

Abstract
The Armitage-Doll multi-stage model of carcinogenesis tremendously refocused cancer science by postulating that carcinogenesis is driven by a sequence of genetic changes in cells. Age-dependent cancer incidence thus has been explained in terms of the time necessary for oncogenic mutations to occur. While the multi-step nature of cancer evolution is well-supported by evidence, the mutation-centric theory is unable to explain a number of phenomena, such as the disproportion between cancer frequency and animal body size or the scaling of cancer incidence to animal lifespan. In this paper, we present a theoretical review of the current paradigm and discuss some fundamental evolutionary theory postulates that explain why cancer incidence is a function of lifespan and physiological, not chronological, aging.
The Armitage-Doll multi-stage paradigm of carcinogenesis in the light of current evidence
Over half a century ago, Carl Nordling postulated that carcinogenesis is driven by mutations that occur in cells [1]. Peter Armitage and Richard Doll generalized these ideas and proposed the multi-stage theory of carcinogenesis in a seminal paper [2], stating that carcinogenesis typically requires 6–7 mutations and/or other cell alteration to malignantly transform a cell, based on the evidence that the age-dependent exponential increase in cancer incidence follows mathematically the 6th power of age. They noted, though, that such a model holds only if the probability of oncogenic events stays the same throughout the human lifespan, which implies linear mutation accumulation with age. Later, Peter Nowell and David Hungerford provided further corroboration of the genetic origins of cancer [3]. Nowell also introduced the concept of clonal evolution of cancers, suggesting that it occurs through rounds of selection in tissues for progressively more advantageous pre-malignant cells and progressively more malignant phenotypes [4]. As pre-neoplastic cells transformed by mutations expand clonally, the likelihood that further oncogenic events will occur in already oncogenically initiated cells increases proportionally to the number of such dividing cells, and such a pattern thus is believed to ultimately provide for the exponential increase in the incidence of cancers with age.
These seminal theoretical concepts and findings tremendously refocused cancer science, leading to extensive discovery efforts for specific oncogenic events, a multitude of which has now been well characterized. Our current understanding of cancer development largely remains within this theoretical framework, with the assumption often made that oncogenic events typically confer certain defined selective (proliferative) advantages to cells over their normal wild-type counterparts (e.g. [5, 6]). However, as new evidence accumulates, it is becoming clear that carcinogenesis is a more complicated process regulated by many forces beyond the occurrence of oncogenic mutations.
The first theoretical problem with prevailing models arises with the recognition that somatic mutations do not accumulate as Armitage and Doll assumed. Data from various murine and human tissues convincingly demonstrate that a substantial portion of somatic mutations (up to 50%) accumulate early in life before full body maturation [7, 8]. The rate of accumulation significantly slows down thereafter (Fig. 1). Evidence from at least some tissues suggests that this slow-down occurs when stem cells transition from body building to body maintenance and thus dramatically reduce their division rates [9]. Within the current theoretical framework, the observations that cancer incidence increases exponentially with age and that mutation accumulation occurs largely during development stand in opposition to each other, which implies that this framework is incomplete. To address this, we suggest that selection for oncogenic clones is highly dependent on tissue context, which changes with age, and necessarily implies that clonal evolution is differentially regulated throughout life. As the probability of sequential mutation accumulation in one cell depends both on cell division rates and the number of dividing cells, early in life when cells cycle rapidly, the lower observed risk of cancer should require that those cells that acquire mutations do not expand into multi-cell clones (which would increase the target size for the next oncogenic mutation) [10]. Late in life, the slow cell division program needs to be compensated by more profound clonal expansions of pre-malignant cells to provide for the elevated risk of cancers that require multiple mutations. Such logic implies that the strength of positive selection in tissues for oncogenically initiated cells should significantly differ between early and late life; otherwise the ultimate cancer incidence curve would resemble more closely the shape of mutation accumulation with age.
Figure 1. Age-dependent physiological events.
