

Abstract

Many 1,2,4-benzotriazine 1,4-dioxides display the ability to selectively kill the oxygen-poor cells found in solid tumors. As a result, there is a desire for synthetic routes that afford access to substituted 1,2,4-benzotriazine 1-oxides that can be used as direct precursors in the synthesis of 1,2,4-benzotriazine 1,4-dioxides. Here we describe the use of Suzuki-Miyaura and Buchwald-Hartwig cross-coupling reactions for the construction of various 1,2,4-benzotriazine 1-oxide analogs bearing substituents at the 3-, 6-, and 7-positions.
INTRODUCTION
Tirapazamine (1) and many other 1,2,4-benzotriazine 1,4-dioxides are selectively toxic to the oxygen poor (hypoxic) cells found within solid tumors.1-3 These compounds undergo intracellular enzymatic one-electron reduction to yield a radical that undergoes relatively harmless back-oxidation to starting material in normally-oxygenated cells. On the other hand, under hypoxic conditions, the neutral radical intermediate decomposes to release a highly oxidizing secondary radical that causes cytotoxic DNA damage.1-16 Promising preclinical results led to the examination of tirapazamine in a large number of clinical trials, but thus far, the results of these studies have not earned FDA approval for the drug.17 As a result, there have been substantial efforts to prepare tirapazamine analogues with improved efficacy.18-27 Accordingly, there is a continuing need for the development of synthetic routes that afford access to tirapazamine analogs.
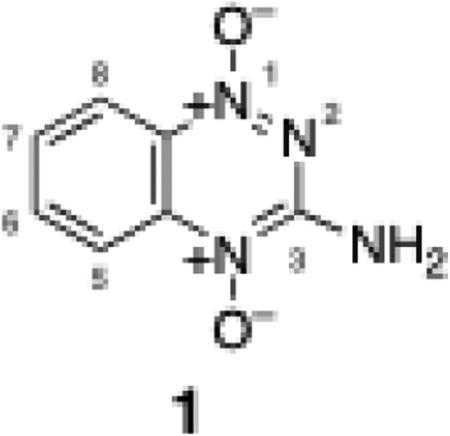
A variety of methods have been developed for the synthesis of tirapazamine and related analogs. Tirapazamine can be prepared by the reaction of benzofuroxan with sodium cyanamide.28 Alternatively, condensation of 2-nitroaniline with cyanamide yields 1,2,4-benzotriazine 1-oxide (2, Scheme 1) which can be oxidized to tirapazamine using H2O2/HOAc, mCPBA, or HOF-CH3CN.14,29-31 The reaction of 1-fluoro-2-nitrobenzene or 1,2-dinitrobenzene with guanidine base provides another route to 2.32 Condensation of various 2-nitroaniline derivatives with cyanamide provides access to a large number of tirapazamine derivatives bearing substituents on the benzo ring.18,20,26,33 Analogues of 2 bearing sulfur or alkoxy substitutents (rather than NH2) at the 3-position have been prepared by diazotization and hydrolysis of 2 to afford the 3-hydroxy-1,2,4-benzotriazine 1-oxide analog, followed by reaction with phosphorus oxyhalide to give the 3-chloro- or 3-bromo-1,2,4-benzotriazine 1-oxide (3a and b), and finally treatment with an appropriate oxygen or sulfur nucleophile.34 Similarly, a variety of tirapazamine analogues bearing alkyl and aryl groups on the 3-amino substituent have been prepared by nucleophilic aromatic substitution involving attack of amines on 3a, followed by oxidation to give the di-N-oxides.35,36 Photochemical methods also may enable preparation of 3-(arylamino)-1,2,4-benzotriazine 1,4-dioxide derivatives from 3-acetamido-1,2,4-benzotriazine 1,4-dioxide.37
Scheme 1.

Synthesis of Halogenated Tirapazamine Derivatives for Use in Pd-Mediated Coupling Reactions.
