

Abstract
Background:
IgG4-related disease (IgG4-RD) is a fibroinflammatory condition characterized by serum IgG4 elevation and tissue infiltration of IgG4-positive plasma cells. Substantial overlap between IgG4-RD and antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitides (AAV) exists in terms of organ involvement and histopathological features. A positive ANCA assay is regarded as a highly specific finding in favor of an AAV, and generally influences away from a diagnosis of IgG4-RD. Recent reports, however, have raised the possibility that some patients with IgG4-RD are ANCA positive, thus suggesting reconsideration of the role of ANCA in the diagnostic workup. In the present work, we describe the first case of concomitant biopsy-proven IgG4-RD and granulomatosis with polyangiitis (GPA), demonstrating antiproteinase 3 (PR3) ANCA of the IgG4 subclass in the patient's serum. We also review the literature in order to provide clinicians with tools for interpreting ANCA positivity in IgG4-RD patients.
Case summary:
A 51-year-old woman was referred for left exopthalmos due to lacrimal gland enlargement and increased serum IgG4 concentration. IgG4-RD was suspected and further imaging studies disclosed multiple pulmonary masses in the right lung. Histological analysis of the left lacrimal gland was diagnostic for IgG4-RD, but lung biopsy showed typical features of GPA. ANCA assay was positive for anti-PR3 antibodies. Further immunofluorescence studies demonstrated anti-PR3 antibodies of IgG1 and IgG4 subclass. Treatment with rituximab induced swift remission of both IgG4-RD and GPA manifestations. We identified 9 other reports of patients with IgG4-RD and positive ANCA in the English literature, 5 cases with biopsy-proven IgG4-RD and 4 cases in whom IgG4-RD was diagnosed presumptively. Four patients had also histological evidence of concomitant AAV.
Conclusion:
The present work demonstrates that ANCA positivity in patients with biopsy-proven IgG4-RD should prompt the exclusion of a concomitant vasculitic process; a positive ANCA does not exclude the diagnosis of IgG4-RD; confirmation through immunoenzymatic assays of the ANCA specificity, clinical-pathological correlation, and histopathological evaluation remain crucial steps for the differential diagnosis between AAV and IgG4-RD.
Keywords: antineutrophil cytoplasmic antibodies, case report, granulomatosis with polyangiitis, IgG4, IgG4-related disease, rituximab, vasculitis
1. Introduction
IgG4-related disease (IgG4-RD) is an emerging fibro-inflammatory condition known to involve potentially every organ system in the body.[1] In view of its recent recognition as a nosological entity, neither diagnostic nor classification criteria have yet been developed for this condition. The approach to diagnosis, therefore, necessarily involves the exclusion of common infectious, neoplastic and inflammatory mimickers, principally through pathological evaluation of biopsy specimens. Histological features characteristic of IgG4-RD are shared by seemingly unrelated organs and include storiform fibrosis, obliterative phlebitis, tissue eosinophilia, and a lymphoplasmacytic infiltrate in which the ratio of IgG4- to IgG-positive plasma cells generally exceeds 0.40.[2]
Substantial overlap in the types of organ involvement and histopathological features occur in IgG4-RD and vasculitides associated with antineutrophil cytoplasmic antibodies (ANCA), that is, granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA)—collectively termed ANCA-associated vasculitis (AAV). As examples, different forms of AAV have predilections for affecting the orbital regions, sinuses, lungs, kidneys, meninges, and even the pituitary gland, all of which are known to be affected by IgG4-RD, as well. Moreover, the AAV can be associated with both peripheral and tissue eosinophilia,[3] and AAV rivals IgG4-RD in its ability to cause IgG4-positive plasma cell infiltration into involved organs.[4]
Elevation of serum IgG4 concentrations has been considered a useful biomarker of IgG4-RD, but broader experience in managing these patients has revealed the shortcomings of this measurement as a diagnostic tool.[5] Multiple non-IgG4-RD conditions can be associated with increased serum IgG4 concentrations. Conversely, a substantial proportion of patients with clinical presentations and organ biopsies consistent with IgG4-RD have normal serum IgG4 concentrations. In contrast, the finding of a positive ANCA assay for either the proteinase 3 (PR3) or myeloperoxidase (MPO) antigen is generally regarded as a highly specific finding in favor of an AAV diagnosis. Recent reports, however, have raised the possibility that some patients with IgG4-RD are ANCA positive, thus suggesting reconsideration of the role of ANCA in the diagnostic workup.[6–14]
In the present work, we describe a patient with concomitant biopsy-proven IgG4-RD and GPA in whom we demonstrated anti-PR3 ANCA of the IgG4 subclass. We also review the literature to provide tools for interpreting ANCA positivity in IgG4-RD patients. To this purpose, we searched the entire PubMed and Google Scholar online databases for variable combinations of the following terms: “ANCA,” “IgG4,” “IgG,” “IgG4-related disease,” “granulomatosis with polyangiitis,” and “vasculitis.” We then considered all the written-English reports of IgG4-RD patients with evidence of a positive ANCA titer. Ethical approval was not necessary because all diagnostic and therapeutic procedures were performed in accordance with international guidelines for the management of GPA and IgG4-RD.