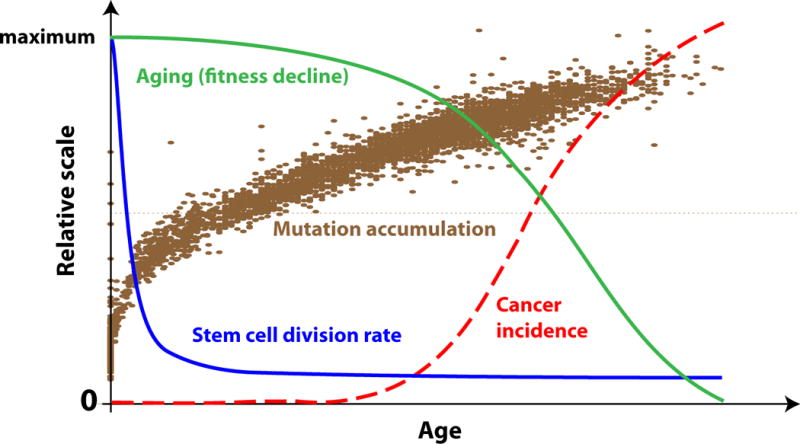
Roughly half of all mutations and epigenetic changes occur before full body maturation [7]. The accumulation of genetic and epigenetic changes significantly slows down later, followed by a slowdown in stem cell division rates when stem cells transition from body maturation to body maintenance (example based on HSC) [9]. Both elevated aging rates and cancer incidence are significantly delayed and do not follow the mutation accumulation curve. However, cancer incidence increase mirrors the curve of physiological decline (aging).
In fact, evidence from the hematopoietic system corroborates the idea that positive selection in tissue for mutant cellular phenotypes is stronger late in life, as selection for particular oncogenic events has been shown to be substantially stronger in old murine hematopoietic microenvironments than in young [11, 12]. Increased oncogenic clonal expansions and clonal hematopoiesis have also been demonstrated in old mice [13, 14]. Moreover, after the age of approximately 40 for humans, exponentially increasing clonality is observed in hematopoiesis, bringing about large expanded clones that are not observed early in life [15–18], and these clonal expansions have been postulated to result from enhanced selection in the aged bone marrow microenvironment [19–21]. Interestingly, increased hematopoietic clonality late in life, whether or not associated with known oncogenic mutations, is associated with increased leukemia development as well as all-cause mortality [16], consistent with these clonal expansions being symptoms of an aged soma which contributes to alterations in oncogenic selection, as well as more general physiological decline.
A second major problem with the prevailing model(s) is the assumption that any mutation can confer a defined effect on a cell’s fitness (see Glossary), a prediction that contradicts evolutionary theory. A cell’s fitness represents the relative ability of stem and progenitor cells of a certain genotype (or epi-genotype) to contract, persist or expand in a cell population. Functional (non-silent) genetic alterations usually alter phenotype in a defined way. However, changes in fitness (selective value) arise only when this phenotype interacts with environment and can vastly differ in an environment-dependent manner. For example, a mutation conferring drug resistance to a cell produces a defined phenotype, but this phenotype will only have a selective advantage over other cells under drug exposure, and the degree of this advantage will also vary depending on the drug regimen. Likewise, a young and fit human tissue represents a dramatically different microenvironment than an aging one typically characterized by increased chronic inflammation, tissue integrity loss and other aging markers. Many experimental studies now demonstrate that selection for oncogenic mutants heavily depends on tissue microenvironmental context, which is influenced by environmental exposure (including carcinogens like cigarette smoke), age, and associated inflammation and altered tissue landscapes [11, 12, 22–25]. Therefore, understanding that selection for mutant cells, including oncogenic mutants, is not a stationary phenomenon but depends on other factors including aging-related factors that operate above the cellular level is crucial to bring the current multi-stage theory of carcinogenesis in accord with evolutionary theory and modern evidence.
The evolution of lifespan as key to understanding aging-related cancer risk
In order to understand how and why physiological aging, not just chronological timing, can be directly implicated in cancer and other disease risk, it is key to consider how lifespans evolve. A substantial emphasis in medical and cancer-related literature is placed on time-dependent accumulation of cellular damage as a major driver of aging, thus positing aging as rate-limited by a certain mutation/damage “clock” [26, 27]. However, as mentioned previously, roughly half of all somatic mutations occur early in life when most cell divisions happen, but physiological aging does not follow this pattern and is significantly delayed, undermining the cell-intrinsic model of aging. The somatic damage accumulation model is also discordant with current views within evolutionary biology [28, 29]. The evolutionary theory of aging and lifespan states that aging and lifespan are determined by the rate at which selection is relaxed during the life of organisms, and is supported by substantial observational and experimental evidence [28, 29]. As proposed by Peter Medawar [30], alleles that increase organismal fitness during the pre- and reproductive period are strongly selected for in populations, while this selection is relaxed later in life because of lower chances of survival and reproduction (Figure 2A). In this way, the evolution of lifespan is dependent on the ecological niche of a species, which determines the likelihood of dying over time for any reason. For example, there are indications that reduced predation alone can substantially slow down the evolved rates of physiological aging in mammals [31], engendering a slower life strategy (delayed reproduction and longer somatic maintenance; see Figure 2A and 2B for a theoretical example). Therefore, evolution at the level of the organism (germline) basically creates two principal periods in mammalian lifespans: 1) the pre- and reproductive period where high body fitness is maintained and 2) post-reproductive period during which body fitness and tissue microenvironment declines progressively faster. Finally, the post-reproductive period does not refer to the ages where an animal is necessarily incapable of reproducing, but simply when reproduction becomes more and more unlikely (typically due to prior death from a variety of causes).
