


Abstract
We have created the Functional Element SNPs Database (FESD) that categorizes functional elements in human genic regions and provides a set of single nucleotide polymorphisms (SNPs) located within each area. In the FESD, the human genic regions were divided into 10 different functional elements, such as promoter regions, CpG islands, 5′-untranslated regions (5′-UTRs), translation start sites, splice sites, coding exons, introns, translation stop sites, polyadenylation signals and 3′-UTRs, and subsequently, all the known SNPs were assigned to each functional element at their respective position. With the FESD web interface, users can select a set of SNPs in the specific functional elements and get their flanking sequences for genotyping experiments, which will help in finding mutations that contribute to the common and polygenic diseases. A web interface for the FESD is freely available at http://combio.kribb.re.kr/ksnp/resd/.
INTRODUCTION
Single nucleotide polymorphisms (SNPs) coupled with high-throughput genotyping technologies have been chosen as the high-density genetic markers for unraveling complex genetic diseases (1). Almost 9 million human SNPs have been deposited into the dbSNP database (2) and many researchers have tried to discover and validate new SNPs. As the number of SNPs deposited in the dbSNP database is increasing exponentially, many researchers who are interested in complex genetic diseases are focusing on candidate gene approach using gene-based haplotypes, which are collections of SNPs located throughout the functional regions of candidate genes (3). Since the number of haplotypes within candidate genes is much smaller than the theoretical number of all possible haplotypes, gene-based haplotypes will give us an effective data-reduction mechanism and enable us to uncover the association of haplotypes with many complex diseases.
A set of SNPs required for constructing haplotypes within the candidate genes can be retrieved from public databases, such as dbSNP, UCSC genome browser or Ensembl browser (2,4). However, since these public databases do not provide user-friendly interfaces for manipulating a set of SNPs, biologists who are not familiar with these public databases have had difficulties in searching a set of SNPs and retrieving their flanking sequences required for genotyping experiments.
There has been a similar program called SNPper, a web-based application designed to automate the tasks of retrieving and extracting SNPs from public databases (5). However, SNPper provides the combined information available only in public resources.
In this study, we have created a Functional Element SNPs Database (FESD) categorizing functional elements in human genic regions and providing their flanking sequences required for genotyping experiments. By using the FESD web interface, researchers can select a set of SNPs for gene-based haplotype study.
MATERIALS AND METHODS
Obtaining the human genome sequences and gene information
The reference sequences of human genome are obtained from the NCBI database and, subsequently, functional element sequences are extracted from those using sequence boundary information derived from the UCSC database.
There are 16 tracks within Genes and Gene Prediction Tracks in the UCSC database, such as Known Genes, RefSeq Genes, MGC Genes, Ensembl Genes, Genscan Genes and so on. The length and position of each gene track is slightly different. Among the gene tracks, the RefSeq Genes track is produced from the mRNA sequence data that are generated and curated by the NCBI RefSeq project (6). In this study, we have used RefSeq Genes (refGene) track for gene information. Although there are 21 222 refSeq IDs in the refGene track, the positions of 51 refSeq IDs are not determined and 12 refSeq IDs are located at the mitochondrial DNA. Since we are not interested in the mitochondrial DNA, we used only the valid 21 159 refSeq IDs.
Categorization of functional elements
Functional elements in the human genes are categorized into 10 different functional regions, such as promoter regions, CpG islands, 5′-untranslated regions (5′-UTRs), translation start sites, splice sites, coding exons, introns, translation stop sites, polyadenylation signals (PASes) and 3′-UTRs (Figure 1).
Figure 1.

Categorization of functional elements in the human gene. Functional elements are divided into 10 different functional regions, such as promoter regions, CpG islands, 5′-UTRs, translation start sites, splice sites, coding exons, introns, translation stop sites, PASes and 3′-UTRs.
Promoter regions
Human promoter sequences are difficult to identify and are poorly annotated in the public databases. We used the Match program, which is a weight matrix-based tool for searching putative transcription factor binding sites in the DNA sequences (7). Match program is closely interconnected and distributed together with the TRANSFAC database, which was developed more than a decade ago to model factor–site interaction (8). The current version of Match program (TRANSFAC Professional 7.4.1) uses 695 matrices and 5401 transcription factors that were collected in the TRANSFAC database, and therefore, provides the great possibility to search for a great deal of different transcription factor binding sites.