2. Case presentation
A 51-year-old nonatopic woman was referred to our outpatient clinic for swelling of the left lacrimal gland, left orbital pain radiating to the left side of her face, mild proptosis, and redness of the left eye (Fig. 1A). She also complained of a mild discomfort in the infra-scapular region. Her medical history was significant for arterial hypertension, gastroesophageal reflux disease, and chronic bronchitis. An ophthalmologic evaluation revealed anterior scleritis of the left eye. No defects in visual acuity or lacrimal gland function were detected. Gadolinium-enhanced magnetic resonance imaging (MRI) of the brain and orbits disclosed an enlarged left lacrimal gland surrounded by an inflammatory tissue in contiguity with the lateral and superior rectus muscles (Fig. 1C). The inflammatory tissue showed intense 18F-fluorodeoxyglucose (18F-FDG) uptake on positron emission tomography/computed tomography (PET/CT) study (Fig. 1E). Both the erythrocyte sedimentation rate (43 mm/h; normal < 20) and total IgE (1798 IU/mL; normal < 100) were elevated, but the C-reactive protein and tests of renal, thyroid, and liver function were within normal range. An assay for antinuclear antibodies was positive in a titer of 1:160 (homogenous pattern) but assays for rheumatoid factor, antibodies to double-stranded DNA, thyroperoxidase, and thyroglobulin were negative. Serum IgG4 level was 253 mg/dL (normal < 135 mg/dL). IgG4-RD was suspected and a lacrimal gland biopsy was planned. The preoperative chest radiograph, however, showed multiple oval lung lesions in the right lung, raising concern for the possibility of malignancy (Fig. 2B). A chest CT scan confirmed 2 round-shaped nodules and 1 cavitary mass in the right lung, and revealed a paravertebral lesion on the right side of the T5 and T6 vertebrae (Fig. 2A, D, G, H). All of these lesions demonstrated intense 18F-FDG uptake on PET/CT (Fig. 2E–H). Biopsies of both the lacrimal gland and lung lesions were therefore performed.
Figure 1.
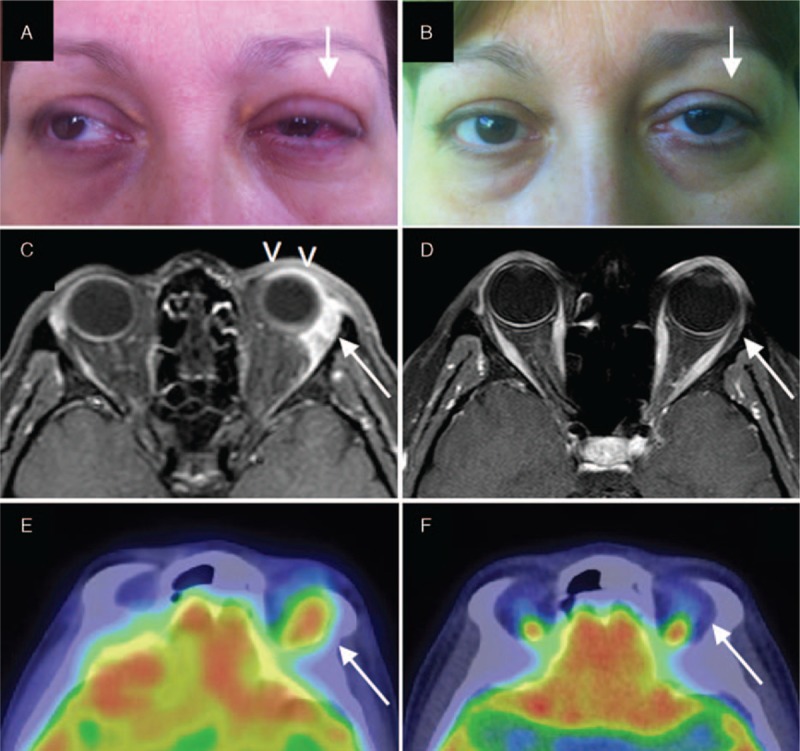
Clinical and radiological findings of orbital IgG4-related disease. (A) Left lacrimal gland enlargement with proptosis and scleritis (arrow). (C) A gadolinium enhanced magnetic resonance imaging showing left scleritis (arrowheads) and enlarged left lacrimal gland surrounded by an inflammatory tissue in contiguity with the lateral and superior rectum muscles (arrow). (E) 18F-fluorodeoxyglucose uptake in the left periorbital tissue on positron emission tomography scan. (B–F) Clinical and radiological remission 6 months after methylprednisolone pulses and rituximab (arrows).
Figure 2.