Figure 2. The evolution of lifespan and cancer incidence.
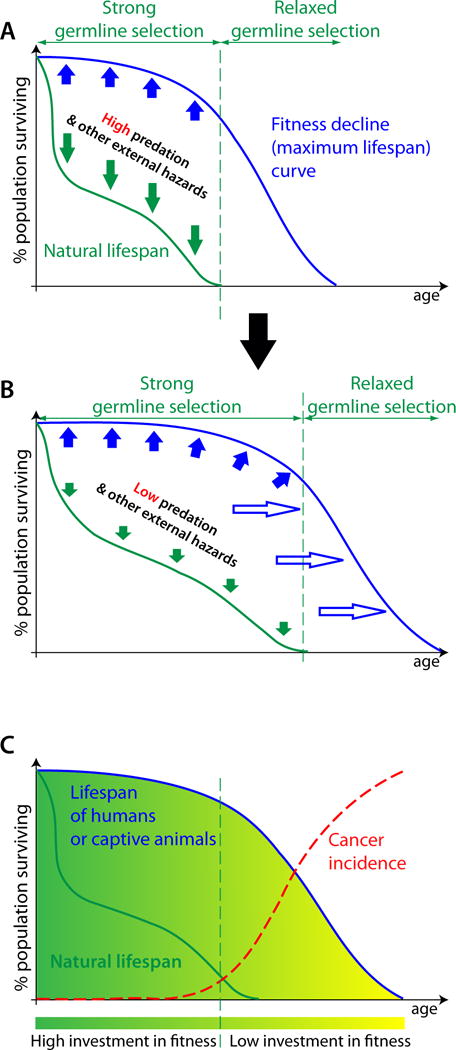
(A) The lifespan of animals in the wild (dark green curve) is usually defined by external hazards, such as predators, food availability, and infectious diseases (dark green arrows). Their maximum lifespan, limited by their rates of physiological aging (blue line), can only be observed in captivity or in modern humans. Germline selection acts to maintain high body fitness (solid blue arrows) for the duration of likely survival and reproduction in the wild. Physiological aging in humans and captive animals accelerates after the time of their likely survival in the wild promoted by progressively relaxing germline selection for body fitness. (B) If external hazards are reduced, animals survive into more advanced ages in the wild and trigger germline selection to extend (hollow blue arrows) their maximum lifespan, pushing the curve of physiological aging rightwards on the age axis and leading to the evolution of a longer potential lifespan. (C) Cancer is rare in wild animals, as well as in humans and captive animals during the period of their likely survival in the wild (natural lifespan), with carcinogenesis being suppressed by higher tissue and stem cell fitness relative to ages past natural lifespan. A dramatic increase in cancer incidence coincides with higher rates of physiological decline.
The evolution of lifespan at the germline level has important implications for the stem and progenitor cells responsible for tissue renewal, which are subject to malignant transformation by oncogenic mutations (Figure 2B). During pre- and reproductive periods, selection for body fitness works to fine-tune the behavior of stem cells and their tissue microenvironment for optimal tissue renewal and function, so that stem cells divide and differentiate in a relatively stable environment to which they are well-adapted. In this environment, there still exists a chance that an oncogenic mutation can improve a cell’s fitness so that the mutated cell out-proliferates its normal counterparts in the stem cell pool. However, the probability and magnitude of such an effect will dramatically increase in the post-reproductive tissue microenvironment, as stem cells have not adapted to this environment through germline evolution (Figure 3). This argument is consistent with evolutionary theory which holds that adaptation increases the strength of purifying selection and also reduces the ability of mutations to increase fitness [32].
Figure 3. The effect of fitness landscapes on selection processes.
