

Abstract
Plastic gene expression underlies phenotypic plasticity and plastically expressed genes evolve under different selection regimes compared with ubiquitously expressed genes. Social insects are well-suited models to elucidate the evolutionary dynamics of plastic genes for their genetically and environmentally induced discrete polymorphisms. Here, we study the evolution of plastically expressed genes in the ant Cardiocondyla obscurior—a species that produces two discrete male morphs in addition to the typical female polymorphism of workers and queens. Based on individual-level gene expression data from 28 early third instar larvae, we test whether the same evolutionary dynamics that pertain to plastically expressed genes in adults also pertain to genes with plastic expression during development. In order to quantify plasticity of gene expression over multiple contrasts, we develop a novel geometric measure. For genes expressed during development, we show that plasticity of expression is positively correlated with evolutionary rates. We furthermore find a strong correlation between expression plasticity and expression variation within morphs, suggesting a close link between active and passive plasticity of gene expression. Our results support the notion of relaxed selection and neutral processes as important drivers in the evolution of adaptive plasticity.
Keywords: ants, gene evolution, expression bias, developmental plasticity, relaxed selection, expression noise.
Introduction
Organisms are able to adaptively adjust gene expression in response to environmental conditions, resulting in changes to the phenotype (Schlichting and Pigliucci 1998; West-Eberhard 2003). This phenotypic plasticity plays a fundamental role in adaptive evolution (West-Eberhard 2005a,b; Schlichting and Wund 2014). Paradoxically, whereas decreasing the necessity for mutation-based genetic adaptation through phenotypic accommodation (West-Eberhard 1998, 2003), phenotypic plasticity (including sexual dimorphism) appears to play a role in accelerating sequence evolution. Genes that are expressed at different levels in different morphs or sexes are referred to as biased or “plastic” genes and tend to evolve faster than uniformly expressed genes (Helanterä and Uller 2014). This correlation also remains significant if potentially confounding factors, such as expression level, expression breadth or DNA methylation patterns, are controlled for (Grath and Parsch 2012; Warnefors and Kaessmann 2013). The positive correlation of gene expression bias and molecular evolutionary rates in the context of phenotypic plasticity has been demonstrated in different study systems. Sex-biased genes in mammals and fruit flies evolve faster than unbiased genes (Ellegren and Parsch 2007; Parsch and Ellegren 2013). Likewise, studies in fire ants (Hunt et al. 2011, 2013), honey bees (Hunt et al. 2010; Harpur et al. 2014), polymorphic aphids (Purandare et al. 2014), and horn beetles (Snell-Rood et al. 2011) have revealed a positive correlation between morph bias and evolutionary rates, and similar patterns have been observed in phenotypically plastic Spea toads (Leichty et al. 2012).
Disentangling the causes and consequences of faster evolution of plastic genes is challenging, due to possible simultaneous contributions from neutral and adaptive processes, and both purifying and positive selection. Accordingly, several mutually nonexclusive processes could explain the observations (Helanterä and Uller 2014). First, it is possible that morph-biased expression patterns in many cases reflect a history of weak purifying selection rather than positive selection on expression regulation. In accordance, morph-biased genes in the fire ant Solenopsis invicta evolved faster due to relaxed selection before the morphs had even evolved (Hunt et al. 2011). Similarly, biased genes in the toad Spea bombifrons were found to have evolved faster in the nonplastic ancestral lineages and other nonplastic extant Spea species (Leichty et al. 2012). The rationale is that genes under relaxed selection in terms of sequence should also evolve under relaxed selection in terms of expression regulation. Thus, they should more likely drift towards biased expression patterns and become co-opted to a plastic trait. Second, plastic expression itself is expected to result in a relaxation of selection. A plastic gene is more likely to be expressed only by a subset of individuals in a population at a given time than a uniformly expressed gene (Snell-Rood et al. 2010; Van Dyken and Wade 2010). Third, where expressed, biased genes can also experience increased positive selection, simply because advantageous mutations will be selected for (Harpur et al. 2014). Finally, a gene can exert different fitness effects in different morphs, potentially resulting in antagonistic directional selection, thus hampering the evolution of the gene (Innocenti and Morrow 2010). For example, an allele increasing wing muscle development in ants may be beneficial for queens but not workers (Hall et al. 2013). By evolving plastic expression patterns, pleiotropic conflicts can be alleviated, allowing directional selection to accelerate sequence evolution (Mank et al. 2008). In general, each of these four factors is expected to contribute to the evolution of plastic genes to some extent, as selection pressures on plastic genes change over evolutionary time and with context.