There is interest in 3-alkyl- and 3-aryl-1,2,4-benzotriazine 1,4-dioxides because these compounds display hypoxia-selective DNA-cleaving properties and cytotoxicities that are comparable to tirapazamine.8,38 In addition, these analogues may possess superior pharmacokinetic properties.19,20 The analogs, 3-methyl- and 3-phenyl-1,2,4-benzotriazine oxide, have been prepared by BF3-catalyzed cyclization of formazan precursors39 or PtO2-catalyzed cyclization of the 2-nitrophenylhydrazone of pyruvic acid,40 followed by N-oxidation using H2O2/TFAA.38
Palladium-catalyzed cross-coupling reactions such as the Suzuki-Miyaura, Buchwald-Hartwig, and Stille reactions are powerful synthetic methods that may enable the synthesis of many diverse tirapazamine analogs.41,42 Along these lines, the palladium-mediated Stille coupling reactions have been employed for the preparation of 3-alkyl, aryl, vinyl and allyl 1,2,4-benzotriazine 1-oxides from 3a.25,33 In addition, there is a single report in which the Suzuki-Miyaura reaction was used to prepare 3-aryl-1,2,4-benzotriazine 1,4-dioxides from 3-halo-1,2,4-benzotriazine 1-oxide precursors.25 The results described herein expand the use of the Suzuki-Miyaura reaction and provide the first uses of the Buchwald-Hartwig reaction for the preparation of 1,2,4-benzotriazine 1-oxide analogs related to the antitumor agent tirapazamine.
RESULTS AND DISCUSSION
Six halogenated 1,2,4-benzotriazine 1-oxides were prepared for use in palladium-catalyzed coupling reactions. Compounds 3 were prepared via reaction of 2 with sodium nitrite in aqueous sulfuric acid, followed by treatment with the appropriate phosphorus oxyhalide (Scheme 2).33,34 Analogs 4 bearing halogens on the benzo-ring were prepared by a well-established route involving condensation of cyanamide with the appropriately substituted 2-nitroaniline.14,29,30
The halogen derivatives 3 were employed as substrates in Suzuki coupling reactions with aryl and cyclopropyl boronic acids (Table 1). In these reactions, compound 3, the boronic acid (1.2 equiv), and the ligand tricyclohexylphosphine or 2-dicyclohexylphosphino-2',6'-dimethoxybiphenyl (PCy3 or SPhos, 10 mol%)43 were mixed with Pd(OAc)2 (5 mol%) in a mixture of toluene or toluene-water (3:1 v/v) containing potassium phosphate or cesium carbonate and heated in a sealed tube at 110 °C for 24 h. Reactions of the chlorinated substrate 3a with cyclopropylboronic acid and 4-cyanophenylboronic acid proceeded in reasonable yields to give the coupling products 5 and 9, respectively.44,45 Use of the brominated derivative 3b did not substantially alter the yields in these cases. Reactions of 3a with 4-bromophenylboronic acid and 4-nitrophenylboronic acid gave very low and modest yields of the products 6 and 7, respectively (Table 1). In these cases, use of the brominated substrate 3b improved the yields of the desired coupling products 6 and 7. This was not unexpected because aryl bromides typically are better substrates than the analogous chlorides in Suzuki coupling reactions.41,42 The reaction between quinolin-2-ylboronic acid and 3a or 3b employing the PCy3 ligand proceeded in very low yields even with the brominated substrate 3b, but use of the electron-rich SPhos ligand46 afforded improved yields of the coupling product 8 (Table 1). Use of the SPhos ligand did not improve the yield of 6 obtained from the coupling of 3b with 4-bromophenylboronic acid using the PCy3 ligand (Table 1). The aryl bromide residue in 6 could be a useful handle for further elaboration via palladium-catalyzed reactions. Reaction of 4-(N-Boc-amino)phenylboronic acid with 3b using the SPhos ligand afforded a reasonable yield of the coupling product 10.