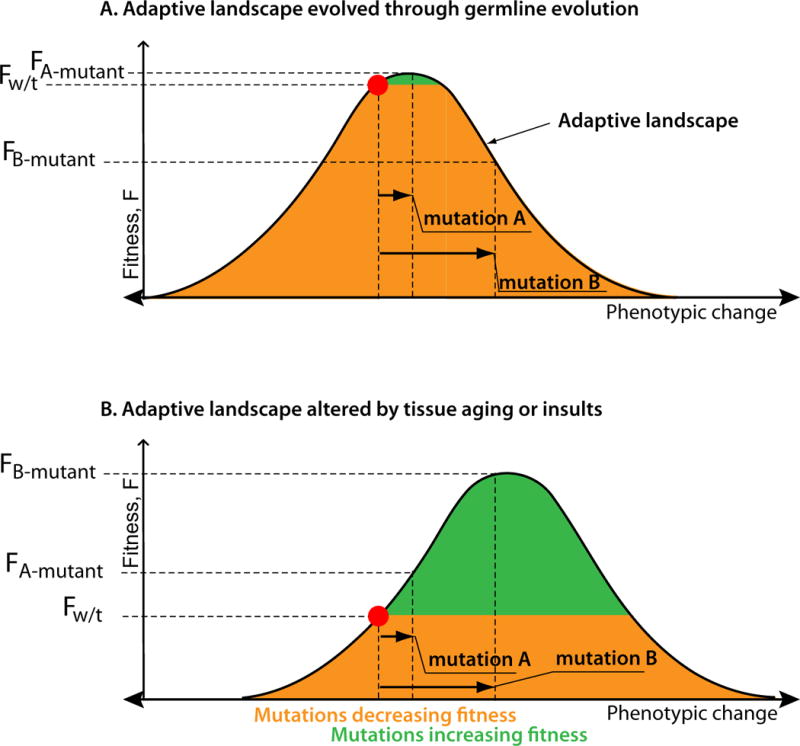
(A) A cellular phenotype (red dot) is driven by the evolution at the germline level to adapt to the tissue microenvironmental fitness landscape (represented here by the simplest Shelford curve of tolerance model [49]), moving the phenotype towards the fitness peak. The strength of selection triggered by a mutation is proportional to the fitness difference the mutation creates. Germline selection reduces the number of mutations capable of increasing fitness (green area), limits the strength of positive selection that such a mutation can confer (compare the fitness differential between wild type versus A-mutant and wild type versus the negatively selected B-mutant), and increases the number of negatively selected mutations (orange area). (B) In an altered environment, the intensity of many environmental factors is changed (represented here with a rightward-shifted Shelford curve). In an altered environment to which a cell is not adapted through germline evolution, the same cellular phenotype results in lower cellular fitness. Also, the number of mutations capable of elevating fitness is greater and the strength of positive selection they can trigger is increased (compare fitness differential between wild type versus A-mutant and wild type versus B-mutant with those in panel A). The directionality of selection triggered by a specific mutation can also change, such as represented here by mutation B which is negatively selected in the normal microenvironment, but strongly positively selected in the altered one.
Lifespans that significantly extend past the reproductive period are a consequence of modernity. Unlike modern humans, non-captive animals typically do not survive into the post-reproductive period, and we can only observe the effects of the “post natural survival” portion of their lifespan in captivity. For example, wild mice rarely survive beyond 6–7 months, but can live for more than 2 years in laboratories, and get the vast majority of cancers during the period that extends beyond their typical survival in nature [33]. As described above, different potential lifespans have evolved in different animals, dependent on environmental conditions, in order to maximize reproductive success. What then accounts for the vast differences in potential lifespans, and thus avoidance of diseases of old age, in different mammals? Is greater longevity achieved by the evolution of lower mutation rates or by enhancing other means of somatic maintenance? A clue is provided by studies of two DNA polymerase δ mutations in mice, L604K and L604G, both of which increase mutation rates more than 5-fold in the heterozygous state [34]. However, only the +/L604K genotype is associated with an accelerated incidence of cancer (occurring several months earlier) that mirrors a several month shortening of lifespan (Figure 4). In contrast, the +/L604G genotype is not associated with either an increase in cancer risk/rate or changes in lifespan. Although the reason underlying this difference is unknown, heterozygosity for L604K shows a (non-significant) trend to higher mutation rates relative to +/L640G. Regardless, increases in mutation rates clearly do not suffice to change the age-dependent cancer incidence curve (as seen in +/L640G mice), but may require other physiological changes (reflected in the shortened lifespan of +/L640K mice).
Figure 4. Rates of aging and cancer incidence in DNA polymerase mutant mice.
