

Abstract
Background
Trimethylamine-N-oxide (TMAO), an atherogenic metabolite species, has emerged as a possible new risk factor for cardiovascular disease. Animal studies have shown that circulating TMAO levels are regulated by genetic and environmental factors. However, large-scale human studies have failed to replicate the observed genetic associations, and epigenetic factors such as DNA methylation have never been examined in relation to TMAO levels.
Methods and Results
We used data from the family-based Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) to investigate the heritable determinants of plasma TMAO in humans. TMAO was not associated with other plasma markers of cardiovascular disease, e.g. lipids or inflammatory cytokines. We first estimated TMAO heritability at 27%, indicating a moderate genetic influence. We used 1000 Genomes imputed data (n=626) to estimate genome-wide associations with TMAO levels, adjusting for age, sex, family relationships, and study site. The genome-wide study yielded one significant hit at the genome-wide level, located in an intergenic region on chromosome 4. We subsequently quantified epigenome-wide DNA methylation using the Illumina Infinium array on CD4+ T-cells. We tested for association of methylation loci with circulating TMAO (n=847), adjusting for age, sex, family relationships, and study site as the genome-wide study plus principal components capturing CD4+ T-cell purity. Upon adjusting for multiple testing, none of the epigenetic findings were statistically significant.
Conclusions
Our findings contribute to the growing body of evidence suggesting that neither genetic nor epigenetic factors play a critical role in establishing circulating TMAO levels in humans.
Keywords: atherosclerosis, cardiovascular disease, genetic, epigenetic, methylation, trimethylamine-N-oxide
Introduction
Trimethylamine-N-oxide (TMAO), a pro-atherogenic metabolite species, has recently emerged as a possible causal risk factor for cardiovascular disease (CVD) [1]. TMAO is synthesized in the liver from trimethylamine (TMA), which in turn is released by the gut flora from TMA-containing dietary phospholipid components such as choline, betaine, lecithin, and L-carnitine. Plasma concentrations of L-carnitine, a nutrient commonly found in red meat and seafood, have been linked to both prevalent and incident CVD in a TMAO-dependent manner [2]. Furthermore, elevated plasma TMAO was associated with increased cardiovascular risk even in low-risk subgroups [3]. Other studies have linked TMAO levels to clinical outcomes in the context of heart failure [4] and chronic kidney disease [5], highlighting its importance in chronic disease pathogenesis.
Animal studies have shown that circulating TMAO levels are regulated by genetic and environmental factors [6]. Specifically, a genome-wide association study conducted in mice identified robust associations between sequence variation and TMAO levels; however, these findings were not replicated in a large-scale human sample [7] and the heritability of TMAO in humans remains to be established. In addition to DNA sequence variants, methylation loci may play a role in TMAO homeostasis because epigenetic processes integrate both genetic and environmental inputs such as diet. For example, betaine—one of the dietary substrates for TMAO production—can serve an alternate methyl source for converting homocysteine to methionine [8], increasing DNA methylation and altering gene expression. Consistent with that hypothesis, a recent human study reported inverse associations between plasma TMAO and methylation capacity, reflected in altered concentrations of S-adenosylhomocysteine and S-adenosylmethionine [9]. Despite the biological plausibility of epigenetic associations with TMAO in humans, such links have not yet been investigated on a genome-wide level. Although TMAO concentrations in urine are an order of magnitude higher than in plasma and easier to measure, using plasma concentrations of TMAO reduces variation due to acute dietary intake, resulting in more reliable measurements [10]. Using family data from the metabolically healthy population of the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN), we present the first heritability estimates of circulating TMAO as well as the first human epigenome-wide study of DNA methylation in relation to this promising biomarker.