Table 1.
Preparation of 3-Aryl and 3-Cyclopropyl Derivatives of 1,2,4-Benzotriazine 1-Oxide.

| |||||
|---|---|---|---|---|---|
| Aryl halide | R2, product | Yield [%] | Ligand | Base | Solvent |
| 3a | 4-cyanophenyl, 5 | 66 | SPhos | Cs2CO3 | Toluene |
| 3b | 74 | SPhos | Cs2CO3 | Toluene | |
| 3a | 4-bromophenyl, 6 | trace | PCy3 | K3PO4 | MeCN/Water |
| 3b | 57 | PCy3 | K3PO4 | MeCN/Water | |
| 3a | 4-nitrophenyl, 7 | 42 | SPhos | Cs2CO3 | Toluene |
| 3b | 65 | SPhos | Cs2CO3 | Toluene | |
| 3a | quinolin-2-yl, 8 | 29 | SPhos | K3PO4 | Toluene/Water |
| 3b | 40 | SPhos | K3PO4 | Toluene/Water | |
| 3a | cyclopropyl, 9 | 76 | SPhos | Cs2CO3 | Toluene |
| 3b | 80 | SPhos | Cs2CO3 | Toluene | |
| 3b | 4-N-Boc-aminophenyl, 10 | 61 | SPhos | K3PO4 | Toluene/Water |
Column chromatography of the reaction between 3a and cyclopropylboronic acid with PCy3 as ligand for palladium, produced a fraction that, upon slow evaporation, gave a low yield (~ 1%) of pale yellow crystals that were characterized by X-ray diffraction. Interestingly, the material proved to be a dinuclear triazine-bridged palladium complex resulting from oxidative addition of 3a to the palladium catalyst. The C1-carbon and N2-nitrogen atoms of two benzotriazine 1-oxides and the two palladium centers form a six-membered ring in a boat conformation in which the nitrogens are trans to the phosphine ligands and carbons are trans to the chlorides (Figure 1). The Pd-Pd interatomic distance is 3.1709(4) Å. This structure is structurally analogous to that of a product previously obtained from the addition of Pd(PPh3)4 to 2-bromopyridine.47,48
Fig. 1.

Crystal Structure of a Complex Resulting from Oxidative Addition of 3a to Palladium.
The success of Suzuki coupling reactions employing 3 led us to extend this reaction to the aryl halides 4a and 4b. In these reactions, 4a or 4b, the boronic acid (1.2 equiv), and the ligand (PCy3 or SPhos, 10 mol%) were combined with Pd(OAc)2 (5 mol%) in a mixture of toluene or toluene-water (3:1 v/v) containing potassium phosphate or cesium carbonate and heated in a sealed tube at 110 °C for 24 h. These reactions afforded reasonable yields (up to 72%) of the products 11-14 (Table 2). Analogous reactions refluxed under nitrogen gas gave yields that were substantially lower than those obtained by heating in sealed tubes.
Table 2.
Preparation of 7-Aryl Derivatives of Tirapazamine.

| |||||
|---|---|---|---|---|---|
| Aryl halide | R2, product | Yield [%] | Ligand | Base | Solvent |
| 4a | 4-methoxyphenyl, 11 | 46 | SPhos | Cs2CO3 | Toluene |
| 4b | 72 | SPhos | Cs2CO3 | Toluene | |
| 4b | 4-aminophenyl, 12 | 30 | PCy3 | K3PO4 | Toluene/Water |
| 4b | 4-cyanophenyl, 13 | 60 | SPhos | Cs2CO3 | Toluene |
| 4b | 2-Naphtyl-6'-methoxy, 14 | 27 | PCy3 | K3PO4 | Toluene/Water |
As part of these studies, we examined whether the boronic acid components of these reactions was stable in the presence of the N-oxide starting materials. We were motivated to examine this issue because Zhu et al. previously showed that alkyl- and aryl-N-oxides have the potential to convert boronic acids and boronic acid pinacol esters to the corresponding alcohols.49 Under our reaction conditions, however, we did not observe significant decomposition of 4-(N-Boc-amino)phenylboronic acid or cyclopropylboronic acid when these materials were heated at 110 °C for three days in toluene/water (3:1) with 3a, 3b, 4, or 4b (2 equiv, in the absence of palladium catalyst).
