accumulation, KATP channel activation and other ischemic factors, the overlying T wave becomes peaked as in systemic hyperkalemia, followed by ST segment elevation. ST segment elevation is a somewhat misleading term because standard electrocardiographic recordings are high-pass filtered to remove baseline drift, obfuscating the contribution of TQ depression to the apparent ST elevation. When DC electrocardiograms are recorded, as much as half of the apparent ST elevation is caused by TQ depression 52. This is a direct reflection of [K+]o accumulation in the ischemic region, which depolarizes the diastolic Em relative to non-ischemic tissue 53. This voltage difference causes a diastolic injury current to flow through gap junctions across the border zone, depressing the TQ segment. During systole, APD shortening in ischemic tissue also generates a systolic injury current of opposite polarity to the diastolic injury current. The combined effect is to depress the TQ segment and elevate the ST segment. The abnormal repolarization in the ischemic area also commonly inverts the T wave.
Arrhythmia mechanisms
In addition to being a marker of acute myocardial ischemia, the injury currents causing ST segment elevation/TQ depression also play a direct causal role in ischemic arrhythmogenesis. Following coronary occlusion, the diastolic injury current caused by difference in resting Em between ischemic and non-ischemic tissue depolarizes the neighboring non-ischemic tissue. In non-ischemic areas where the injury current density is highest, the depolarization may be sufficient to trigger a PVC, potentially initiating reentry, especially if the CV is depressed in the ischemic region 52 (Fig. 8A). In addition, as [K+]o accumulation and KATP channel activation increase repolarization reserve, the subepicardial tissue layer, with its greater intrinsic repolarization reserve due to Ito, becomes susceptible to all-or-one repolarization and phase 2 reentry 42, which can precipitate VF (Fig. 5 and 8B).
Figure 8.
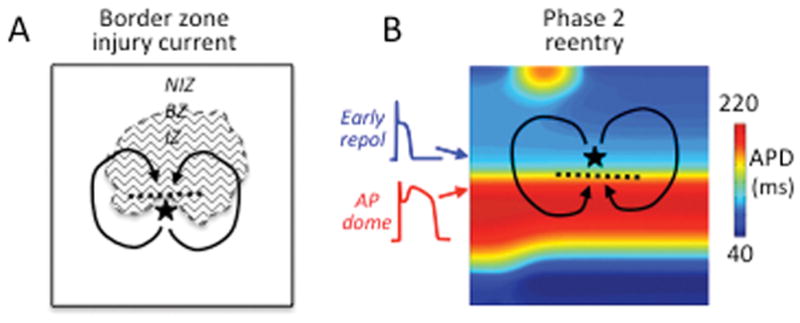
Initiation of reentry during acute ischemia. A. Injury current across the border zone (BZ) excites adjacent repolarized tissue in the non-ischemic zone (NIZ) to trigger a PVC (|) which propagates through the NIZ and reenters the ischemic zone (IZ) to initiate reentry. B. In ischemic subepicardial tissue, regions of all-or-none early repolarization with very short APD (blue) are juxtaposed to adjacent regions with normal APD and accentuated AP domes (red). The AP dome propagates into the repolarized region to trigger a closely coupled PVC (|) that propagates laterally until the normal AP region repolarizes and then reenters this region to initiate phase 2 reentry.
In summary, the interaction between the depolarized ischemic region with elevated [K+]o and normally polarized non-ischemic regions creates marked electrophysiological dispersion in resting Em, APD, ERP and CV. These changes generate both triggers and an arrhythmogenic substrate promoting initiation of reentry, resulting in a high incidence of ventricular ectopy, VT and VF during the first 25–30 min after acute coronary occlusion (called Harris phase 1 arrhythmias 54). These early arrhythmias are the major cause of mortality in patients with acute myocardial infarction who die before reaching the hospital (around 60 % of all deaths from acute myocardial infarction 55). After 20–30 min of ischemia, however, gap junctions between ischemic myocytes become dephosphorylated and close, electrically isolating ischemic myocytes from their neighbors. At this point, corresponding to the secondary rise in [K+]o (Fig. 6B), the ischemic region becomes inexcitable and no longer capable of transmitting injury currents across the border zone into non-ischemic tissue, causing the arrhythmias to cease. However, arrhythmias often return 6–8 hours later due to automaticity arising from surviving but hypoxic subendocardial Purkinje fibers (called Harris phase 2 arrhythmias 49, 54).