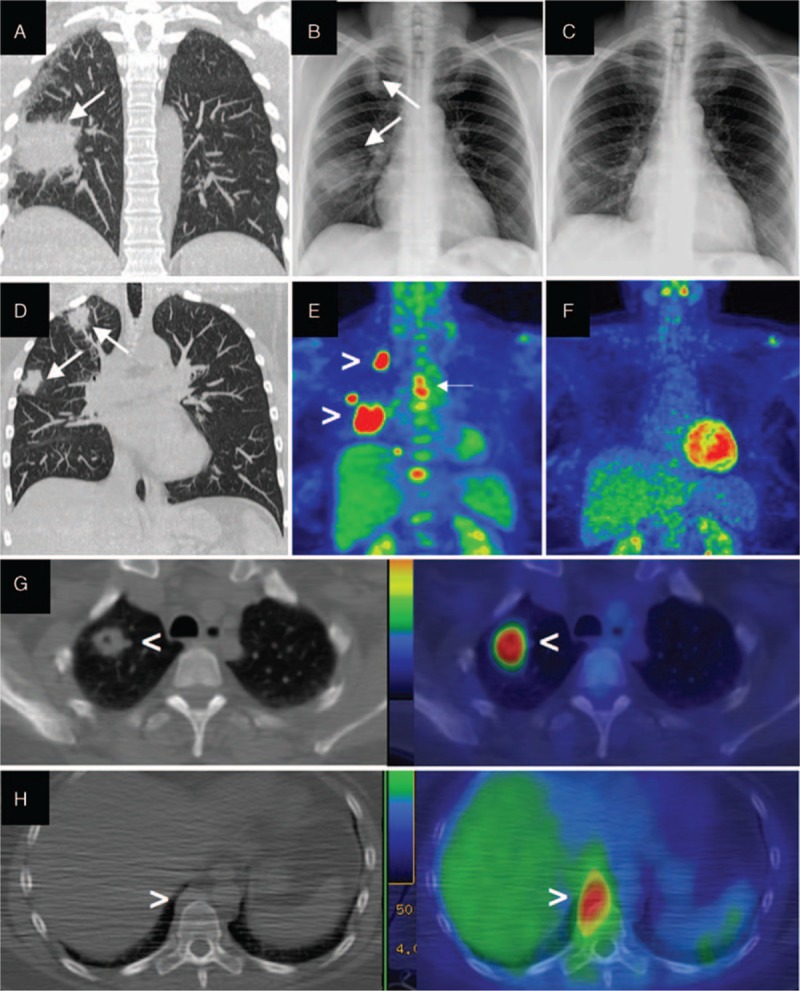
Radiological findings of pulmonary granulomatosis with polyangiitis. (A, D, G) Thoracic CT scan and (B) chest radiograph showing right lung masses (arrows), 1 of them (G) with cavitation (arrowhead), all showing intense 18F-FDG uptake on PET/CT scan (E, G) (arrowheads). (H) Thoracic CT scan revealing a right paravertebral mass with 18F-FDG uptake on PET/CT scan (arrowheads). Disappearance of lung masses 6 months after rituximab on radiograph (C) and PET/CT scan (F). 18F-FDG = 18F-fluorodeoxyglucose, PET/CT = positron emission tomography/computed tomography.
Examination of the left lacrimal gland tissue revealed areas of storiform fibrosis with abundant IgG4-positive plasma cells (IgG4+/IgG+ plasma cells ratio > 40%), consistent with the suspicion of IgG4-related dacryoadenitis (Fig. 3C–F). Neutrophilic infiltration, vasculitis, granulomatous inflammation, and necrosis were all not present in the lacrimal gland, examined in multiple sections. However, the lung tissue showed chronic granulomatous inflammation, leukocytoclastic vasculitis, and areas of geographic necrosis, features highly suggestive of GPA (Fig. 4A–E). Moreover, numerous IgG4-positive plasma cells were distributed throughout the GPA lesions (Fig. 4B–F). The patient's serum tested positively for ANCA by both immunofluorescence (cytoplasmic ANCA pattern) and enzyme immunoassay, with specificity for PR3 (423 AU; normal value < 20 AU). The ANCA appeared to consist of both the IgG1 and IgG4 subclasses (Fig. 3A and B). In addition, the counts of circulating plasmablasts—defined as CD19 + CD20–CD27 + CD38 + bright cells on flow cytometry—were dramatically increased (50,000 cells/mL; normal < 635 cells/mL).[5] The patient was started on prednisone (1 mg/kg) followed by 2 infusions of rituximab 1000 mg, administered 15 days apart.