Although the range of transcription factors may be huge, it is reported that most of the human promoters are located within 2 kb upstream of the transcription start site (7). Therefore, the length of input sequences for the Match program is restricted to 2 kb upstream of the transcription start site. To find transcription factor binding sites with the help of Match program, it is very important to choose appropriate options of the cut-off for core and matrix similarity. We used the minimizing the false positive (FP) errors option for cut-offs and chose the vertebrate option for group of matrices.
CpG islands
CpG islands are associated with genes and are typically common near transcription start sites in vertebrates. CpG islands in the UCSC database are predicted by searching the sequence and scoring each dinucleotide. We downloaded the complete tracks and filtered them out only if they were located within the 2000 bp upstream. Only 12 916 refSeq IDs out of 21 159 had CpG islands in their 2000 bp upstream region.
5′-UTRs, 3′-UTRs, coding exons, introns, start codon and stop codon
In refGene track of the UCSC database, there are position information of exon, intron boundaries and coding sequence (CDS) boundaries (6). The 5′-UTRs are upstream sequences from the CDS start sites, whereas the 3′-UTRs are downstream sequences from the CDS stop sites. In this study, coding exons are considered as CDSs within the transcripts, which mean that 5′-UTRs are not coding exons. In contrast, introns are non-transcribed sequences from the DNA sequences; therefore, introns can exist between UTRs and coding exons. The start and end sites of translations are the first and the last 3 bp of CDS, respectively.
Splice sites
We considered the first and the last 2 bp in introns as splice sites (9). The positions of introns are obtained from refGene track in the UCSC database. When any SNPs are found within the splice sites, we place them in the database.
Polyadenylation signal sites
There are several software programs that have been developed to detect PASes in human DNA and mRNA sequences. In one of the early studies, Tabaska and Zhang (10) developed a program named Polyadq, which finds PASes using a pair of quadratic discriminant functions. Recently, Legendre and Gautheret (11) developed Erpin program, which uses 2 g position-specific nucleotide acid patterns to analyze 300 bases upstream and downstream region of a candidate PAS.
In this study, we have used the Erpin program to find PASes from 3′-UTR region of the gene. The Linux version of Erpin program was downloaded and installed at a local Linux PC. The training set file was also downloaded from the website and the query database that contains only 3′-UTR was subtracted from refGene track in the UCSC database. The recommended optimal parameters are as follows: ‘erpin polya_signal.epn <input database> 11,7 -umask 11 -umask 11 4 5 6 7 -cut-off 70% 74% -fwd -unifstat’. The first-level search is for the hexameric PAS with a cut-off score of 70%, and the second-level search is for the 46 nt region immediately downstream of the PAS with a cut-off score of 74% (11).
Assigning SNPs to each functional element
This analysis used 8 431 426 SNPs from the dbSNP database (build 120). The position of SNPs was obtained from the flat file that was downloaded from the NCBI ftp site (ftp://ftp.ncbi.nih.gov/snp/human/). Known SNPs are assigned to each functional element, if the SNP is located at the corresponding element.
Flanking sequence search program: SNPflank
The SNPflank database consists of two types of data: complete genome sequence from the RefSeq genome contigs and the positional information of refSNPs from the dbSNP database (12). Both data were completely downloaded from the NCBI ftp site, parsed with Perl scripts, and subsequently imported into a MySQL relational database.
RESULTS AND DISCUSSION
The distribution of SNPs in the genic–intergenic regions
Table 1 shows the distribution of SNPs in the genic–intergenic regions of the human genome. The genic region contained 3 446 791 SNPs (40.8%), whereas the intergenic region contained 4 984 635 SNPs (59.2%). There are more SNPs in the intergenic region; however, the density of the intergenic region is slightly lower than the genic region.
Table 1. The distribution of SNPs in the genic–intergenic regions.