We next examined whether the Buchwald-Hartwig reaction could be applied the construction of substituted 1,2,4-benzotriazine 1-oxides.50 In these reactions, 4a or c, the ligand 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (X-Phos, 10 mol%), Pd(OAc)2 (5 mol%), K3PO4, and the desired amine (1.2 equiv) were dissolved in a solvent mixture composed of t-BuOH/water (3:1 v/v), placed in a sealed tube, and stirred for 24 h or subjected to microwave irradiation for 45 min (300 W, 60 PSI). Reactions of aniline or 6-aminoquinoline with 4a or 4c gave good yields of the coupling products 15-18 using either conventional or microwave heating (Table 3). Microwave heating has been used successfully in other palladium-mediated reactions51 and, in this case, allowed the use of shorter reaction times. Diphenylamine gave no product under these reaction conditions, presumably due to steric crowding. These palladium-catalyzed C-N bond forming reactions offer a useful alternative to the previously described nucleophilic aromatic substitution reactions involving the reaction of amines with 6- and 7-fluoro derivatives of tirapazamine that give low yields in some cases.12,18
Table 3.
Preparation of 6- and 7-Aminoaryl Tirapazamine Derivatives.

| ||||
|---|---|---|---|---|
| Aryl halide | Amine (R3, R4) | Product (R5, R6) | Yield [%] reflux | Yield [%] microwave |
| 4c | R3 = H, R4 = Ph | R5 = H, R6 = NHPh, 15 | 58 | 76 |
| 4a | R3 = H, R4 = Ph | R5 = NHPh, R6 = H, 16 | 81 | 75 |
| 4c | R3 = H, R4 = quinoline | R5 = H, R6=6-aminoquinoline, 17 | 73 | 61 |
| 4a | R3 = H, R4 = quinoline | R5=6-aminoquinoline, R6=H, 18 | 70 | 45 |
| 4a | R3 = Ph, R4 = Ph | R5 = NPh2, R6 = H | 0 | 0 |
| 4c | R3 = H, R4 = Ph | R5=H, R6 = NPh2 | 0 | 0 |
CONCLUSION
In summary, we examined the utility of palladium-mediated reactions for the synthesis of 1,2,4-benzotriazine derivatives related to the antitumor agent tirapazamine. Our work expands the use Suzuki-Miyaura-type reactions for the construction of 3-aryl-1,2,4-benzotriazines and presents the first use of Suzuki-Miyaura and Buchwald-Hartwig reactions for the functionalization of the 6- and 7-positions of the 1,2,4-benzotriazine ring system. Given the large number of commercially available aryl boronic acids and arylamines, our results should enable the preparation of many structurally diverse tirapazamine analogs. Nitrogen-rich heterocycles sometimes present challenges in palladium-catalyzed coupling reactions52,53 perhaps because of their propensity to coordinate the palladium catalyst. Indeed, we isolated and crystallographically characterized a dinuclear triazine-bridged palladium complex resulting from oxidative addition of 3a to the palladium catalyst. Nonetheless, the yields of the coupling products obtained here are of practical utility in the short, 3-4 step synthetic routes used to prepare the 1,2,4-benzotriazine 1,4-dioxide antitumor agents.