Methods
Study population
The GOLDN study [11] recruited families of European descent with at least two siblings at two centers of the NHLBI Family Heart Study (Minneapolis and Salt Lake City). The primary aim of the study was to characterize genetic and epigenetic predictors of variability in lipid response to two interventions, namely a high-fat meal and a 3-week fenofibrate challenge. Both DNA and plasma TMAO for the current study were quantified on pre-intervention (baseline) samples to exclude potential effects of the diet and drug interventions. All participants provided written informed consent. Institutional Review Boards at University of Minnesota, University of Utah, and Tufts University/New England Medical Center approved the study protocol. GOLDN screened ~1,350 individuals and excluded those with age < 18 years; fasting triglycerides ≥1500 mg/dL; recent history of myocardial infarction, coronary bypass surgery, or coronary angioplasty; self-report of a positive history of liver, kidney, pancreas, or gall bladder disease, or a history of nutrient malabsorption; current use of insulin; abnormal liver or kidney function; in women of childbearing potential, pregnancy, breastfeeding, not using an acceptable form of contraception, yielding a net sample of 1,048 individuals that consented to the use of their DNA in research.
TMAO measurements
We measured TMAO levels by proton nuclear magnetic resonance (NMR) spectroscopy using a Vantera® NMR Clinical Analyzer at LipoScience (now LabCorp, Raleigh, NC). Briefly, plasma was diluted with citrate/phosphate buffer (3:1 v/v) to lower the pH to 5.3 in order to move the TMAO signal away from the overlapped signal from betaine. The diluted specimen was placed in a barcoded sample vial, from which 200μL was then automatically injected with preheating to 47°C into the flowcell of a 400 MHz superconducting magnet. Spectra were acquired using a Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence by signal averaging 48 transients with a total acquisition time of 5.5 minutes per sample. Free induction decay (FID) signals were multiplied by an exponential window function with a 0.1 Hz line broadening, Fourier transformed, and automatically phased and baseline corrected. The TMAO methyl signal at ca. 3.30 ppm was quantified using a proprietary non-negative linear least squares analysis that models the line shape as a mix of Gaussian and Lorentzian peak shapes. The derived TMAO signal amplitudes were then transformed into μmol/L concentrations using a conversion factor determined from analysis of dialyzed plasma samples spiked with known amounts of TMAO. NMR-derived TMAO concentrations are highly correlated (r2 = 0.98) with those measured using the liquid chromatography/mass spectrometry assay developed at the Cleveland Clinic [2].
Epigenetic phenotyping
We measured DNA methylation in GOLDN on the epigenome-wide scale using the Illumina Infinium HumanMethylation450 Beadchip (Illumina, San Diego, CA) as previously described [12, 13]. Briefly, to reduce the effect of cell type, we restricted the measurements to CD4+ T-cells that were isolated from peripheral blood frozen buffy coat samples. We isolated DNA using commercially available DNeasy kits (Qiagen, Venlo, Netherlands). We quantified methylation using β scores (proportion of total signal from the methylation-specific probe or color channel) and detection P-values (probability that the total intensity for a given probe falls within the background signal intensity). We estimated both β scores and detection P-values using the GenomeStudio software (Illumina, San Diego, CA). Quality control exclusion criteria were: β scores with an associated detection p-value greater than 0.01, samples with more than 1.5% missing data points across ~470,000 autosomal CpGs, or probes where 10% of samples or more failed to yield adequate intensity [13]. After exclusions, we normalized β scores (separately for Infinium I and II chemistries) using the ComBat package to address batch effects [14]. Finally, we removed methylation loci where the probe sequence mapped to a location that did not match the annotation file or to more than one locus. The final set of CpGs included 463,995 loci.
Genotyping
We genotyped GOLDN participants at 906,600 loci using the Affymetrix Genome Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA) as described in prior publications [15]. We called genotypes using the Birdseed algorithm [16]. We removed 53,530 monomorphic loci and 82,462 SNPs with a call rate below 96% (1,556 SNPs overlapped on these two criteria). Additionally, we removed any SNPs based on the number of families with Mendel errors as follows: 1,486 SNPs with minor allele frequency (MAF)>20% and Mendel errors in 3+ families, 1,338 SNPs with 20%≥MAF>10% and Mendel errors in 2+ families, 1,767 SNPs with 20%≥MAF>10% and Mendel errors in 1+ family, and 9,592 SNPs with MAF<5% and any Mendel errors. In families with remaining Mendel errors, the erroneous SNPs were set to missing (31,595 loci). 718,542 SNPs remained in the analysis following the quality control procedures described above. Of those, only 12 failed the Hardy-Weinberg equilibrium test at P-value < 10−6. After removal of the 64,908 SNPs with MAF<1%, 654,634 SNPs were available for imputation.