Another difficulty in unraveling the evolutionary history and trajectory of plastic genes is the lack of a formal model for neutrality of gene expression evolution. Genetic drift is a mathematically well-understood process, and synonymous sites offer a reasonable benchmark for neutrality in sequence evolution of protein-coding genes. Defining a comparable model for the evolution of expression regulation is however difficult, particularly in the context of polymorphic species with intraspecific adaptive expression differences. A possible approach towards a neutral evolutionary model of gene expression is to assess the level of expression variation across biological replicates. This is based on the assumption that a large proportion of the observed variation in gene expression between individuals is a result of neutral processes, when environmental and genetic factors have been controlled for (Nuzhdin 2004; Gilad et al. 2006; Whitehead and Crawford 2006; Romero et al. 2012; Rohlfs et al. 2014). In this context, individual-level gene expression data is crucial to approximate neutral rates of expression variation. Such data is particularly scarce in polymorphic animals due to the common practice of pooling samples, which has hampered empirical studies of evolutionary consequences of phenotypic plasticity and morph-biased expression.
In eusocial insects, the foundation of phenotypic differentiation into discrete castes lies in different developmental trajectories (Wheeler 1986), making them a prime example for studying the effects of developmental expression bias on the molecular evolution of genes. In the ant Cardiocondyla obscurior, reproductive queens (QU) and sterile workers (WO) occur next to winged (WM) and ergatoid (i.e., from Greek “worker-like” and wingless) males (EM), providing a complex tetraphenic system of four distinct morphs (Schrader et al. 2015). The female castes (QU and WO) develop from fertilized diploid eggs. QU are long-lived (average life span ∼24 weeks (Oettler and Schrempf 2016)) and fully devoted to the production of WO and sexual offspring. In contrast, WO have a much lower life expectancy (∼8 weeks) during which they perform a wide range of tasks related to colony maintenance (e.g., foraging, nest construction, defense, and brood care). In Cardiocondyla, two discrete male morphs with opposing life histories have evolved (Oettler et al. 2010). The WM resemble males of most other ant species, with fully developed wings and large eyes for dispersal and a rather short life span (∼2 weeks). The smaller EM have small eyes, no ocelli, and in contrast to WM exhibit lifelong spermatogenesis (Heinze and Hölldobler 1993). They furthermore have highly specialized mandibles that are deployed in mortal conflicts with other EM over the reproductive monopoly in a colony.
Both differences (Schrader et al. 2015) and similarities (Klein et al. 2016) between morphs during the plastic ontogenesis of C. obscurior have been studied on a transcriptomic level. Here, we address the relationship between plastic gene expression during larval development and molecular evolution in C. obscurior, using data from individually sequenced third instar larvae of known developmental trajectory (Schrader et al. 2015). Most studies on the correlation of gene expression bias and sequence evolution in polymorphic insects have been conducted on adults and only a single study has so far focused on larvae (Vojvodic et al. 2015). Extending such studies to the developmental life stages is important because larval expression bias can have a different effect on evolutionary rates of genes than adult expression bias. For example, in Drosophila, genes with female-biased expression in larvae evolve more rapidly than genes that are female-biased in adults (Perry et al. 2014). Focusing on the early developmental stage offers an additional, substantial advantage for our transcriptomic study. Using gene expression data from individuals sampled at the onset of developmental divergence (Schrempf and Heinze 2006), reduces confounding effects of morphological differences during later life stages which can introduce biases in whole-body transcriptomic comparisons (Harrison et al. 2015).
Based on sequence divergence between two populations of C. obscurior and sequence divergence between C. obscurior and the closest related sequenced ant species (Monomorium pharaonis), we determine evolutionary rates of protein-coding genes in C. obscurior and show that these are positively correlated with gene expression plasticity during ontogenesis. We furthermore reveal a strong correlation of expression plasticity between morphs and expression variation within morphs, suggesting a strong link between active and passive plasticity of gene expression. We discuss our findings and argue in favor of the importance of relaxed selection in the evolution of adaptive plasticity from ancestrally merely passively plastic traits.
New Approaches
Differences in gene expression between two groups are usually quantified as logarithmic ratios of expression level (“logFC”), providing a pairwise measure of expression bias. For comparing expression variability across more than two groups, other methods have to be applied that quantify and summarize gene expression differences across multiple samples (Kryuchkova-Mostacci and Robinson-Rechavi 2016). These methods are most commonly used to specifically identify genes that are overexpressed in a single tissue and they could similarly be used for assessing morph-specific expression bias in polymorphic species. Abandoning this focus on specificity, we aimed to apply a method to quantify the general plasticity and versatility of gene expression across multiple contrasts. We use a geometric approach that describes a gene’s expression pattern across n contrasts as a vector in Euclidean n-space. For summarizing the versatility and plasticity of expression in a single measure for each gene, we then calculate the Euclidean length of each gene’s vector (Formula 1) resulting in the plasticity index Pi (π), a single combined measure of gene expression plasticity (supplementary fig. S1, Supplementary Material online). We here use π to quantify gene expression plasticity across larvae of the four different morphs of C. obscurior. For our experimental design with the four groups QU, WO, EM, and WM and thus six possible pairwise contrasts, we constructed the following six dimensions in Euclidean space: logFC QU/WO, logFC QU/EM, logFC QU/WM, logFC EM/WM, logFC EM/WO, and logFC WO/WM.