EXPERIMENTAL
Representative Procedure for the Synthesis of 6- and 7-Halo-1,2,4-benzotriazine 1-Oxides: 6-Chloro-1,2,4-benzotriazine 1-oxide (4c)
The compound 4-chloro-2-nitroaniline (3.02 g, 36 mmol) and cyanamide (6.20 g, 72 mmol) were mixed, melted by heating at 100 °C, and then cooled to room temperature. Concentrated HCl (30 mL) was added dropwise and the resulting mixture heated to 100 °C and stirred for 2 h (Caution: exotherm). The red-orange solution was then cooled to room temperature and NaOH (30 mL of a 16 M solution in water) was added over 15 min with stirring. The reaction mixture was heated to 100 °C for 3.5 h and then cooled to room temperature. Water (25 mL) was added and the resulting solid collected by filtration and washed with a solution of ethyl acetate-hexane (3:1) to give 6 in 38% yieild. In cases where purification was required, column chromatography on silica gel eluted with a gradient of 5%→20% methanol in CH2Cl2. 1H-NMR (DMSO-d6, 300 MHz): δ 8.12 (d, J = 2 Hz, 1H), 7.77 (dd, J = 9 Hz, J = 2 Hz, 1H), 7.54 (d, J = 9 Hz, 1H), 7.47 (s, 2H); 13C-NMR (DMSO-d6, 300 MHz): δ 160.4, 147.7, 136.1, 130.0, 128.3, 128.0, 119.0. HRMS (ESI, [M+H+]) m/z calcd for C10H10N3O 197.0230, found 197.0228.
Representative Procedure for Coupling of 3-Halo-1,2,4-benzotriazine 1-Oxides with Boronic Acids: 3-Cyclopropyl-1,2,4-benzotriazine 1-Oxide (9)
The compound 3-chloro-1,2,4-benzotriazine 1-oxide33,34 (3a, 60 mg, 0.33 mmol), cyclopropyl boronic acid (1.2 equiv, 34 mg), potassium phosphate (203 mg), PCy3 (10 mol%), and Pd(OAc)2 (5 mol%) were placed in a nitrogen-purged flask, dissolved in a mixture of toluene (2 mL) and water (100 μL), and refluxed for 24 h. The reaction was cooled to room temperature, water was added (5 mL), the mixture extracted with dichloromethane, the combined organic extracts dried over Na2SO4, and rotary evaporated. Column chromatography on silica gel eluted with a gradient of 5→25% ethyl acetate in hexane gave 9 as a pale yellow solid in 47% yield. 1H-NMR (300 MHz, CDCl3): δ 8.41 (1H, d, 8.7 Hz), 7.89-7.87 (2H, m), 7.62 (ddd, J = 9 Hz, 6 Hz, 2 Hz, 1H), 2.31 (tt, J = 8 Hz, J = 4 Hz, 1H), 1.36-1.31 (2H, m), 1.22-1.16 (2H, m); 13C-NMR (125 MHz, CDCl3): δ 167.7, 146.9, 134.7, 133.5, 128.4, 127.6, 119.4, 16.0, 10.42; HRMS (ESI) m/z calc for C10H10N3O (M+H+) 188.0824, found 188.0823.
Representative Procedure for Coupling of 6- and 7-Halo-1,2,4-benzotriazine 1-Oxides with Arylamines: 7-Aminophenyl-1,2,4-benzotriazine 1-Oxide (15)
The compound 7-chloro-1,2,4-benzotriazine 1-oxide (80 mg, 0.4 mmol), Pd(OAc)2 (5 mol%, 0.02 mmol, 4.6 mg), XPhos (10 mol%, 0.04 mmol), K3PO4 (3 equiv, 1.2 mmol, 254 mg) and aniline (75 mg, 0.8 mmol, 2 equiv) were placed in a sealed tube equipped with a stir bar and suspended in a solvent mixture composed of t-butanol:water (20 mL, 9:1). The mixture was stirred while heated at 110 °C for 24 h. The mixture was evaporated to dryness, taken up in methanol or tetrahydrofuran, filtered through celite, slurried with silica gel, evaporated, and the resulting powder dry-loaded on top of a silica gel column. Elution with a gradient of 0→50% ethyl acetate in hexane gave 15 as a red solid in 78% yield. 1H-NMR (500 MHz, DMSO-d6) δ 6.94 (s, 2 H) 7.00 (t, J=7 Hz, 1 H) 7.21 (d, J=8 Hz, 2 H) 7.36 (t, J=9 Hz, 2 H) 7.50 (d, J=9 Hz, 1 H) 7.58 (d, J=9 Hz, 2 H) 7.63 (s, 1 H) 8.77 (s, 1 H); 13C-NMR (126 MHz, DMSO-d6) δ 159.3, 144.6, 142.1, 141.9,130.6 129.8, 129.5, 127.4, 122.1, 118.9, 98.4; HRMS(ESI) m/z calc for C13H11N3O 254.1036, found 254.1046.