We performed imputations in two stages: pre-phasing using the MACH software/library and imputation using Minimac software (Abecasis Lab, Ann Arbor, MI) [17, 18]. The original Phase 1 release of 1000 Genomes reference panel used for imputation contained ~38 million single nucleotide variants (SNVs). After removing the singletons and monomorphic sites and merging the typed and imputed data, 27,449,496 variants on 821 participants were available for the genome-wide association study. We subsequently removed SNPs with poor imputation quality (r2<0.3) or MAF<0.01, yielding 9,432,837 variants. Of all genotyped participants, 626 had valid TMAO measurements and were included in the analysis.
Statistical analysis
We used Kruskal-Wallis rank sum tests to evaluate trends in demographic and clinical characteristics across quartiles of the untransformed TMAO distribution. For subsequent analyses, we log-transformed the TMAO variable to achieve normality. We estimated heritability of TMAO in GOLDN using the variance component approach implemented in the SOLAR program as previously described [19]. All models run in SOLAR included age and sex. For the genome- and epigenome-wide analyses, we used normal inverse transformed residuals obtained by regressing log-transformed TMAO on age and sex. We assessed epigenome-wide associations between TMAO residuals and DNA methylation variants using linear mixed models, adjusted for age, sex, center, and four principal components capturing T-cell purity as fixed effects and pedigree as a random effect [20]. We conducted sensitivity analyses, additionally adjusting for current smoking and alcohol intake. For genome-wide associations, we fit linear mixed models, adjusting for only pedigree (random effect) as well as age, sex, and center (fixed effects) because there was no evidence of confounding by population stratification in the genetically homogeneous GOLDN populations. We used HaploReg (Broad Institute, Cambridge, MA) to investigate the functional annotation of the top signal from the genome-wide study. Furthermore, we interrogated top genetic and epigenetic signals for potential overlap with other functional marks (e.g. histone modifications) or gene expression in biologically relevant tissues, e.g. liver and the gastrointestinal tract, using publicly available bioinformatics resources implemented in the UCSC Genome Browser. Statistical significance was assessed at the Bonferroni corrected thresholds of 0.05/9,432,837=5.3×10−9 and 0.05/463,995= 1.1×10−7 for the genome- and epigenome-wide studies, respectively. All genome- and epigenome-wide analyses were implemented in R, using the lmekin function in the kinship package to adjust for family relationships. We estimated genomic control parameters (λ) at 1.01 and 1.17 for the genome- and epigenome-wide analyses, respectively. We constructed quantile-quantile (Supplemental Figures 1 and 2) and Manhattan (Figures 1 and 2) plots to visualize the results.
Figure 1.
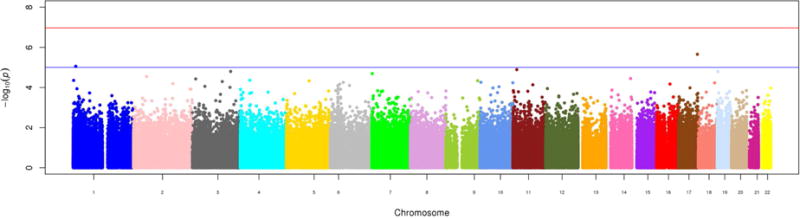
Manhattan plot of epigenome-wide results of testing for association between methylation at > 450,000 cytosine-phosphate-guanine sites and circulating trimethylamine-N-oxide. The X-axis displays the chromosome on which the site is located, the Y-axes display −log10(P-value). The red horizontal line indicates the threshold for epigenome-wide statistical significance after a Bonferroni correction. [Format double column; color figure]
Figure 2.