| Mean density (SNP/kb) | Mean spacing (bp/SNP) | SNP count (SNP) | Total length (bp) | |
|---|---|---|---|---|
| Genic region | 2.853 | 350.493 | 3 446 791 | 1 208 074 845 |
| Intergenic region | 2.784 | 359.210 | 4 984 635 | 1 790 531 349 |
| Total genome | 2.814 | 355.646 | 8 431 426 | 2 998 606 194 |
Table 2 shows the distribution of SNPs along with the functional element categories. The proportion of SNPs occurring in the promoter, CpG, 5′-UTR, start codon, splice site, coding exon, intron, stop codon, PAS and 3′-UTR was 3.68, 1.57, 0.37, 0.002, 0.03, 2.26, 90.04, 0.003, 0.002 and 2.03%, respectively. This is because the proportion of SNPs depends on the length of the regions. If we considered the length of each region, the density of SNPs in each functional region would have been 3.269, 2.956, 3.233, 1.119, 1.326, 2.453, 2.847, 1.985, 2.105 and 3.841, respectively. The density of SNPs in the promoter and UTRs was significantly higher than the average density of genome whose density was 2.814. The density of SNPs in the 3′-UTR was slightly higher than that in the 5′-UTR regions. Among the functional elements, the density of start codon was the lowest at 1.119.
Table 2. The distribution of SNPs in the functional elements.
| Mean density (SNP/kb) | Mean spacing (bp/SNP) | SNP count (SNP) | Total length (bp) | |
|---|---|---|---|---|
| Promoter | 3.269 | 305.882 | 133 327 | 40 782 381 |
| CpG | 2.956 | 338.278 | 56 829 | 19 223 987 |
| 5′-UTR | 3.233 | 309.317 | 13 997 | 4 329 509 |
| Start codon | 1.119 | 894.042 | 71 | 63 477 |
| Splice site | 1.326 | 754.223 | 1 067 | 804 756 |
| Coding exon | 2.453 | 407.634 | 81 963 | 33 410 932 |
| Intron | 2.847 | 351.230 | 3 264 275 | 1 146 509 847 |
| Stop codon | 1.985 | 503.786 | 126 | 63 477 |
| PAS | 2.105 | 475.034 | 87 | 41 328 |
| 3′-UTR | 3.841 | 260.373 | 73 638 | 19 173 314 |
The density of SNPs in the functional elements is depicted in Figure 2. Critical functional elements such as start codon, splice site and stop codon are likely to possess rare SNP density, whereas the density of other functional elements are similar to average density of the genome.
Figure 2.

The density of SNPs in the functional elements. The density is calculated as the average number of SNPs within 1000 bp of each functional region. The density of start codon is the lowest at 1.119.
Table 3 shows the density of SNPs along with the functional element categories separated by chromosome. According to Table 3, the density of SNPs in chr7 is significantly higher than that in others and the density of SNPs in chr21 and chr22 is slightly higher than that in others. In contrast, the density of SNPs in chrX and chrY is slightly lower than that in others. These density differences may be attributable to differences in the input SNP/chromosome from the dbSNP database. For example, the NCBI is hosting two versions of chromosome 7.
Table 3. The density of SNPs in the gene region by chromosome.
| Promoter | CpG | 5′-UTR | Start | Splice | Exon | Intron | Stop | PAS | 3′-UTR | |