Supplementary Material
Acknowledgements
We are grateful to the National Institutes of Health for support of this work (CA 100757). We thank Steven R. Tannenbaum and John S. Wishnok for access to the ESI-TOF mass spectrometer (Agilent Technologies) in the Department of Biological Engineering, Massachusetts Institute of Technology.
Footnotes
Additional Supporting Information may be found in the online version of this article
Supplementary data
Supplementary data (descriptions of experimental procedures and characterization of all compounds, 50 pages) related to this article can be found at http://dx.doi.org/10.1016/j.tet. The crystal structure shown in Figure 1 can be accessed from the Cambridge Crystallographic Data Centre deposition number CCDC 718512.
REFERENCES AND NOTES
- 1.Brown JM. Cancer Res. 1999;59:5863–5870. [PubMed] [Google Scholar]
- 2.Brown JM, Wilson WR. Nature Rev. Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 3.Zeman EM, Brown JM, Lemmon MJ, Hirst VK, Lee WW. Int. J. Radiat. Oncol. Biol. Phys. 1986;12:1239–1242. doi: 10.1016/0360-3016(86)90267-1. [DOI] [PubMed] [Google Scholar]
- 4.Birincioglu M, Jaruga P, Chowdhury G, Rodriguez H, Dizdaroglu M, Gates KS. J. Am. Chem. Soc. 2003;125:11607–11615. doi: 10.1021/ja0352146. [DOI] [PubMed] [Google Scholar]
- 5.Daniels JS, Gates KS. J. Am. Chem. Soc. 1996;118:3380–3385. [Google Scholar]
- 6.Kotandeniya D, Ganley B, Gates KS. Bioorg. Med. Chem. Lett. 2002;12:2325–2329. doi: 10.1016/s0960-894x(02)00468-7. [DOI] [PubMed] [Google Scholar]
- 7.Chowdhury G, Junnutula V, Daniels JS, Greenberg MM, Gates KS. J. Am. Chem. Soc. 2007;129:12870–12877. doi: 10.1021/ja074432m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Junnotula V, Sarkar U, Sinha S, Gates KS. J. Am. Chem. Soc. 2009;131:1015–1024. doi: 10.1021/ja8049645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinde SS, Hay MP, Patterson AV, Denny WA, Anderson RF. J. Am. Chem. Soc. 2009;131:14220–14221. doi: 10.1021/ja906860a. [DOI] [PubMed] [Google Scholar]
- 10.Anderson RF, Yadav P, Patel DJ, Reynisson J, Tipparaju SR, Guise CP, Patterson AV, Denny WA, Maroz A, Shinde SS, Hay MP. Org. Biomol. Chem. 2014;12:3386–3392. doi: 10.1039/c4ob00236a. [DOI] [PubMed] [Google Scholar]
- 11.Shen X, Rajapakse A, Galazzi F, Junnotula V, Fuchs-Knotts T, Glaser R, Gates KS. Chem. Res. Toxicol. 2014;27:111–118. doi: 10.1021/tx400356y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson KM, Parsons ZD, Barnes CL, Gates KS. J. Org. Chem. 2014;79:7520–7531. doi: 10.1021/jo501252p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yadov P, Marshall AJ, Reynisson J, Denny WA, Hay MP, Anderson RF. Chem. Comm. 2014;50:13729–13731. doi: 10.1039/c4cc05657d. [DOI] [PubMed] [Google Scholar]