Synopsis
Up to 20% of patients admitted to the hospital exhibit hypokalemia 56 and 3.5% exhibit hyperkalemia 57. Both have powerful electrophysiological effects promoting cardiac arrhythmias. Hypokalemia ([K+]o<3.5 mmol/l) reduces repolarization reserve by directly inhibiting K+ channel conductances and indirectly by suppressing Na+-K+ ATPase. The latter results in intracellular Na+ and Ca2+ loading activating CaMK signaling whose targets include Na+ and Ca2+ channels, initiating positive feedback cascades that further reduce repolarization reserve to the range promoting EADs, DADs and afterdepolarization-mediated arrhythmias (Fig. 4). The pro-arrhythmic effects of Class III antiarrhythmic drugs are increased by elevated CaMK activity during hypokalemia and heart failure. In animal models, blocking CaMK or the late Na+ current activated by CaMK are effective strategies to suppress afterdepolarization-mediated arrhythmias induced by hypokalemia and class III antiarrhythmic drugs.
In contrast, systemic hyperkalemia ([K+]o>5.5 mmol/l) enhances repolarization reserve by increasing K+ channel conductance, shortening APD and inducing post-repolarization refractoriness, manifested electrocardiographically by peaked T waves. Hyperkalemia also depolarizes resting membrane potential, which first accelerates but then slows CV at [K+]o >8 mmol/l, manifested electrocardiographically by broadening and disappearance of the P wave (sinoventricular conduction) and QRS widening. Hyperkalemia also accentuates CV restitution, even when CV is accelerated, such that premature extrasystoles conduct more slowly and are more likely to develop conduction block. Accentuated CV restitution, together with post-repolarization refractoriness, may also make hyperkalemic tissue more susceptible to arrhythmogenic spatially discordant repolarization alternans. When combined with hypoxia and acidosis to simulate global ischemia, hyperkalemia can promote phase 2 reentry by further increasing repolarization reserve in epicardial ventricular tissue (Fig. 542). Whether hyperkalemia alone causes phase 2 reentry remains to be determined.
Systemic and global interstitial hyperkalemia are less arrhythmogenic than regional interstitial hyperkalemia following coronary occlusion. Following coronary occlusion, interstitial [K+]o rises rapidly at a rate of 1.0–1.5 mmol/l/min in the central ischemic zone. APD shortening and resting membrane potential depolarization cause both diastolic and systolic injury currents to flow across the border zone. These injury currents appear electrocardiographically as ST elevation, but actually represent a combination of TQ depression and ST elevation 52. Injury currents flowing across the border zone can reexcite non-ischemic recovered tissue to induce extrasystoles that initiate reentry (Fig. 8A). Interstitial hyperkalemia also promotes all-or-none early repolarization in the ischemic subepicardium, inducing phase 2 reentry and VT/VF (Fig. 8B), analogous to Brugada Short QT and Early Repolarization Syndromes 41. These arrhythmogenic effects of interstitial hyperkalemia play a critical role in the majority of sudden cardiac deaths due to VT/VF in patients who never reach the hospital after an acute coronary occlusion 55.
Acknowledgments
Sources of Funding: Supported by NIH/NHLBI grant P01 HL079831 and the Laubisch and Kawata Endowments (JNW).
Footnotes
Disclosures: None.