|---|---|---|---|---|---|---|---|---|---|---|
| Chr1 | 3.051 | 2.790 | 3.196 | 1.112 | 1.183 | 2.325 | 2.710 | 1.906 | 1.195 | 3.780 |
| Chr2 | 2.506 | 2.336 | 2.521 | 1.705 | 0.507 | 1.824 | 2.235 | 2.436 | 2.419 | 3.395 |
| Chr3 | 2.673 | 2.437 | 3.103 | 1.123 | 0.970 | 2.052 | 2.523 | 1.404 | 1.827 | 3.343 |
| Chr4 | 2.817 | 2.545 | 2.858 | 0.430 | 0.675 | 1.959 | 2.294 | 1.291 | 2.639 | 3.032 |
| Chr5 | 2.804 | 2.837 | 3.001 | 0 | 0.481 | 2.016 | 2.423 | 0.676 | 1.610 | 3.247 |
| Chr6 | 3.756 | 3.576 | 3.581 | 0.904 | 1.177 | 2.651 | 3.012 | 2.111 | 4.284 | 4.126 |
| Chr7 | 5.030 | 5.071 | 5.767 | 4.462 | 3.590 | 4.717 | 4.861 | 3.432 | 5.078 | 6.337 |
| Chr8 | 3.075 | 2.817 | 3.606 | 1.801 | 0.926 | 2.343 | 2.537 | 0.900 | 1.996 | 3.388 |
| Chr9 | 3.331 | 3.057 | 3.291 | 0.834 | 1.422 | 2.340 | 3.063 | 0.834 | 0 | 3.644 |
| Chr10 | 3.308 | 3.271 | 3.514 | 0.805 | 1.024 | 2.638 | 3.173 | 1.610 | 0.874 | 3.844 |
| Chr11 | 3.354 | 3.270 | 3.565 | 1.112 | 1.291 | 2.581 | 3.086 | 3.338 | 4.079 | 3.980 |
| Chr12 | 3.101 | 2.831 | 3.089 | 0.894 | 1.203 | 2.211 | 2.890 | 1.788 | 3.875 | 3.989 |
| Chr13 | 3.265 | 2.939 | 2.975 | 0.960 | 0.592 | 2.072 | 2.944 | 0 | 3.100 | 3.263 |
| Chr14 | 2.901 | 2.658 | 2.791 | 0.516 | 1.016 | 1.944 | 2.416 | 1.550 | 1.494 | 3.460 |
| Chr15 | 2.501 | 2.212 | 2.357 | 0 | 0.769 | 2.102 | 2.375 | 2.683 | 0.931 | 3.450 |
| Chr16 | 3.097 | 2.751 | 2.747 | 0 | 1.062 | 2.540 | 3.212 | 1.114 | 0 | 3.530 |
| Chr17 | 4.588 | 2.513 | 2.784 | 0.792 | 1.360 | 2.384 | 2.532 | 2.377 | 0.837 | 4.013 |
| Chr18 | 3.112 | 3.149 | 2.743 | 3.300 | 1.272 | 2.244 | 2.632 | 0 | 3.067 | 3.432 |
| Chr19 | 3.106 | 2.850 | 3.382 | 1.216 | 1.313 | 2.865 | 2.825 | 2.433 | 1.048 | 4.250 |
| Chr20 | 4.160 | 3.835 | 4.067 | 0.917 | 5.198 | 3.487 | 4.167 | 3.668 | 0.793 | 5.137 |
| Chr21 | 3.709 | 2.999 | 2.691 | 1.082 | 1.291 | 2.982 | 4.065 | 2.164 | 0 | 4.329 |
| Chr22 | 4.178 | 3.956 | 4.304 | 0.649 | 2.489 | 3.339 | 3.969 | 4.548 | 0 | 5.160 |
| ChrX | 2.145 | 1.853 | 2.251 | 1.481 | 1.259 | 1.780 | 1.822 | 1.481 | 1.644 | 2.614 |
| ChrY | 2.491 | 2.514 | 1.857 | 0 | 1.396 | 2.442 | 1.424 | 0 | 0 | 3.479 |
DATABASE ACCESS
Searching for genes
The FESD website provides capabilities for searching for genes in several ways: (i) gene name, mRNA accession number and protein accession number; (ii) LocusLink ID and OMIM ID; (iii) chromosome position and band position; (iv) transcription factor name; and (v) disorder or clinical synopsis (Figure 3A).
Figure 3.
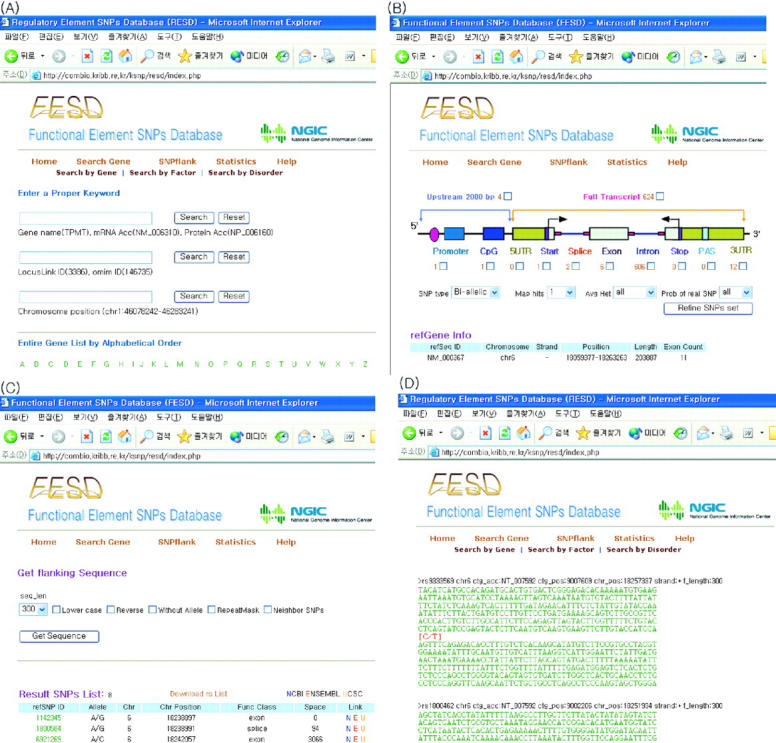
Web interface of FESD. (A) Gene search interface. Users can search genes using gene name, mRNA accession number, protein accession number, LocusLink ID, OMIM ID, chromosome position, band position, transcription factor name and disorder or clinical synopsis. (B) Graphical view of sets of SNPs along with each functional element. (C) A refined list of SNPs and their hyper links to public databases. (D) FASTA formatted flanking sequences for selected SNPs.