- 14.Fuchs T, Chowdhary G, Barnes CL, Gates KS. J. Org. Chem. 2001;66:107–114. doi: 10.1021/jo001232j. [DOI] [PubMed] [Google Scholar]
- 15.Yin J, Glaser R, Gates KS. Chem. Res. Toxicol. 2012;25:634–645. doi: 10.1021/tx200546u. [DOI] [PubMed] [Google Scholar]
- 16.Yin J, Glaser R, Gates KS. Chem. Res. Toxicol. 2012;25:620–633. doi: 10.1021/tx2005458. [DOI] [PubMed] [Google Scholar]
- 17.Marcu L, Olver I. Curr. Clin. Oncol. 2006;1:71–79. doi: 10.2174/157488406775268192. [DOI] [PubMed] [Google Scholar]
- 18.Hay MP, Gamage SA, Kovacs MS, Pruijn FB, Anderson RF, Patterson AV, Wilson WR, Brown JM, Denny WA. J. Med. Chem. 2003;46:169–182. doi: 10.1021/jm020367+. [DOI] [PubMed] [Google Scholar]
- 19.Hay MP, Hicks KO, Pruijn FB, Pchalek K, Siim BG, Wilson WR, Denny WA. J. Med. Chem. 2007;50:6392–6404. doi: 10.1021/jm070670g. [DOI] [PubMed] [Google Scholar]
- 20.Hay MP, Pchalek K, Pruijn FB, Hicks KO, Siim BG, Anderson MM, Shinde SS, Denny WA, Wilson WR. J. Med. Chem. 2007;50:6654–6664. doi: 10.1021/jm701037w. [DOI] [PubMed] [Google Scholar]
- 21.Hay MP, Pruijn FB, Gamage SA, Liyanage HDS, Kovacs MS, Patterson AV, Wilson WR, Brown JM, Denny WA. J. Med. Chem. 2004;47:475–488. doi: 10.1021/jm030399c. [DOI] [PubMed] [Google Scholar]
- 22.Solano B, Junnotula V, Marin A, Villar R, Burguete A, Vicente E, Perez-Silanes S, Monge A, Dutta S, Sarkar U, Gates KS. J. Med. Chem. 2007;50:5485–5492. doi: 10.1021/jm0703993. [DOI] [PubMed] [Google Scholar]
- 23.Ganley B, Chowdhury G, Bhansali J, Daniels JS, Gates KS. Bioorg. Med. Chem. 2001;9:2395–2401. doi: 10.1016/s0968-0896(01)00163-8. [DOI] [PubMed] [Google Scholar]
- 24.Chowdhury G, Kotandeniya D, Barnes CL, Gates KS. Chem. Res. Toxicol. 2004;17:1399–1405. doi: 10.1021/tx049836w. [DOI] [PubMed] [Google Scholar]
- 25.Hay MP, Denny WA. Tet. Lett. 2002;43:9569–9571. [Google Scholar]
- 26.Hay MP, Hicks KO, Pchalek K, Lee HH, Blaser A, Pruijn FB, Anderson RF, Shinde SS, Wilson WR, Denny WA. J. Med. Chem. 2008;51:6853–6865. doi: 10.1021/jm800967h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuchs T, Gates KS, Hwang J-T, Greenberg MM. Chem. Res. Toxicol. 1999;12:1190–1194. doi: 10.1021/tx990149s. [DOI] [PubMed] [Google Scholar]
- 28.Seng F, Ley K. Angew. Chem. Int. Ed. Eng. 1972;11:1009–1010. [Google Scholar]
- 29.Mason JC, Tennant GJ. Chem. Soc. B. 1970:911–916. [Google Scholar]
- 30.Arndt F. Ber. Dtsch. Chem. Ges. 1914;46:3522–3530. [Google Scholar]
- 31.Gatenyo J, Johnson K, Rajapakse A, Gates KS, Rozen S. Tetrahedron. 2012;68:8942–8944. [Google Scholar]
- 32.Suzuki H, Kawakami T. Synthesis. 1997 [Google Scholar]