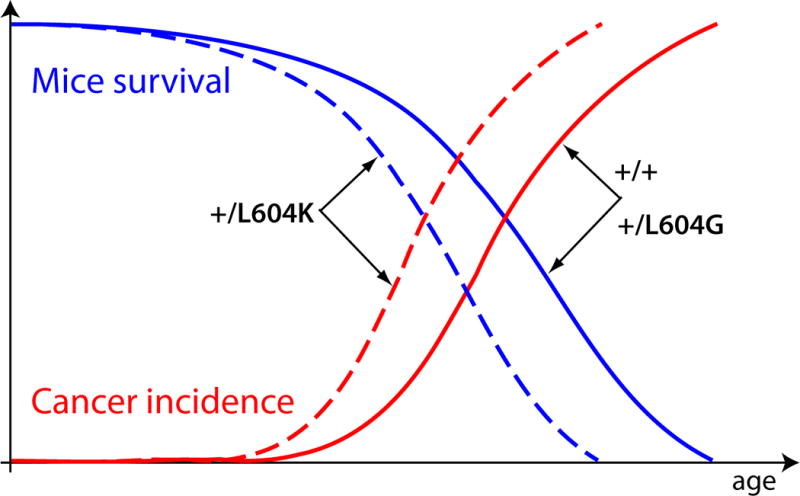
Mutations L604K and L604G in the heterozygous state both increase mutations rates more than 5 times in mice. Mutation L604G does not have any effect on lifespan and cancer incidence. Mutation L604K, however, causes a shortening of the lifespan and a symmetrical acceleration of the cancer incidence curve relative to +/+ (wild type) and +/L604G [34].
Without considering the effects of lifespan evolution on age-dependent cancer and other disease risk, the current multi-stage model of cancer creates seemingly unresolved paradoxes. The well-known Peto’s paradox is the inability of the current model to explain why animals with larger bodies and longer lifespans (both effectively increasing the target size of dividing stem cell pools for mutations) do not have proportionally higher incidence of cancers [35]. Multiple explanations have been proposed focusing mostly on the evolution of intracellular mechanisms that reduce the risk of cell transformation [36], but the paradox remains unresolved. Explanations have often focused on changes in mutation rates for different species, and yet the evolution of complex multicellularity has not been accompanied by reductions in mutation rate (in fact, mammals have higher mutation rates than many single-cell eukaryotes and bacteria [37]), and as described above, substantial increases in mutation rate can be engineered in mice without increasing cancer risk or decreasing lifespan. If carcinogenesis is viewed simply as a process depending on time and number of cell divisions, this paradox might indeed have no solutions. However, considering that lifespan and body size primarily evolve based on the species ecological niche, and that cancer and other disease risk is lifespan-dependent, resolves this paradox. Similar sized animals within very closely related groups, such as rodents, often have very different lifespans. Evolutionary theory holds that this occurs because of different species-specific probabilities of late-life survival and reproduction. This evolution of divergent lifespans is unlikely to be caused by a very rapid evolution of mutation rates, extra tumor suppressor genes or other cancer-related factors that are often proposed [36]. Very long lifespans, such as those of whales, Greenland sharks, Galapagos tortoises and elephants, appear to typically evolve in animals that are under reduced risk of predation or death by other external factors as adults. There is a common life strategy that is acted upon by natural selection when these external risk factors change - somatic maintenance; the “tuning” of this investment will either increase or decrease the period of somatic maintenance depending on environmental selective forces in order to maximize reproductive success in the new environment (Figure 2). This somatic maintenance strategy has implications for the timing of diseases of old age like cancer, should an animal somehow manage to survive beyond periods typical in nature (as humans do today).
From this perspective it is not lifespan that depends on cancer-free survival, but rather that the age-dependence of cancer incidence should be viewed as a function of lifespan, which itself is dependent on the curve of body fitness decline (Figure 2B). This perspective helps explain why animals rarely have cancer in the wild, since many animals simply do not survive to the age when cancer becomes a risk factor (Figure 2C). Senescence appears to be common in some groups of wild animals [38,39], however cancer demography data are lacking from wild populations and more research is needed to elucidate if cancer rates are higher in aged wild animals. It is also unclear how senescence in wild populations affects the overall odds of reproductive success, which is the key regulator of the age-dependent strength of germline selection. Recent evidence also indicates that different tissues in the body have different rates of aging [50], which might potentially be a contributor to the differential tissue-specific cancer risk. Finally, while still speculative, understanding cancer as being determined by evolved life history may help explain the earlier incidence of a number of cancers, such as brain, bowel, chronic myeloid leukemia, chronic lymphocytic leukemia and acute myeloid leukemia, in males relative to females (cancers which are neither sex-specific or substantially due to smoking were queried; Cancer Research UK; http://www.cancerresearchuk.org). Males (for humans and many other species) age earlier, and this earlier decline in tissue microenvironments could potentially elicit earlier somatic oncogenic adaptation relative to females.