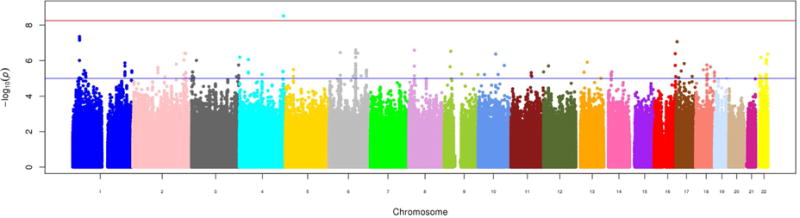
Manhattan plot of genome-wide results of testing for association between >9,400,000 genetic variants and circulating trimethylamine-N-oxide. The X-axis displays the chromosome on which the site is located, the Y-axes display −log10(P-value). The red horizontal line indicates the threshold for genome-wide statistical significance after a Bonferroni correction. [Format double column; color figure]
We estimated statistical power a priori using a combination of computer simulation and asymptotic distributions and a pedigree structure identical to GOLDN. We generated CpG methylation proportions with varying heritability and phenotypic data for each individual in the pedigree. For the epigenome-wide study, our simulations project the statistical power ranging from 0.71 for h2locus= 0.02 to 1.00 for h2locus= 0.05 or above—a realistic assumption given methylation heritabilities as high as 0.6 previously observed in our epigenetic data [21]. For the genome-wide association analysis, we had sufficient statistical power to detect the effect of SNPs with moderate heritability at the 5.3×10−9 (0.05/9,432,837 variants) significance level: from 0.89 to detect at least 3 of 10 causal loci for h2locus= 0.05 to 0.92 to detect all 10 causal loci for h2locus= 0.10.
Results
Clinical, demographic, and lifestyle characteristics by quartile of TMAO are summarized in Table 1. Circulating TMAO levels exhibited a strong positive association with age and weaker associations with sex (fewer women were represented in higher quartiles), hypertension, plasma triglycerides, and diabetes status. Adjustment for age (data not shown) rendered all observed associations statistically insignificant. Other risk factors for chronic disease, including intake of animal products, were not associated with circulating TMAO in the GOLDN population.
Table 1.
| 1st Quartile | 2nd Quartile | 3rd Quartile | 4th Quartile | P-value for Trend | |
|---|---|---|---|---|---|
| Median TMAO, μM | 1.43 | 2.43 | 4.39 | 10.475 | 2.2×10−16 |
| Range, μM | 1.43–1.70 | 1.71–3.24 | 3.25–6.06 | 6.08–42.31 | |
| Age, years | 45 ±14 | 48±17 | 51±16 | 51±17 | 1.5×10−7 |
| Sex, n (%) female | 141 (59.7) | 113 (47.9) | 105 (44.4) | 102 (43.2) | 0.002 |
| Smoker, n (%) current | 20 (8.47) | 15 (6.36) | 20 (8.47) | 17 (7.2) | 0.78 |
| Body mass index, kg/m2 | 28±6 | 28±5 | 28±6 | 29±6 | 0.22 |
| Hypertension, n (%) | 49 (21) | 57 (24) | 62 (26) | 78 (33) | 0.02 |
| Cholesterol, mg/dl | |||||
| Low-density lipoprotein | 117±29 | 124±32 | 123±34 | 123±30 | 0.07 |
| High-density lipoprotein | 48±13 | 46±12 | 46±13 | 48±14 | 0.19 |
| Triglycerides, mg/dl | 121±73 | 140±85 | 140±87 | 148±128 | 0.02 |
| C-reactive protein, mg/dl | 0.23±0.33 | 0.20±0.31 | 0.27±0.69 | 0.27±0.35 | 0.22 |
| Diabetes, n (%) | 14 (6) | 20 (8) | 12 (5) | 28 (12) | 0.03 |
| Intake of animal products‡, servings/day | 7±5 | 8±5 | 8±5 | 8±5 | 0.39 |
TMAO, trimethylamine-N-oxide
We present continuous variables other than TMAO quartile medians as means (standard deviations) and categorical variables as absolute numbers (percentages of total sample).
Includes eggs, beef, pork, lamb, poultry, fish, and other seafood.
We estimated circulating TMAO to be moderately heritable (h2= 0.27, P-value=3×10−6). We present the results of the epigenome-wide association study in Table 2 and Figure 1 and the results of the genome-wide association study in Table 3 and Figure 2. Only one SNP and no CpG loci reached genome-wide significance in our population. Notably, the top genome-wide association hits, including the significant locus rs114755225 on chromosome 4, were mostly rare variants (MAF <0.05). We reached out to several TMAO studies to attempt replication, however, neither rs114755225 nor its proxies were available in other populations. Subsequent lookup of previously suggested7 TMAO candidate regions (the FMO cluster, 1q23.3, and 2p12) in genetic and epigenetic results from GOLDN failed to uncover any significant associations. There was no notable overlap in genomic position between top SNP and CpG site signals.