Figure 3.
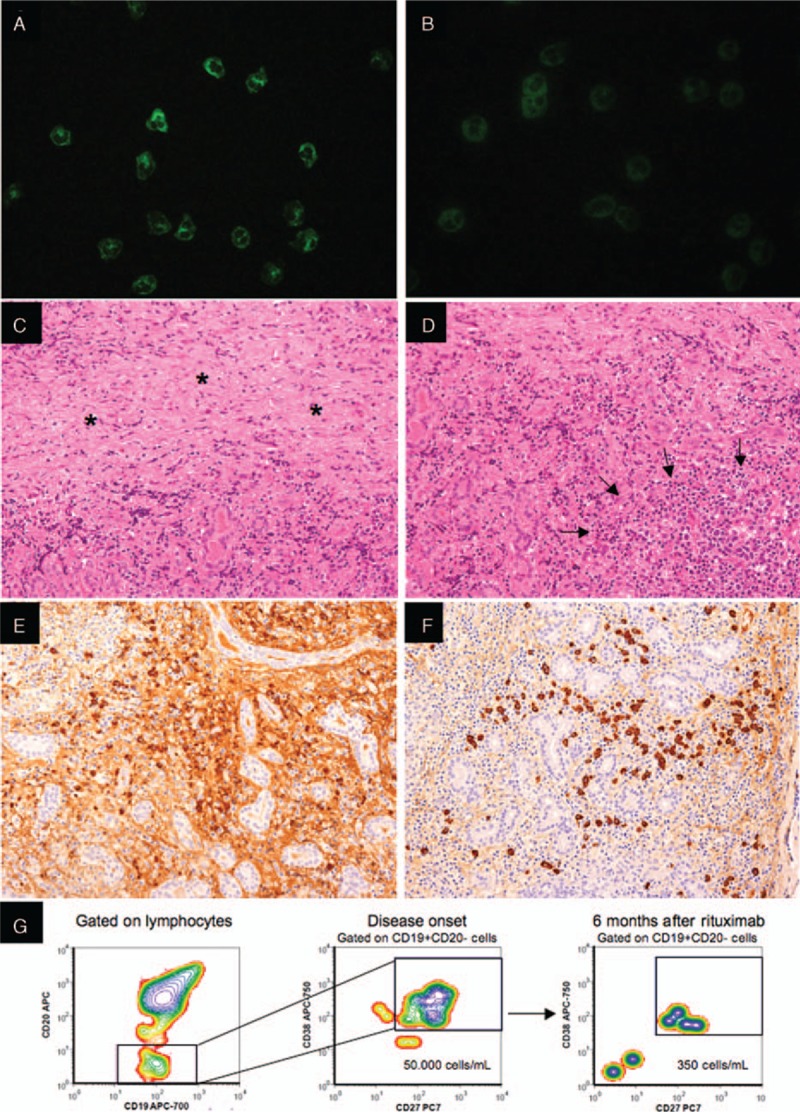
Immunofluorescence studies, flow cytometry analysis, and histological evaluation of the left lacrimal gland affected by IgG4-related disease. (A and B) Immunofluorescence assays performed on commercially available slides (Nova Lite ANCAkit/Substrated Slides—Inova Diagnostic San Diego, CA) demonstrates antineutrophil cytoplasmic antibodies of IgG1 (A) and IgG4 (B) subclass in the patient's serum (serum dilution 1:20; secondary anti-IgG1 (mouse antihuman IgG1-Alexa Fluor 488 conjugate—Thermofisher, St Waltham, MA) and anti-IgG4 (mouse antihuman IgG4-FITC conjugate—Alpha Diagnostics International, San Antonio, TX) antibodies dilution 1:50 and 1:20, respectively). (C–F) Histological examination of the left lacrimal gland reveals areas of storiform fibrosis (C—asterisks), and lymphoplasmocytic infiltrate (D—arrows) without granulomas or vascultis. Immunohistochemistry for total IgG (E) and IgG4 (F) shows abundant IgG4 positive plasma cells with an IgG4/IgG ratio > 40%. (G) Circulating plasmablasts (CD19 + CD20–CD27 + CD38bright cells) are expanded at disease onset and diminish following B cell depletion therapy with rituximab.
Figure 4.
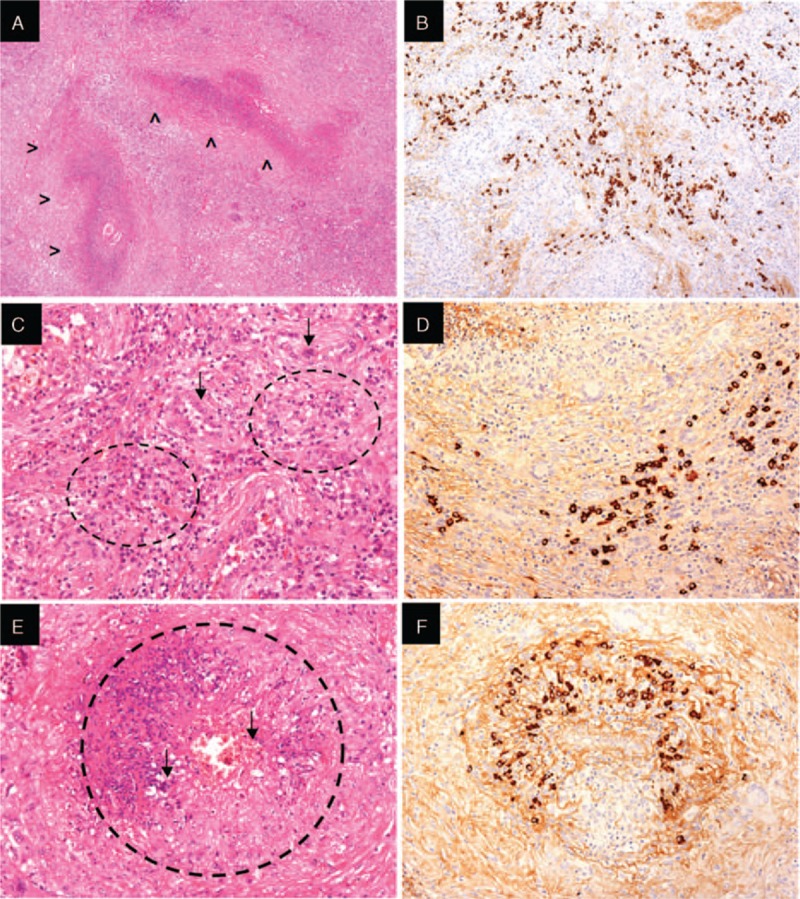
Histological analysis of the pulmonary lesion affected by granulomatosis with polyangiitis. Geographic necrosis (A—arrowheads), granulomas (C—circles) with giant cells (C—arrows), and necrotizing vasculitis (E—circle) with extravasation of red blood cells and neutrophils (E—arrows). Immunohistochemistry (B, D, F) reveals IgG4 positive plasma cells infiltrate in A, C, and E.