Understanding carcinogenesis as rate-limited by physiological aging-dependent processes
Carcinogenesis is currently understood as a series of oncogenic mutations that increase the relative fitness of the cells in which they occur and lead to a series of clonal expansions of these oncogenically-initiated cells [5]. Stem and progenitor cells have been shown to compete for niche space [40], so fitness can be considered as the ability of dividing cells to remain in the competitive stem and progenitor pool and expand their progeny instead of differentiating, dying or senescing. If a mutation increases the probability for a cellular genotype to become more frequent in a competitive population, then this mutation increases cell fitness. In this light, carcinogenesis is recognized as a Darwinian process of mutation and selection for the fittest among dividing somatic cells. Within the current multi-stage paradigm, oncogenic mutations are deemed to provide immediate and measurable fitness advantage to the affected cells and thus drive carcinogenesis [5, 6]. However, multiple lines of evidence indicate that this process is not so simple, and oncogenic mutations experience very different fates depending on the tissue environmental context in which they occur, and the evolutionary forces, including drift and selection, to which they are subjected [20]. For example, in small populations (of cells or animals), drift can be the dominant evolutionary force. Indeed this is the case in the gut epithelium, where epithelial stem cells are split into small clusters of 15–20 cells and their clonal dynamics in the gut crypts have been shown to be essentially neutral [41, 42]. Even potent oncogenic mutations, such as those in p53, do not confer significant selective advantage to gut stem cells, unless they occur in an inflamed environment [23]. On the other hand, in large competitive stem cell pools, such as hematopoietic stem cells (HSC), selection has been shown to dominate clonal dynamics [12, 43, 44]. Studies indicate that selection acts differentially on oncogenic mutations in young and healthy versus old and damaged tissue contexts [11, 12, 45], with the former being dominated by stabilizing selection that prevents most genetic changes from conferring fitness advantages to cells. As noted above, because of the co-evolution at the germline level of stem cells with their tissue microenvironment that regulates cell fates, stem cells in young tissues are closer to their peak fitness than cells in aged or damaged tissues (Figure 3). This adaptation to specific tissue environments leads to the increased role of stabilizing selection which reduces the probability that a random genetic mutation can be advantageous. In adult stem cell pools of a stable size, the average frequency of differentiation per stem cell division is close to 0.5, and stabilizing selection acts to increase the rate of differentiation of cells bearing phenotype-altering mutations, as shown for multiple oncogenic mutations engineered in HSC in mice [46]. In this case regardless of the effect of oncogenic mutations on cell cycle, differentiation rates above 0.5 per cell division will always lead to the elimination of the mutant clone from the pool. For example, deletion of the PTEN gene substantially reduces the self-renewal of mouse HSC [47, 48]; while deletion of PTEN simultaneously in all HSC leads to leukemia, the more natural context of gene disruption in a single HSC would be expected to result in clonal exhaustion by differentiation. Evidence from the human hematopoietic system also shows that during early life (until the age of 40) clonality above a few percent of the pool is practically non-existent, regardless of the fact that roughly half of all mutations that could accumulate in a full lifetime have already occurred by the age of ~18 years [15–18]. This suggests that positive selection for particular mutant phenotypes in general only has a major presence in late life during post-reproductive ages.
Concluding remarks
The fate of somatic mutations (including oncogenic ones) should be governed by the same basic evolutionary principles that govern natural populations, whereby adaptation leads to higher fitness and increased stabilizing and purifying selection, while positive selection is driven by environmental change and lower population fitness [20, 46]. From this perspective, carcinogenesis should be viewed as a function of physiological aging, whereby aging and altered tissue microenvironments lead to selection for previously accumulated random mutations, some of which confer a fitness advantage under these altered conditions. This framework explains many previously unexplained phenomena, such as the rarity of cancers among wild animals (which do not typically live beyond their reproductive ages), the scaling of cancer incidence curve to lifespan, and the fact that longer-lived and larger animals do not have proportionally more cancers. It is also easier to explain from this perspective why cancers that require different numbers of oncogenic mutations and originate from tissues with different numbers of stem cells and cell division profiles all follow a similar age-dependent timing of incidence increase, including cancers that appear to require just one mutation, such as chronic myeloid leukemia. In this regard, while solid evidence supports the general paradigm that carcinogenesis is a Darwinian multi-stage process of rounds of mutation and positive selection, the current paradigm oversimplifies the evolutionary forces that drive these selection steps and determine their timing. It thus lacks key theoretical foundations of modern evolutionary theory and requires revision (see Outstanding Questions Box). Understanding somatic evolution in the context of the evolution of animal life histories and from a tissue-level ecological perspective provides immense explanatory power to better understand a very complex disease: cancer.
Outstanding Questions Box.
Would expanding field data to different groups of mammals and other vertebrates significantly enhance our understanding for how lifespan and age-related disease risk, and the risk of cancer in particular, are linked?