Table 2.
Top epigenome-wide associations for circulating TMAO in GOLDN (n=847).*
| Marker | Chromosome | Region | Gene | β ± SE | P-value |
|---|---|---|---|---|---|
| cg08040395 | 17 | – | ENGASE | 0.01±0.002 | 2.2×10−6 |
| cg19731194 | 1 | – | Intergenic | 0.01±0.002 | 8.8×10−6 |
| cg21066735 | 11 | Island | Intergenic | 0.002±0.0004 | 1.3×10−5 |
| cg00089486 | 3 | South Shore | SHOX2 | 0.003±0.0007 | 1.6×10−5 |
| cg01515960 | 19 | Island | Intergenic | 0.01±0.003 | 1.6×10−5 |
| cg25778892 | 7 | North Shore | Intergenic | 0.01±0.002 | 2.0×10−5 |
| cg27427369 | 2 | South Shelf | ERLEC1; LOC10030265 | −0.003±0.0006 | 2.8×10−5 |
| cg01767862 | 14 | – | SNORD114-28 | −0.003±0.0008 | 3.6×10−5 |
| cg00810908 | 3 | Island | FBLN2 | 0.002±0.0004 | 3.7×10−5 |
| cg08055924 | 4 | Island | MIR574; FAM114A1 | 0.003±0.0008 | 4.4×10−5 |
TMAO, trimethylamine-N-oxide
Table 3.
Top genome-wide associations for circulating TMAO in GOLDN (n=626).*
| SNP | Chromosome | Gene | Minor Allele Frequency | β ± SE | P-value |
|---|---|---|---|---|---|
| rs114755225 | 4 | Intergenic | 0.02 | −1.20±0.20 | 3.1×10−9 |
| rs148553452 | 1 | EYA3 | 0.01 | −1.70±0.31 | 4.4×10−8 |
| rs146552658 | 1 | EYA3 | 0.01 | −1.69±0.31 | 5.6×10−8 |
| rs114145653 | 1 | PHACTR4 | 0.01 | −1.89±0.34 | 7.0×10−8 |
| rs148992889 | 1 | EYA3 | 0.01 | −1.63±0.30 | 7.1×10−8 |
| rs75116832 | 17 | UBE2G1 | 0.01 | −1.82±0.34 | 8.6×10−8 |
| rs143831173 | 6 | Intergenic | 0.03 | −0.89±0.17 | 2.5×10−7 |
| rs114858855 | 6 | Intergenic | 0.03 | −0.89±0.17 | 2.5×10−7 |
| rs6557607 | 8 | RHOBTB2 | 0.06 | −0.66±0.13 | 2.6×10−7 |
| rs143482172 | 9 | MOB3B | 0.01 | −1.57±0.30 | 3.0×10−7 |
| rs58180025 | 6 | Intergenic | 0.04 | −0.87±0.17 | 3.4×10−7 |
| rs138865076 | 6 | Intergenic | 0.04 | −0.87±0.17 | 3.5×10−7 |
| rs146839869 | 6 | ENPP4 | 0.01 | −1.39±0.27 | 3.5×10−7 |
| rs75363923 | 6 | Intergenic | 0.01 | −1.56±0.30 | 3.8×10−7 |
TMAO, trimethylamine-N-oxide
Discussion
We conducted the first family-based, population-level investigation of genetic and epigenetic determinants of TMAO in humans. Despite evidence of significant heritability, we did not identify either DNA sequence variants or methylation markers that significantly contribute to circulating TMAO levels. Interestingly, we also did not replicate known associations between circulating TMAO and other cardiovascular risk factors, or consumption of animal products. The observed lack of association between diet and plasma TMAO contrasts other studies linking intake of meat, seafood, dairy, and eggs to elevated levels of atherogenic metabolites [2, 22–24]. As most studies used similar TMAO measurement protocols, reasons for this discrepancy may include differences in habitual dietary patterns, cohort composition, or diet ascertainment methods. Specifically, dietary variation in GOLDN participants was quite limited (e.g. there were few vegetarians), we did not have sufficient power to explore potential modifying effects of the habitual diet. Furthermore, because TMAO is synthesized by gut microbiota, it is also likely that the metagenomic composition of the participants was both influenced by habitual diet and impacted circulating TMAO levels, confounding the observed relationships.