Six months later, the orbital and lung lesions had resolved. In particular, a whole body PET/CT scan showed normalization of the 18F-FDG uptake findings (Figs. 1F and 2F). Follow-up ophthalmologic and orbital MRI evaluations confirmed inactive scleritis and substantial regression of the orbital lesion, respectively (Fig. 1B–D). Lung masses were no longer evident on either chest radiography or CT (Fig. 2C). Laboratory and serological studies revealed normalization of the inflammatory markers and of the serum IgG4 (67 mg/dL), normalization of the circulating plasmablast level (350 cells/mL) (Fig. 3G), a negative PR-3 ANCA assay, and marked reduction of serum IgE levels (200 IU/mL).
3. Review of the literature
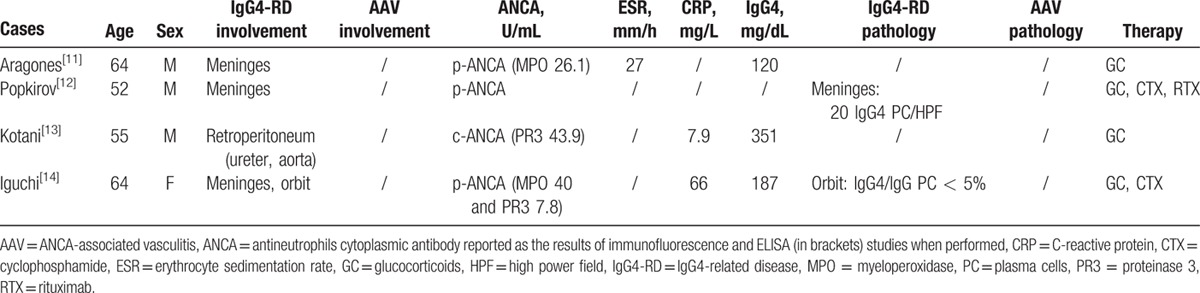
We identified 9 other reports of patients with IgG4-RD and positive ANCA in the English literature (Tables 1 and 2). Five cases had biopsy-proven IgG4-RD and were included in our analysis (Table 1). The remaining 4 cases, in whom IgG4-RD was diagnosed presumptively (based on elevated serum IgG4 levels and compatible organ involvement), are listed separately in Table 2 but not discussed further, because they lacked histopathological investigation.
Table 1.
Cases reported in the literature of patients with biopsy-proven IgG4-RD and positive ANCA.
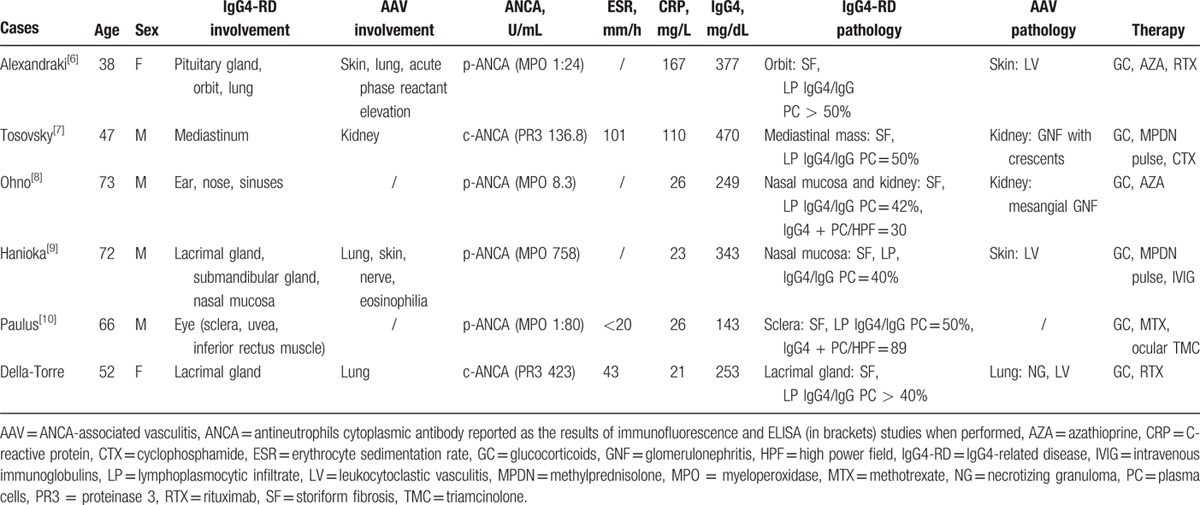
Table 2.
Cases reported in the literature of patients with possible IgG4-RD and positive ANCA.
The male to female ratio of biopsy-proven IgG4-RD cases was 4:2, with a median age at diagnosis of 59 years (range 38–73 years) (Table 1). Organs involved by IgG4-RD were the lacrimal glands and the periorbital tissue (2 cases each), the hypophysis, the mediastinum, the salivary glands, the meninges, the nose, and the paranasal sinuses (1 case each). Four out of 6 patients had also histological evidence of concomitant AAV in other organs, namely neutrophilic abscesses, leukocytoclastic vasculitis, fibrinoid necrosis, granulomatous inflammation, and crescentic glomerulonephritis. Organ involvement of AAV included the lung (3 cases), the skin and the kidney (2 cases each), and the peripheral nerves and the inner ear (1 case each). Biopsies of organs involved by AAV were obtained in 4 cases and histology revealed leukocytoclastic vasculitis (3 cases), and granulomas and crescentic glomerulonephritis (1 case each). Among these 4 patients, 1 was diagnosed with EGPA based on a history of long-standing asthma, multiple mononeuritis, peripheral blood eosinophilia, ANCA positivity with a specificity for MPO, and histological evidence of cutaneous leukocytoclastic vasculitis with abundant eosinophils.[9] Two patients were diagnosed with AAV in the absence of specific findings to diagnose GPA, MPA, or EGPA definitively.[6,7] One patient was diagnosed with GPA (present case).