References
- 1.Schulman M, Narins RG. Hypokalemia and cardiovascular disease. Am J Cardiol. 1990;65:4E–9. doi: 10.1016/0002-9149(90)90244-u. E; discussion 22E–23E. [DOI] [PubMed] [Google Scholar]
- 2.Pezhouman A, Singh N, Song Z, Nivala M, Eskandari A, Cao H, Bapat A, Ko CY, Nguyen TP, Qu Z, Karagueuzian HS, Weiss JN. Molecular basis of hypokalemia-induced ventricular fibrillation. Circulation. 2015;132:1528–1537. doi: 10.1161/CIRCULATIONAHA.115.016217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poelzing S, Veeraraghavan R. Heterogeneous ventricular chamber response to hypokalemia and inward rectifier potassium channel blockade underlies bifurcated T wave in guinea pig. Am J Physiol Heart Circ Physiol. 2007;292:H3043–3051. doi: 10.1152/ajpheart.01312.2006. [DOI] [PubMed] [Google Scholar]
- 4.Sato D, Xie LH, Sovari AA, Tran DX, Morita N, Xie F, Karagueuzian H, Garfinkel A, Weiss JN, Qu Z. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:2983–2988. doi: 10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–1899. doi: 10.1016/j.hrthm.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss JN, Garfinkel A, Karagueuzian HS, Nguyen TP, Olcese R, Chen PS, Qu Z. Perspective: A dynamics-based classification of ventricular arrhythmias. J Mol Cell Cardiol. 2015;82:136–152. doi: 10.1016/j.yjmcc.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang MG, Sato D, de Lange E, Lee JH, Karagueuzian HS, Garfinkel A, Weiss JN, 1Qu Z. Bi-stable wave propagation and early afterdepolarization-mediated cardiac arrhythmias. Heart Rhythm. 2012;9:115–122. doi: 10.1016/j.hrthm.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimoni Y, Clark RB, Giles WR. Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J Physiol. 1992;448:709–727. doi: 10.1113/jphysiol.1992.sp019066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanguinetti MC, Jurkiewicz NK. Role of external Ca2+ and K+ in gating of cardiac delayed rectifier K+ currents. Pflugers Arch. 1992;420:180–186. doi: 10.1007/BF00374988. [DOI] [PubMed] [Google Scholar]
- 10.Yang T, Snyders DJ, Roden DM. Rapid inactivation determines the rectification and [K+]o dependence of the rapid component of the delayed rectifier K current in cardiac cells. Circ Res. 1997;80:782–789. doi: 10.1161/01.res.80.6.782. [DOI] [PubMed] [Google Scholar]
- 11.Firek L, Giles WR. Outward currents underlying repolarization in human atrial myocytes. Cardiovasc Res. 1995;30:31–38. [PubMed] [Google Scholar]
- 12.Killeen MJ, Thomas G, Gurung IS, Goddard CA, Fraser JA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CL. Arrhythmogenic mechanisms in the isolated perfused hypokalaemic murine heart. Acta Physiol (Oxf) 2007;189:33–46. doi: 10.1111/j.1748-1716.2006.01643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 14.Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N, Zhang S. Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest. 2009;119:2745–2757. doi: 10.1172/JCI39027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cordeiro JM, Spitzer KW, Giles WR. Repolarizing K+ currents in rabbit heart Purkinje cells. J Physiol. 1998;508:811–823. doi: 10.1111/j.1469-7793.1998.811bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han F, Tucker AL, Lingrel JB, Despa S, Bers DM. Extracellular potassium dependence of the Na+-K+-ATPase in cardiac myocytes: isoform specificity and effect of phospholemman. Am J Physiol Cell Physiol. 2009;297:C699–705. doi: 10.1152/ajpcell.00063.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonough AA, Zhang Y, Shin V, Frank JS. Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am J Physiol. 1996;270:C1221–1227. doi: 10.1152/ajpcell.1996.270.4.C1221. [DOI] [PubMed] [Google Scholar]