Prior studies reported no associations between common genetic variation and plasma TMAO in humans [7]. The enrichment of top SNP signals for rare variants in the GOLDN data, however, may offer a clue to the genetic architecture of circulating TMAO. To date, the only validated genetic determinant of TMAO in humans is a cluster of rare missense mutations in the FMO3 gene, which has been linked to the ‘fish odor syndrome’ (trimethylaminuria) in several families [25]. It is possible that other rare variants also contribute to circulating TMAO, accounting for at least part of the observed heritability. We present preliminary evidence implicating one such variant, rs114755225 on chromosome 4, in TMAO homeostasis. The rs114755225 polymorphism is located in the intergenic region and has not been previously linked to physiological traits. While bioinformatic analyses suggest colocalization of rs114755225 with a H3K4me3 promoter peak in duodenum cells, the interpretation of this finding is challenged by the lack of nearby genes. Future rare variant studies are warranted to validate our preliminary finding and potentially identify novel rare mutations with functional relevance to the TMAO metabolic pathway. One potential region for follow-up investigation is the EYA3 gene, implicated in circadian functioning and represented among our top, albeit not statistically significant GWAS findings; a recent report found relationships between circadian rhythms and urinary TMAO concentrations [26].
We also hypothesized that genome-wide DNA methylation patterns, which reflect inputs from both sequence variation and environment (particularly diet), may be associated with plasma TMAO in humans. We did not find support for our hypothesis in the GOLDN cohort. There are several potential explanations for the observed null associations. First, the Illumina HumanMethylation450 array covers a limited portion of the genome, with a bias towards coding and promoter regions; future investigations using bisulfite sequencing or recently developed higher resolution methylation arrays may uncover novel signals of interest. Second, the overall associations may be obscured by confounding factors such as smoking or alcohol intake. However, sensitivity analyses (data not shown) adjusting for both lifestyle variables did not appreciably alter our results. Third, it is possible that the assumptions of methylation site heritability underlying our statistical power calculations were violated in our data, resulting in suboptimal ability to detect any effects. Fourth, the choice of tissue (blood, specifically CD4+ T-cells) may not be optimal for capturing the epigenetic correlates of TMAO metabolism. Originally, CD4+ T-cells were selected for quantifying epigenetic patterns in GOLDN due to their 1) role in the inflammatory processes and thus cardiometabolic disease, 2) availability (CD4+ T-cells are the most abundant lymphocyte in the blood), and 3) control of confounding by cell or tissue type compared to whole blood samples. Despite the relevance of CD4+ T-cells, the lack of liver tissue samples or other more proximal biological tissues is a clear limitation of our study, we have supplemented insights obtained from CD4+ T-cells with lookups of the same variants in other tissues using public databases. Finally, other factors—most importantly the gut microbiota composition—may represent stronger determinants of circulating TMAO than either DNA sequence or methylation variants. Future studies of inter-individual variability in diet-derived metabolites would benefit from incorporating metagenomic data in their approach, ideally integrating it with other–omics layers for a comprehensive picture of TMAO metabolism in humans.
Supplementary Material
Highlights.
GOLDN study data was used to investigate heritable determinants of plasma TMAO.
TMAO was not associated with other markers of CVD; heritability was 27%.
Genome-wide analysis identified one significant association on chromosome 4.
Epigenome-wide analysis found no significant associations.
Findings suggest neither genetics nor epigenetics influence TMAO levels in humans.
Acknowledgments
The authors thank Catherine M. Sreenan for her assistance in preparation of the manuscript.
Funding
This work was funded by the American Heart Association (14CRP18060003, PI: Aslibekyan) and the National Institutes of Health (R01HL104135, PI: Arnett). Funding agencies played no role in the study design; in data collection, analysis, and interpretation; in the writing of the manuscript; nor in the decision to submit the manuscript for publication.