- 33.Pchalek K, Hay MP. J. Org. Chem. 2006;71:6530–6535. doi: 10.1021/jo060986g. [DOI] [PubMed] [Google Scholar]
- 34.Jiu J, Mueller GP. J. Org. Chem. 1959;24:813–818. [Google Scholar]
- 35.Jiang F, Yang B, Fan L, He Q, Hu Y. Bioorganic Med. Chem. Lett. 2006;16:4209–4213. doi: 10.1016/j.bmcl.2006.05.095. [DOI] [PubMed] [Google Scholar]
- 36.Jiang F, Weng Q, Sheng R, Xia Q, He Q, Yang B, Hu Y. Arch. Pharm. Chem. Life Sci. 2007;340:258–263. doi: 10.1002/ardp.200600201. [DOI] [PubMed] [Google Scholar]
- 37.Daniels JS, Chatterji T, MacGillivray LR, Gates KS. J. Org. Chem. 1998;63:10027–10030. [Google Scholar]
- 38.Kelson AB, McNamara JP, Pandey A, Ryan KJ, Dorie MJ, McAfee PA, Menke DR, Brown JM, Tracy M. Anti-Cancer Drug Design. 1998;13:575–592. [PubMed] [Google Scholar]
- 39.Attallah RH, Nazer MZ. Tetrahedron. 1982;38:1793–1796. [Google Scholar]
- 40.Khodja M, Moulay S, Boutoumi H, Wilde H. Heteroatom Chem. 2006;17:166–172. [Google Scholar]
- 41.Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633–9695. [Google Scholar]
- 42.Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. Eng. 2005;44:4442–4489. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]
- 43.Shen W. Tetrahedron Lett. 1997;38:5575–5578. [Google Scholar]
- 44.In contrast, cyclopentyl and cyclohexylboronic acids do not provide coupling products under these conditions. Previous work (ref 45) has shown cyclopropylboronic acid is superior to other secondary alkylboronic acids as a substrate in Suzuki coupling reactions.
- 45.van den Hoogenband A, Lange JHM, Terpstra JW, Koch M, Visser GM, Korstanje TJ, Jastrzebski JTBH. Tetrahedron Lett. 2008;49:4122–4124. [Google Scholar]
- 46.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 47.Beeby A, Bettington S, Fairlamb IJS, Goeta AE, Kapdi AR, Niemela EH, Thompson AL. New J. Chem. 2004;28:600–605. [Google Scholar]
- 48.Bedford RB, Cazin CSJ. JCS Chem. Comm. 2001:1540–1541. doi: 10.1039/b105394a. [DOI] [PubMed] [Google Scholar]
- 49.Zhu C, Wang R, Falck JR. Org. Lett. 2012;14:3494–3497. doi: 10.1021/ol301463c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fors BP, Krattiger P, Strieter E, Buchwald SL. Org. Lett. 2008;10:3505–3508. doi: 10.1021/ol801285g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Baelen G, Maes BUW. Tetrahedron. 2008;64:5604–5619. [Google Scholar]
- 52.Thompson AE, Hughes G, Batsanov AS, Bryce MR, Parry PR, Tarbit B. J. Org. Chem. 2005;70:388–390. doi: 10.1021/jo0402226. [DOI] [PubMed] [Google Scholar]
- 53.Guram AS, Wang X, Bunel EE, Faul MM, Larsen RD, Martinelli MJ. J. Org. Chem. 2007;72:5104–5112. doi: 10.1021/jo070341w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.