- 18.Weiss JN. Palpitations, potassium and the pump. J Physiol. 2015;593:1387–1388. doi: 10.1113/jphysiol.2014.285924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aronsen JM, Skogestad J, Lewalle A, Louch WE, Hougen K, Stokke MK, Swift F, Niederer S, Smith NP, Sejersted OM, Sjaastad I. Hypokalaemia induces Ca2+ overload and Ca2+ waves in ventricular myocytes by reducing Na+,K+-ATPase alpha2 activity. J Physiol. 2015;593:1509–1521. doi: 10.1113/jphysiol.2014.279893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swaminathan PD, Purohit A, Hund TJ, Anderson ME. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–1677. doi: 10.1161/CIRCRESAHA.111.243956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late INa augmentation leading to cellular Na+ and Ca2+ overload. Circ Res. 2011;108:555–565. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.January CT, Riddle JM, Salata JJ. A model for early afterdepolarizations: induction with the Ca2+ channel agonist Bay K 8644. Circ Res. 1988;62:563–571. doi: 10.1161/01.res.62.3.563. [DOI] [PubMed] [Google Scholar]
- 23.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. 2. Afterdepolarizations, triggered activity, and potentiation. Circ Res. 1994;74:1097–1113. doi: 10.1161/01.res.74.6.1097. [DOI] [PubMed] [Google Scholar]
- 24.Madhvani RV, Angelini M, Xie Y, Pantazis A, Suriany S, Borgstrom NP, Garfinkel A, Qu Z, Weiss JN, Olcese R. Targeting the late component of the cardiac L-type Ca2+ current to suppress early afterdepolarizations. J Gen Physiol. 2015;145:395–404. doi: 10.1085/jgp.201411288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu MB, Ko CY, Song Z, Garfinkel A, Weiss JN, Qu Z. A dynamical threshold for cardiac delayed afterdepolarization-mediated triggered activity. Biophys J. 2016;111:2523–2533. doi: 10.1016/j.bpj.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zehender M, Hohnloser S, Just H. QT-interval prolonging drugs: mechanisms and clinical relevance of their arrhythmogenic hazards. Cardiovasc Drugs Ther. 1991;5:515–530. doi: 10.1007/BF03029779. [DOI] [PubMed] [Google Scholar]
- 27.Wenzel-Seifert K, Wittmann M, Haen E. QTc prolongation by psychotropic drugs and the risk of Torsade de Pointes. Dtsch Arztebl Int. 2011;108:687–693. doi: 10.3238/arztebl.2011.0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang T, Roden DM. Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93:407–411. doi: 10.1161/01.cir.93.3.407. [DOI] [PubMed] [Google Scholar]
- 29.Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca2+/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- 30.Mazur A, Roden DM, Anderson ME. Systemic administration of calmodulin antagonist W-7 or protein kinase A inhibitor H-8 prevents torsade de pointes in rabbits. Circulation. 1999;100:2437–2442. doi: 10.1161/01.cir.100.24.2437. [DOI] [PubMed] [Google Scholar]
- 31.Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C, Roden DM. Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation. 2014;130:224–234. doi: 10.1161/CIRCULATIONAHA.113.007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia. A mechanistic investigation of action potential conduction and conduction failure. Circ Res. 1997;80:124–138. doi: 10.1161/01.res.80.1.124. [DOI] [PubMed] [Google Scholar]
- 33.Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–566. doi: 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- 34.Medbo JI, Sejersted OM. Plasma potassium changes with high intensity exercise. J Physiol. 1990;421:105–122. doi: 10.1113/jphysiol.1990.sp017935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dinari G, Aygen MM. Sinoventricular conduction. N Engl J Med. 1973;289:1238–1239. doi: 10.1056/NEJM197312062892309. [DOI] [PubMed] [Google Scholar]
- 36.Kim EM, Choy Y, Vassalle M. Mechanisms of suppression and initiation of pacemaker activity in guinea-pig sino-atrial node superfused in high [K+]o. J Mol Cell Cardiol. 1997;29:1433–1445. doi: 10.1006/jmcc.1997.0382. [DOI] [PubMed] [Google Scholar]
- 37.Cohen HC, Gozo EG, Jr, Pick A. The nature and type of arrhythmias in acute experimental hyperkalemia in the intact dog. Am Heart J. 1971;82:777–85. doi: 10.1016/0002-8703(71)90199-2. [DOI] [PubMed] [Google Scholar]
- 38.Martynyuk AE, Morey TE, Belardinelli L, Dennis DM. Hyperkalemia enhances the effect of adenosine on IK,ADO in rabbit isolated AV nodal myocytes and on AV nodal conduction in guinea pig isolated heart. Circulation. 1999;99:312–318. doi: 10.1161/01.cir.99.2.312. [DOI] [PubMed] [Google Scholar]
- 39.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. doi: 10.1161/01.cir.99.10.1385. [DOI] [PubMed] [Google Scholar]