The median C-reactive protein at diagnosis was 26.2 mg/dL (range 21–167 mg/dL). The erythrocyte sedimentation rate, measured in 3 cases, ranged from normal to 100 mm/h. Serum IgG4 concentration was increased in all 6 patients (median 298 mg/dL; range 143–470 mg/dL). All patients also had positive ANCA, 3 with a specificity for MPO, 2 with PR-3 specificity, and 1 with unknown specificity.
Glucocorticoids represented the first-line treatment for remission induction. Early recurrence after steroid tapering or partial clinical responses, however, prompted the addition of second-line immunosuppressive agents in all 6 cases. Ohno et al[8] and Alexandraki et al[6], for instance, added azathioprine to oral prednisolone at disease relapse but did not observe any further benefit. Alexandraki et al obtained disease remission with rituximab as third-line therapy. Paulus at al[10] achieved prompt clinical remission in a patient with scleritis and orbital myositis by combining methotrexate and intraocular triamcinolone to oral prednisone. In the case reported by Hanioka et al,[9] prednisolone led to a reduction of lacrimal gland swelling, but peripheral neuropathy improved only with intravenous immunoglobulins and methyprednisolone pulses. Similarly, intravenous cyclophosphamide and methylprednisolone pulses were required in addition to oral prednisone in order to prevent renal failure in a patient with rapidly progressive glomerulonephritis.[7]
4. Discussion
Our results indicate that patients with biopsy-proven IgG4-RD can also have positive ANCA with specificity for either MPO or PR3, with or without the concomitant presence of an overt AAV. This finding is supported by a growing body of literature and suggests caution in the interpretation of ANCA positivity, a finding traditionally viewed as highly specific for AAV if confirmed by both immunofluorescence and enzyme immunoassay.[6–10,15]
ANCA assays require precise interpretation according to well-established algorithms.[16–18] This process is based, first, upon the confirmation of MPO or PR3 specificity through either ELISA or chemiluminescent assays. The specificity of immunofluorescence for ANCA testing is suboptimal, as immunofluorescence assays are associated with positive results in a number of conditions other than small vessel vasculitides.[19] Most ANCA target antigens other than MPO or PR3; that is, they target an array of granulocyte or monocyte granule components and often have multiple or unknown specificities. Once confirmation by enzyme immunoassay or chemiluminescence is obtained; however, a positive ANCA result in and of itself is not diagnostic of AAV but rather requires clinical-pathological correlation and histological confirmation.
The case we describe here is emblematic in this sense because, based on the sole assumption that pulmonary pseudo-tumors are well-described manifestations of IgG4-RD, the histological evidence of IgG4-related dacryoadenitis appeared sufficient for diagnostic purposes and did not require confirmation with a lung biopsy. Because cavitary lesions are not typical of IgG4-RD, however, we proceeded to lung biopsy, which ultimately led to the concomitant diagnosis of AAV. The fact of a paravertebral thoracic mass in our patient further complicated the differential diagnosis, as it is an atypical manifestation of either GPA or IgG4-RD.[20,21] In summary, the finding of a positive ANCA—even with a specificity for PR3—should not lead to premature closure of the diagnostic evaluation, particularly if the pattern of organ involvement is not strictly typical of AAV. In our patient, the presence of lacrimal gland disease and cavitary pulmonary lesions urged more detailed investigations.
On the other hand, positive ANCA might precede overt manifestations of AAV.[22] As described in our aggregate analysis, this was actually the case of 3 patients with biopsy-proven IgG4-RD and positive ANCA who developed clinical manifestations of AAV shortly after the diagnosis. This observation suggests interesting considerations with regard to the pathogenic relevance of ANCA antibodies. Indeed, both MPO- and PR3-ANCA of patients with AAV have been previously shown to induce respiratory burst, calcium flux, and degranulation of neutrophils, thus directly contributing to the pathophysiology of AAV.[23–25] IgG4 ANCA are known to elicit the vigorous generation of reactive oxygen species in vitro, comparable to those elicited by IgG3 ANCA, and greater than those induced by IgG1 ANCA.[26] The ANCA in AAV are most often IgG1 and IgG4—a finding that we confirmed in our patient.[23,27] It is therefore tempting to speculate that the prominent IgG1 and IgG4 production that is characteristic of IgG4-RD might foster the development of AAV in some patients with an appropriate genetic background.[28,29] Indeed, the abundant IgG4 positive plasma cell infiltrate that we found in GPA lesions may further support this hypothesis.
IgG4-RD in patients with positive ANCA was limited to the head and neck, and did not involve other commonly affected organs, such as the lung, the pancreas, the biliary tree, and the retroperitoneum. In contrast, AAV in patients with established IgG4-RD presented with systemic manifestations beyond the ear/nose/throat region, consistent with the notion of that ANCA positivity is more common in the former clinical setting than in the latter.[30] These observations suggest that patients with IgG4-RD and positive ANCA might be more at risk of developing a systemic form of AAV. Indeed, head and neck manifestations of IgG4-RD are typically ANCA negative[31–33] raising the possibility that ANCA in these patients might sustain an underlying vasculitic process rather than being primarily linked to IgG4-RD. Additional cases and a longer follow-up are needed in order to confirm this hypothesis.