List of Abbreviations
- CpG
5′—cytosine—phosphate—guanine—3′ DNA site
- CPMG
Carr-Purcell-Meiboom-Gill (pulse sequence)
- CVD
cardiovascular disease
- FID
free induction decay (signal)
- GOLDN
Genetics of Lipid Lowering Drugs and Diet Network (Study)
- MAF
minor allele frequency
- NMR
nuclear magnetic resonance
- SNP
single nucleotide polymorphism
- TMA
trimethylamine
- TMAO
trimethylamine-N-oxide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations
Ethics and consent to participate: All participants provided written informed consent. Institutional Review Boards at University of Minnesota, University of Utah, and Tufts University/New England Medical Center approved the study protocol.
Competing interests: The authors declare that they have no competing interests
Author contributions: Authors SA, PNH, MAP, JMO, DMA, and DKA made substantial contributions to the conception and design of the study. Authors EJJ, EG, IS, PNH, JMO, DMA, and DKA made substantial contributions to the acquisition of data. All authors made substantial contributions to the analysis and/or interpretation of data and the drafting and/or critical revision of the manuscript. All authors approved the final, submitted version of the manuscript.
References
- 1.Mayr M. Recent highlights of metabolomics in cardiovascular research. Circ Cardiovasc Genet. 2011;4(4):463–4. doi: 10.1161/CIRCGENETICS.111.961003. http://dx.doi.org/10.1161/circgenetics.111.961003. [DOI] [PubMed] [Google Scholar]
- 2.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–85. doi: 10.1038/nm.3145. http://dx.doi.org/10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368(17):1575–84. doi: 10.1056/NEJMoa1109400. http://dx.doi.org/10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang WH, Wang Z, Fan Y, Levison B, Hazen JE, Donahue LM, Wu Y, Hazen SL. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 2014;64(18):1908–14. doi: 10.1016/j.jacc.2014.02.617. http://dx.doi.org/10.1016/j.jacc.2014.02.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, Li XS, Levison BS, Hazen SL. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116(3):448–55. doi: 10.1161/CIRCRESAHA.116.305360. http://dx.doi.org/10.1161/circresaha.116.305360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL, Lusis AJ. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17(1):49–60. doi: 10.1016/j.cmet.2012.12.011. http://dx.doi.org/10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartiala J, Bennett BJ, Tang WH, Wang Z, Stewart AF, Roberts R, McPherson R, Lusis AJ, Hazen SL, Allayee H. Comparative genome-wide association studies in mice and humans for trimethylamine N-oxide, a proatherogenic metabolite of choline and L-carnitine. Arterioscler Thromb Vasc Biol. 2014;34(6):1307–13. doi: 10.1161/ATVBAHA.114.303252. http://dx.doi.org/10.1161/atvbaha.114.303252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loscalzo J. Lipid metabolism by gut microbes and atherosclerosis. Circ Res. 2011;109(2):127–9. doi: 10.1161/RES.0b013e3182290620. http://dx.doi.org/10.1161/RES.0b013e3182290620. [DOI] [PubMed] [Google Scholar]
- 9.Obeid R, Awwad HM, Rabagny Y, Graeber S, Herrmann W, Geisel J. Plasma trimethylamine N-oxide concentration is associated with choline, phospholipids, and methyl metabolism. Am J Clin Nutr. 2016;103(3):703–11. doi: 10.3945/ajcn.115.121269. http://dx.doi.org/10.3945/ajcn.115.121269. [DOI] [PubMed] [Google Scholar]
- 10.Walsh MC, Brennan L, Malthouse JP, Roche HM, Gibney MJ. Effect of acute dietary standardization on the urinary, plasma, and salivary metabolomic profiles of healthy humans. Am J Clin Nutr. 2006;84(3):531–9. doi: 10.1093/ajcn/84.3.531. [DOI] [PubMed] [Google Scholar]
- 11.Corella D, Arnett DK, Tsai MY, Kabagambe EK, Peacock JM, Hixson JE, Straka RJ, Province M, Lai CQ, Parnell LD, Borecki I, Ordovas JM. The −256T>C polymorphism in the apolipoprotein A-II gene promoter is associated with body mass index and food intake in the genetics of lipid lowering drugs and diet network study. Clin Chem. 2007;53(6):1144–52. doi: 10.1373/clinchem.2006.084863. http://dx.doi.org/10.1373/clinchem.2006.084863. [DOI] [PubMed] [Google Scholar]