- 40.Weiss JN, Qu Z, Chen P-S, Lin S-F, Karagueuzian HS, Hayashi H, Garfinkel A, Karma A. The dynamics of cardiac fibrillation. Circulation. 2005;112:1232–1240. doi: 10.1161/CIRCULATIONAHA.104.529545. [DOI] [PubMed] [Google Scholar]
- 41.Antzelevitch C, Yan GX. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm. 2015;12:1852–1866. doi: 10.1016/j.hrthm.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res. 1996;32:593–603. [PubMed] [Google Scholar]
- 43.Patel C, Yan GX, Antzelevitch C. Short QT syndrome: from bench to bedside. Circ Arrhythm Electrophysiol. 2010;3:401–408. doi: 10.1161/CIRCEP.109.921056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kazbanov IV, Clayton RH, Nash MP, Bradley CP, Paterson DJ, Hayward MP, Taggart P, Panfilov AV. Effect of global cardiac ischemia on human ventricular fibrillation: insights from a multi-scale mechanistic model of the human heart. PLoS Comput Biol. 2014;10:e1003891. doi: 10.1371/journal.pcbi.1003891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shivkumar K, Deutsch NA, Lamp ST, Khuu K, Goldhaber JI, Weiss JN. Mechanism of hypoxic K loss in rabbit ventricle. J Clin Invest. 1997;100:1782–1788. doi: 10.1172/JCI119705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hill JL, Gettes LS. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation. 1980;61:768–778. doi: 10.1161/01.cir.61.4.768. [DOI] [PubMed] [Google Scholar]
- 47.Weiss J, Shine KI. Extracellular K+ accumulation during myocardial ischemia in isolated rabbit heart. Am J Physiol. 1982;242:H619–628. doi: 10.1152/ajpheart.1982.242.4.H619. [DOI] [PubMed] [Google Scholar]
- 48.Weiss J, Shine KI. [K]o accumulation and electrophysiological alterations during early myocardial ischemia. Am J Physiol. 1982;243:H318–327. doi: 10.1152/ajpheart.1982.243.2.H318. [DOI] [PubMed] [Google Scholar]
- 49.Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischaemia and infarction. Physiol Rev. 1989;69:1049–1168. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- 50.Harris AS, Bisteni A, Russell RA, Brigham JC, Firestone JE. Excitatory factors in ventricular tachycardia resulting from myocardial ischemia; potassium a major excitant. Science. 1954;119:200–203. doi: 10.1126/science.119.3085.200. [DOI] [PubMed] [Google Scholar]
- 51.Gilmour RF, Jr, Zipes DP. Different electrophysiological responses of canine endocardium and epicardium to combined hyperkalemia, hypoxia, and acidosis. Circ Res. 1980;46:814–825. doi: 10.1161/01.res.46.6.814. [DOI] [PubMed] [Google Scholar]
- 52.Janse MJ, van Capelle FJ, Morsink H, Kleber AG, Wilms-Schopman F, Cardinal R, d’Alnoncourt CN, Durrer D. Flow of “injury” current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial ischemia in isolated porcine and canine hearts. Evidence for two different arrhythmogenic mechanisms. Circ Res. 1980;47:151–165. doi: 10.1161/01.res.47.2.151. [DOI] [PubMed] [Google Scholar]
- 53.Coronel R, Fiolet JWT, Wilms-Schopman FJG, Schaapherder AFM, Johnson TA, Gettes LS, Janse MJ. Distribution of extracellular potassium and its relation to electrophysiologic changes during acute myocardial ischemia in the isolated perfused porcine heart. Circulation. 1988;77:1125–1138. doi: 10.1161/01.cir.77.5.1125. [DOI] [PubMed] [Google Scholar]
- 54.Harris AS, Guevara Rojas A. The initiation of ventricular fibrillation due to coronary occlusion. Exper Med & Surg. 1943;1:105. [Google Scholar]
- 55.Wallace WA, Yu PN. Sudden death and the pre-hospital phase of acute myocardial infarction. Annu Rev Med. 1975;26:1–7. doi: 10.1146/annurev.me.26.020175.000245. [DOI] [PubMed] [Google Scholar]
- 56.Paice BJ, Paterson KR, Onyanga-Omara F, Donnelly T, Gray JM, Lawson DH. Record linkage study of hypokalaemia in hospitalized patients. Postgrad Med J. 1986;62:187–191. doi: 10.1136/pgmj.62.725.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Einhorn LM, Zhan M, Hsu VD, Walker LD, Moen MF, Seliger SL, Weir MR, Fink JC. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Int Med. 2009;169:1156–1162. doi: 10.1001/archinternmed.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]