Selecting sets of SNPs
Once a gene is searched and submitted to the FESD server, sets of SNPs can be chosen by two different ways (Figure 3B). First, by using the hyperlinks that present the number of SNPs located within that area, users can get the sets of SNPs (Figure 3B). Second, users can merge different sets of SNPs by simply clicking checkboxes in the graphic view. When multiple functional elements are selected, only distinct SNPs derived from overlapped elements will be displayed. At the same time, users will also be able to filter the selected sets of SNPs by certain thresholds such as SNP type (bi-allelic or indel), number of hits in genome, average heterozygosity and probability of real SNPs.
Getting flanking sequences of selected SNPs
FESD provide flanking sequences of selected SNPs for genotyping experiments. There are many options for getting flanking sequences such as length range, alternating case and reverse complement change (Figure 3C). Flanking sequences of multiple SNPs are provided with the FASTA format (Figure 3D). Details of FESD are given in the online help page.
FUTURE DIRECTIONS
FESD will be updated regularly to reflect the newly discovered SNPs in the dbSNP database and the change of track information in the UCSC database, and also to improve the methods used for categorizing the functional elements. We also plan to give users more options for choosing sets of SNPs and refining them with the help of validation information of SNPs.
Acknowledgments
ACKNOWLEDGEMENTS
This research was supported by a grant from the KRIBB Research Initiative Program and in part by the Korean HapMap Project from the Korea Ministry of Science & Technology.
REFERENCES
- 1.Gray I.C., Campbell,D.A. and Spurr,N.K. (2000) Single nucleotide polymorphisms as tools in human genetics. Hum. Mol. Genet., 9, 2403–2408. [DOI] [PubMed] [Google Scholar]
- 2.Sherry S.T., Ward,M.H., Kholodov,M., Baker,J., Phan,L., Smigielski,E.M. and Sirotkin,K. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ring H.Z. and Kroetz,D.L. (2002) Candidate gene approach for pharmacogenetic studies. Pharmacogenomics, 3, 47–56. [DOI] [PubMed] [Google Scholar]
- 4.Kent W.J., Sugnet,C.W., Furey,T.S., Roskin,K.M., Pringle,T.H., Zahler,A.M. and Haussler,D. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riva A. and Kohane,I.S. (2002) SNPper: retrieval and analysis of human SNPs. Bioinformatics, 18, 1681–1685. [DOI] [PubMed] [Google Scholar]
- 6.Pruitt K.D. and Maglott,D.R. (2001) RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res., 29, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kel A.E., Gossling,E., Reuter,I., Cheremushkin,E., Kel-Margoulis,O.V. and Wingender,E. (2003) MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res., 31, 3576–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matys V., Fricke,E., Geffers,R., Gossling,E., Haubrock,M., Hehl,R., Hornischer,K., Karas,D., Kel,A.E., Kel-Margoulis,O.V. et al. (2003) TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res., 31, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang M.Q. (1998) Statistical features of human exons and their flanking regions. Hum. Mol. Genet., 7, 919–932. [DOI] [PubMed] [Google Scholar]
- 10.Tabaska J.E. and Zhang,M.Q. (1999) Detection of polyadenylation signals in human DNA sequences. Gene, 231, 77–86. [DOI] [PubMed] [Google Scholar]
- 11.Legendre M. and Gautheret,D. (2003) Sequence determinants in human polyadenylation site selection. BMC Genomics, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benson D.A., Karsch-Mizrachi,I., Lipman,D.J., Ostell,J. and Wheeler,D.L. (2004) GenBank: update. Nucleic Acids Res., 32, D23–D26. [DOI] [PMC free article] [PubMed] [Google Scholar]