How do the processes of aging determine the strength and directionality of selection driven by oncogenic mutations?
What are the key changes in tissue microenvironments with old age (or in smokers, etc.) that affect the fate of somatic mutations? Can strategies be developed that restore more youthful microenvironments, thus dampening selection for oncogenic events?
Trends Box.
While the increasing cancer incidence with age is currently understood from the perspective of time-dependent mutation accumulation, this model fails to account for key observations, such as early life mutation accumulation and the scaling of cancer incidence to the lifespans of different animals.
Animal evolution has selected for somatic maintenance strategies that maximize reproductive success, and these strategies determine the evolution of maximal lifespans
External conditions, including food availability, disease and predators, influence somatic maintenance strategies, as higher odds of earlier death will promote strategies that reduce long-term somatic maintenance and thus lead to earlier senescence when removed from these hazards.
Somatic maintenance strategies have a major impact on cancer incidence, as well-maintained tissues in young animals promote stabilizing selection (disfavoring oncogenic mutations), while tissue decline in post-reproductive periods promotes positive selection for adaptive mutations (including oncogenic ones).
Acknowledgments
We would like to thank Hannah Scarborough, Roberta Pelanda, Eric Pietras and Richard Davis of the University of Colorado for critical review of the manuscript. These studies were supported by National Cancer Institute grants (R01CA180175 and R21CA179501) to J.D.
Glossary
- Adaptive
Increases fitness (e.g. a mutation that increases fitness is adaptive).
- Allele
a variant of a gene. Different alleles for the same gene will be largely homologous, but can modestly differ in sequence and can encode for proteins with different activities
- Drift
random assortment of genetic alleles. The influence of drift becomes greater as population size decreases.
- Fitness
a measure of reproductive success; classically, the ability of an organism to pass its genes on to future generations of that organism. For somatic tissues, a measure of the ability of a cell to pass its genotype or epigenotype on to future cell generations.
- Germline
the genetic lineage that is maintained in the next generation following reproduction.
- Lifespan evolution
changes in somatic maintenance programs that are selected for in different environments to maximize reproductive success (part of Life History Theory).
- Microenvironment
the tissue environment for somatic cells.
- Phenotype
the characteristics of an organism or a cell that are determined by its genotype.
- Selection
the process of differential survival of individuals in a population that impacts on allele frequency based on their fitness; depends on genetic and environmental contexts.
- Positive selection
increases the frequency of alleles that increase fitness
- Purifying (or negative) selection
decreases the frequency of alleles that reduce fitness
- Stabilizing selection
maintenance of a well-adapted phenotype, as mutations will mostly reduce fitness
- Somatic
cells and tissues of an organism (the soma), excluding gametes (e.g. sperm and eggs)
- Somatic maintenance
the maintenance of cells, tissues and organs through replacement and repair.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nordling CO. A new theory on the cancer-inducing mechanism. British Journal of Cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowell P, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497. doi: 10.1126/science.144.3623.1229. [DOI] [PubMed] [Google Scholar]
- 4.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 6.Bozic I, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A. 2010;107:18545–18550. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vijg J, et al. Aging and genome maintenance. Ann N Y Acad Sci. 2005;1055:35–47. doi: 10.1196/annals.1323.007. [DOI] [PubMed] [Google Scholar]
- 9.Bowie MB, et al. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest. 2006;116:2808–2816. doi: 10.1172/JCI28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rozhok AI, et al. Stochastic modeling reveals an evolutionary mechanism underlying elevated rates of childhood leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2016 doi: 10.1073/pnas.1509333113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henry CJ, et al. Aging-associated inflammation promotes selection for adaptive oncogenic events in B cell progenitors. J Clin Invest. 2015;125:4666–4680. doi: 10.1172/JCI83024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henry CJ, et al. Declining lymphoid progenitor fitness promotes aging-associated leukemogenesis. Proc Natl Acad Sci U S A. 2010;107:21713–21718. doi: 10.1073/pnas.1005486107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vas V, et al. Contribution of an aged microenvironment to aging-associated myeloproliferative disease. PLoS One. 2012;7:e31523. doi: 10.1371/journal.pone.0031523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vas V, et al. Aging of the microenvironment influences clonality in hematopoiesis. PLoS One. 2012;7:e42080. doi: 10.1371/journal.pone.0042080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKerrell T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015;10:1239–1245. doi: 10.1016/j.celrep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodell MA, Rando TA. Stem cells and healthy aging. Science. 2015;350:1199–1204. doi: 10.1126/science.aab3388. [DOI] [PubMed] [Google Scholar]