- 12.Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, Chatham WW, Kimberly RP. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet. 2013;9(8):e1003678. doi: 10.1371/journal.pgen.1003678. http://dx.doi.org/10.1371/journal.pgen.1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aslibekyan S, Demerath EW, Mendelson M, Zhi D, Guan W, Liang L, Sha J, Pankow JS, Liu C, Irvin MR, Fornage M, Hidalgo B, Lin LA, Thibeault KS, Bressler J, Tsai MY, Grove ML, Hopkins PN, Boerwinkle E, Borecki IB, Ordovas JM, Levy D, Tiwari HK, Absher DM, Arnett DK. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring) 2015;23(7):1493–501. doi: 10.1002/oby.21111. http://dx.doi.org/10.1002/oby.21111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27. doi: 10.1093/biostatistics/kxj037. http://dx.doi.org/10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 15.Feinstein AR, Horwitz RI. Choosing cases and controls: the clinical epidemiology of “clinical investigation”. J Clin Invest. 1988;81(1):1–5. doi: 10.1172/JCI113279. http://dx.doi.org/10.1172/jci113279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, Lee C, Nizzari MM, Gabriel SB, Purcell S, Daly MJ, Altshuler D. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40(10):1253–60. doi: 10.1038/ng.237. http://dx.doi.org/10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–9. doi: 10.1038/ng.2354. http://dx.doi.org/10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–34. doi: 10.1002/gepi.20533. http://dx.doi.org/10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62(5):1198–211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hidalgo B, Irvin MR, Sha J, Zhi D, Aslibekyan S, Absher D, Tiwari HK, Kabagambe EK, Ordovas JM, Arnett DK. Epigenome-Wide Association Study of Fasting Measures of Glucose, Insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network Study. Diabetes. 2014;63(2):801–7. doi: 10.2337/db13-1100. http://dx.doi.org/10.2337/db13-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aslibekyan S, Frazier-Wood AC, Absher DM, Sha J, Zhi D, Kabagambe EK, Straka RJ, Tiwari HK, Tsai MY, Hopkins PN, Borecki IB, Ordovas JM, Arnett DK. DNA Methylation Patterns are Associated with Genetic Variation and Systemic Inflammation, American Heart Association’s Epidemiology and Prevention/Physical Activity, Nutrition and Metabolism 2013 Scientific Sessions. American Heart Association; New Orleans, LA: 2013. [Google Scholar]
- 22.Hazen SL, Brown JM. Eggs as a dietary source for gut microbial production of trimethylamine-N-oxide. Am J Clin Nutr. 2014;100(3):741–3. doi: 10.3945/ajcn.114.094458. http://dx.doi.org/10.3945/ajcn.114.094458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohrmann S, Linseisen J, Allenspach M, von Eckardstein A, Muller D. Plasma Concentrations of Trimethylamine-N-oxide Are Directly Associated with Dairy Food Consumption and Low-Grade Inflammation in a German Adult Population. J Nutr. 2016;146(2):283–9. doi: 10.3945/jn.115.220103. http://dx.doi.org/10.3945/jn.115.220103. [DOI] [PubMed] [Google Scholar]
- 24.Zhang AQ, Mitchell SC, Smith RL. Dietary precursors of trimethylamine in man: a pilot study, Food and chemical toxicology. an international journal published for the British Industrial Biological Research Association. 1999;37(5):515–20. doi: 10.1016/s0278-6915(99)00028-9. [DOI] [PubMed] [Google Scholar]
- 25.Teresa E, Lonardo F, Fiumara A, Lombardi C, Russo P, Zuppi C, Scarano G, Musumeci S, Gianfrancesco F. A spectrum of molecular variation in a cohort of Italian families with trimethylaminuria: identification of three novel mutations of the FM03 gene. Mol Genet Metab. 2006;88(2):192–5. doi: 10.1016/j.ymgme.2006.02.014. http://dx.doi.org/10.1016/j.ymgme.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Giskeodegard GF, Davies SK, Revell VL, Keun H, Skene DJ. Diurnal rhythms in the human urine metabolome during sleep and total sleep deprivation. Sci Rep. 2015;5:14843. doi: 10.1038/srep14843. http://dx.doi.org/10.1038/srep14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.