Table 2 describes the clinical characteristics of 4 additional patients with positive ANCA and a diagnosis of possible IgG4-RD with meningeal (3 cases) and retroperitoneal (1 case) involvement. ANCA specificity was confirmed by ELISA in all 4 patients, but no diagnostic procedures were performed in these patients. For these reasons, despite serum IgG4 increase, we did not include these cases in our aggregate analysis. However, as opposite to pachymeningeal involvement, which might occurs in up to 60% of patients with GPA,[34] retroperitoneal fibrosis is an uncommon manifestation of AAV. Therefore, while those cases of ANCA positive pachymeningitis are more likely patients with GPA, ANCA positive retroperitoneal fibrosis with elevated serum IgG4 could represent a case of IgG4-RD. Patients with positive ANCA and retroperitoneal fibrosis, indeed, have been already reported in the literature, raising the possibility of a novel immune-mediated condition associated with retroperitoneal inflammation.[35,36] Histological findings of the retroperitoneal tissue, however, have not been reported in these case series, thus making impossible to drive definitive conclusions.
5. Conclusions
In conclusion, the present work demonstrates that ANCA positivity in patients with biopsy-proven IgG4-RD should prompt the exclusion of a concomitant vasculitic process. In addition, a positive ANCA does not exclude the diagnosis of IgG4-RD absolutely, particularly in cases with head and neck involvement that is characteristic of IgG4-RD. The hypothesis that ANCA might predict the development of AAV in patients with IgG4-RD deserves larger case series with a longer follow-up. Confirmation through immunoenzymatic assays of the ANCA specificity, clinical-pathological correlation, and histopathological evaluation remain crucial steps for the differential diagnosis between AAV and IgG4-RD.
Acknowledgment
The authors thank Dr Raffaella Milani (Immunohematology and Transfusion Medicine Unit, IRCCS San Raffaele Scientific Institute) for performing flow cytometry studies.
Footnotes
Abbreviations: 18F-FDG = 18F-fluorodeoxyglucose, AAV = ANCA-associated vasculitis, ANCA = antineutrophil cytoplasmic antibodies, EGPA = eosinophilic granulomatosis with polyangiitis, GPA = granulomatosis with polyangiitis, IgG4-RD = IgG4-related disease, MPA = microscopic polyangiitis, MPO = myeloperoxidase, MRI = magnetic resonance imaging, PET/CT = positron emission tomography/computed tomography, PR3 = proteinase 3.
ED-T and ML have contributed equally to this manuscript.
This work was supported in part by a grant from the “Fondazione Italiana per la Ricerca sull’ Artrite” (FIRA Onlus 2014).
The authors have no conflicts of interest to disclose.
References
- 1.Della-Torre E, Lanzillotta M, Doglioni C. Immunology of IgG4-related disease. Clin Exp Immunol 2015; 181:191–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012; 25:1181–1192. [DOI] [PubMed] [Google Scholar]
- 3.Seo P, Stone JH. The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med 2004; 117:39–50. [DOI] [PubMed] [Google Scholar]
- 4.Chang SY, Keogh KA, Lewis JE, et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener's): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol 2013; 44:2432–2437. [DOI] [PubMed] [Google Scholar]
- 5.Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis 2015; 74:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexandraki KI, Kaltsatou M, Chatzellis E, et al. Hypophysitis in IgG4-related disease associated with p-ANCA vasculitis. Am J Med 2016; 129:25–27. [DOI] [PubMed] [Google Scholar]
- 7.Tosovský M, Bradna P, Laco J, et al. Case 1-2012: ANCA associated glomerulonephritis in combination with IgG4-positive mediastinal mass in a patient with ankylosing spondylitis treated with TNF alpha inhibitors. Acta Medica 2012; 55:42–46. [DOI] [PubMed] [Google Scholar]
- 8.Ohno K, Matsuda Y, Arai T, et al. Myeloperoxidase-antineutrophil cytoplasmic antibody-positive otitis media and rhinosinusitis with pathological features of immunoglobulin G4-related disease: a case report. Ann Otol Rhinol Laryngol 2016; 125:516–521. [DOI] [PubMed] [Google Scholar]
- 9.Hanioka Y, Yamagami K, Yoshioka K, et al. Churg-Strauss syndrome concomitant with chronic symmetrical dacryoadenitis suggesting Mikulicz's disease. Intern Med 2012; 51:2457–2461. [DOI] [PubMed] [Google Scholar]
- 10.Paulus YM, Cockerham KP, Cockerham GC, et al. IgG4-positive sclerosing orbital inflammation involving the conjunctiva: a case report. Ocul Immunol Inflamm 2012; 20:375–377. [DOI] [PubMed] [Google Scholar]
- 11.Aragonès JM, Arias-Rivero M, García-Barrionuevo JM, et al. IgG4- and MPO-ANCA-associated hypertrophic pachymeningitis. Rev Neurol 2015; 61:454–457. [PubMed] [Google Scholar]
- 12.Popkirov S, Kowalski T, Schlegel U, et al. Immunoglobulin-G4-related hypertrophic pachymeningitis with antineutrophil cytoplasmatic antibodies effectively treated with rituximab. J Clin Neurosci 2015; 22:1038–1040. [DOI] [PubMed] [Google Scholar]