- 20.Rozhok AI, DeGregori J. Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations. Proc Natl Acad Sci U S A. 2015;112:8914–8921. doi: 10.1073/pnas.1501713112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKerrell T, Vassiliou GS. Aging as a driver of leukemogenesis. Sci Transl Med. 2015;7:306fs338. doi: 10.1126/scitranslmed.aac4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fleenor CJ, et al. Contrasting Roles for C/EBPalpha and Notch in Irradiation-Induced Multipotent Hematopoietic Progenitor Cell Defects. Stem Cells. 2014 doi: 10.1002/stem.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vermeulen L, et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995–998. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
- 24.Gatenby RA, Vincent TL. An evolutionary model of carcinogenesis. Cancer Res. 2003;63:6212–6220. [PubMed] [Google Scholar]
- 25.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 26.Lopez-Otin C, et al. The hallmarks of ageing. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossi DJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gavrilov LA, Gavrilova NS. Evolutionary theories of aging and longevity. ScientificWorldJournal. 2002;2:339–356. doi: 10.1100/tsw.2002.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose M, Mueller L. Evolution of the human lifespan: past, future, and present. American Journal of Human Biology. 1998;10:409–420. doi: 10.1002/(SICI)1520-6300(1998)10:4<409::AID-AJHB2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 30.Medawar P. An unsolved problem of biology. H.K.Lewis; 1952. [Google Scholar]
- 31.Austad SN. Retarded senescence in an insular population of virginia opossums (Didelphis virginiana) Journal of Zoology. 1993;229:695–708. [Google Scholar]
- 32.Orr HA. The population genetics of adaptation: The distribution of factors fixed during adaptive evolution. Evolution. 1998;52:935–949. doi: 10.1111/j.1558-5646.1998.tb01823.x. [DOI] [PubMed] [Google Scholar]
- 33.Frith CH, et al. The morphology, immunohistochemistry, and incidence of hematopoietic neoplasms in mice and rats. Toxicologic pathology. 1993;21:206–218. doi: 10.1177/019262339302100213. [DOI] [PubMed] [Google Scholar]
- 34.Venkatesan RN, et al. Mutation at the polymerase active site of mouse DNA polymerase delta increases genomic instability and accelerates tumorigenesis. Mol Cell Biol. 2007;27:7669–7682. doi: 10.1128/MCB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peto R, et al. Cancer and ageing in mice and men. Br J Cancer. 1975;32:411–426. doi: 10.1038/bjc.1975.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caulin AF, Maley CC. Peto’s Paradox: evolution’s prescription for cancer prevention. Trends Ecol Evol. 2011;26:175–182. doi: 10.1016/j.tree.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch M. Evolution of the mutation rate. Trends in Genetics. 2010;26:345–352. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nussey DH, et al. Senescence in natural populations of animals: widespread evidence and its implications for bio-gerontology. Ageing research reviews. 2013;12:214–225. doi: 10.1016/j.arr.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayward AD, et al. Asynchrony of senescence among phenotypic traits in a wild mammal population. Exp Gerontol. 2015;71:56–68. doi: 10.1016/j.exger.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bondar T, Medzhitov R. p53-Mediated Hematopoietic Stem and Progenitor Cell Competition. Cell Stem Cell. 2010;6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez-Garcia C, et al. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822–825. doi: 10.1126/science.1196236. [DOI] [PubMed] [Google Scholar]
- 42.Snippert HJ, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 43.Abkowitz JL, et al. Evidence that hematopoiesis may be a stochastic process in vivo. Nat Med. 1996;2:190–197. doi: 10.1038/nm0296-190. [DOI] [PubMed] [Google Scholar]
- 44.Rozhok AI, et al. Stochastic modeling indicates that aging and somatic evolution in the hematopoietic system are driven by non-cell-autonomous processes. Aging. 2014;6:e1–16. doi: 10.18632/aging.100707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fleenor CJ, et al. Contrasting roles for C/EBPalpha and Notch in irradiation-induced multipotent hematopoietic progenitor cell defects. Stem Cells. 2015;33:1345–1358. doi: 10.1002/stem.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeGregori J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene. 2012;32:1869–1875. doi: 10.1038/onc.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yilmaz OH, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 49.Shelford VE. Some concepts in bioecology. Ecology. 1931;12:455–467. [Google Scholar]
- 50.Thomas F, Nesse RM, Gatenby R, Gidoin C, Renaud F, Roche B, Ujvari B. Evolutionary Ecology of Organs: A Missing Link in Cancer Development? Trends in Cancer. 2016;2:409–415. doi: 10.1016/j.trecan.2016.06.009. [DOI] [PubMed] [Google Scholar]