- 13.Kotani S, Wakamatsu R, Itoh A, et al. Proteinase 3 anti-neutrophil cytoplasmic antibody (PR3-ANCA) positive IgG4-related retroperitoneal fibrosis: utility of PET-CT with 18F-fluorodeoxy glucose (FDG). Intern Med 2012; 51:755–758. [DOI] [PubMed] [Google Scholar]
- 14.Iguchi A, Wada Y, Kobayashi D, et al. A case of MPO- and PR3-ANCA-positive hypertrophic cranial pachymeningitis with elevated serum IgG4. Mod Rheumatol 2013; 23:151–155. [DOI] [PubMed] [Google Scholar]
- 15.Radice A, Sinico RA. Antineutrophil cytoplasmic antibodies (ANCA). Autoimmunity 2005; 38:93–103. [DOI] [PubMed] [Google Scholar]
- 16.Watts R, Lane S, Hanslik T, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis 2007; 66:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lutalo PMK, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener's granulomatosis). J Autoimmun 2014; 48–49:94–98. [DOI] [PubMed] [Google Scholar]
- 18.Sørensen SF, Slot O, Tvede N, et al. A prospective study of vasculitis patients collected in a five year period: evaluation of the Chapel Hill nomenclature. Ann Rheum Dis 2000; 59:478–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radice A, Bianchi L, Sinico RA. Anti-neutrophil cytoplasmic autoantibodies: methodological aspects and clinical significance in systemic vasculitis. Autoimmun Rev 2013; 12:487–495. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura H, Hisatomi K, Koga T, et al. Successful treatment of a patient with IgG4-related disease with a paravertebral mass lesion. Mod Rheumatol 2011; 21:524–527. [DOI] [PubMed] [Google Scholar]
- 21.Ramirez GA, Della-Torre E, Campochiaro C, et al. Juxta-vertebral lesions in granulomatosis with polyangiitis. Semin Arthritis Rheum 2016; http://dx.doi.org/10.1016/jhttp://dx.doi.org/10.1016/j. semarthrit.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Jayne DR, Gaskin G, Pusey CD, et al. ANCA and predicting relapse in systemic vasculitis. QJM 1995; 88:127–133. [PubMed] [Google Scholar]
- 23.Segelmark M, Wieslander J. IgG subclasses of antineutrophil cytoplasm autoantibodies (ANCA). Nephrol Dial Transplant 1993; 8:696–702. [DOI] [PubMed] [Google Scholar]
- 24.Kallenberg CGM. Pathogenesis of ANCA-associated vasculitides. Ann Rheum Dis 2011; 70:59–63. [DOI] [PubMed] [Google Scholar]
- 25.Harper L, Radford D, Plant T, et al. IgG from myeloperoxidase-antineutrophil cytoplasmic antibody-positive patients stimulates greater activation of primed neutrophils than IgG from proteinase 3-antineutrophil cytosplasmic antibody-positive patients. Arthritis Rheum 2001; 44:921–930. [DOI] [PubMed] [Google Scholar]
- 26.Holland M, Hewins P, Goodall M, et al. Anti-neutrophil cytoplasm antibody IgG subclasses in Wegener's granulomatosis: a possible pathogenic role for the IgG4 subclass. Clin Exp Immunol 2004; 138:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brouwer E, Tervaert JW, Horst G, et al. Predominance of IgG1 and IgG4 subclasses of anti-neutrophil cytoplasmic autoantibodies (ANCA) in patients with Wegener's granulomatosis and clinically related disorders. Clin Exp Immunol 1991; 83:379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamisawa T, Zen Y, Pillai S, et al. IgG4-related disease. Lancet 2015; 385:1460–1471. [DOI] [PubMed] [Google Scholar]
- 29.Mahajan VS, Mattoo H, Deshpande V, et al. IgG4-related disease. Ann Rev Pathol 2014; 9:315–347. [DOI] [PubMed] [Google Scholar]
- 30.Millet A, Pederzoli-Ribeil M, Guillevin L, et al. Antineutrophil cytoplasmic antibody-associated vasculitides: is it time to split up the group? Ann Rheum Dis 2013; 72:1273–1279. [DOI] [PubMed] [Google Scholar]
- 31.Reder L, Della-Torre E, Stone JH, et al. Clinical manifestations of IgG4-related disease in the pharynx: case series and review of the literature. Ann Otol Rhinol Laryngol 2015; 124:173–178. [DOI] [PubMed] [Google Scholar]
- 32.Della-Torre E, Mattoo H, Mahajan VS, et al. IgG4-related midline destructive lesion. Ann Rheum Dis 2014; 73:1434–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanzillotta M, Campochiaro C, Trimarchi M, et al. Deconstructing IgG4-related disease involvement of midline structures: comparison to common mimickers. Mod Rheumatol [Under Review]. [DOI] [PubMed] [Google Scholar]
- 34.De Luna G, Terrier B, Kaminsky P, et al. Central nervous system involvement of granulomatosis with polyangiitis: clinical-radiological presentation distinguishes different outcomes. Rheumatology (Oxford) 2015; 54:424–432. [DOI] [PubMed] [Google Scholar]
- 35.Mavragani CP, Voulgarelis M. Retroperitoneal fibrosis and c-ANCA positivity. Clin Rheumatol 2007; 26:115–116. [DOI] [PubMed] [Google Scholar]
- 36.Vaglio A, Manenti L, Allegri L, et al. ANCA-positive periaortic vasculitis: does it fall within the spectrum of vasculitis? J Intern Med 2002; 251:268–271. [DOI] [PubMed] [Google Scholar]