| (1) |
Using the equation for the Euclidean distance across all six contrasts, we provide a simple and coherent way for calculating π. Including all and not only a subset of pairwise comparisons in the calculation of π results in a balanced design and an unbiased measure, even though the different contrasts are not independent. However, because the six different logFC measures are interrelated (supplementary equations 1–3, Supplementary Material online), π can in principle also be calculated using only three of the six logFC measures (supplementary equations 4–13, Supplementary Material online).
Results
The “specificity index” Tau (τS) (Yanai et al. 2005) is the most commonly used method to quantify expression specificity and it outperforms most other measures of expression specificity (Kryuchkova-Mostacci and Robinson-Rechavi 2016). τS and π are moderately correlated (Kendall’s rank correlation, P < 2e−16, τKendall = 0.765, fig. 1), indicating that the two measures overlap in their performance to quantify expression variability. However, we manually inspected expression patterns of four genes most strongly deviating from the correlation, indicating that their expression pattern is considered highly variable based on π and less so based on τS. These genes show patterns of morph-specific underexpression (Cobs_02055, Cobs_12086, and Cobs_12224) and sex-specific overexpression (Cobs_17732) (fig. 2), demonstrating that π captures more complex patterns of expression than τS. The low expression levels of the first three examples preclude assumptions about the biological significance of the observed differences in expression variation but adequately illustrate differences between π and τS as measures of expression variation. We have screened more strongly expressed genes revealing similar yet less pronounced differences between π and τS (e.g., Cobs_04700, Cobs_06687, Cobs_09630, Cobs_12556, Cobs_17048, Cobs_18086, Supplementary Table.tsv). τS is explicitly designed to detect overexpression in a single group (i.e., expression specificity) and it is thus not surprising that it performs less well in detecting underexpression and that it is less sensitive to more versatile patterns of expression.
Fig. 1.
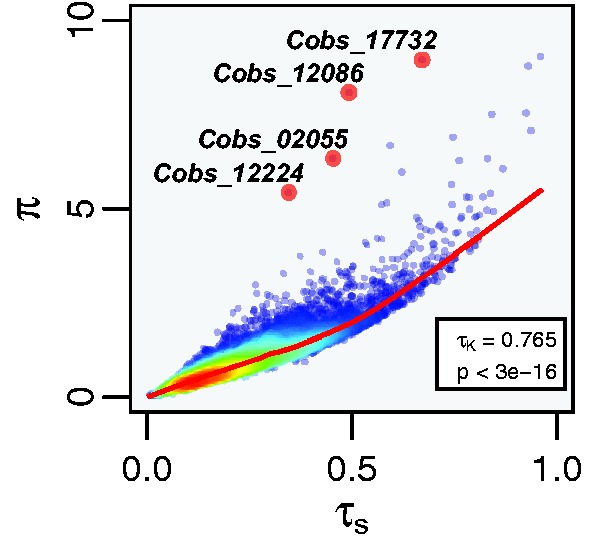
Correlation of the specificity index τS and the plasticity index π. The specificity index τS and the plasticity index π are moderately correlated for the analyzed dataset. However, several genes have a relatively higher π than τS, indicating differences in the performance of the two measures. The top four genes showing the strongest deviation from the correlation are labeled (larger red dots, see fig. 2). τK = Kendall’s correlation coefficient, p = P-value of Kendall’s correlation between τS and π.
Fig. 2.

Comparison of the performance of τS and π to dissect complex gene expression patterns over multiple contrasts. Gene expression (in log2 normalized read counts) in each morph and corresponding specificity index τS and plasticity index π values for four genes expressed in C. obscurior. Genes were selected based on their strong deviation from the mean ratio of τS and π to assess performance differences of the two measures (independent of their average expression level). Three of the selected genes show morph-specific downregulation (Cobs_02055, Cobs_12086, Cobs_12224) and Cobs_17732 shows female-biased overexpression. Boxplots show the median, inter-quartile ranges (IQR) and 1.5 × IQR and red dots show mean expression across seven replicates. QU = queens, WO = workers, EM = ergatoid males, WM = winged males.
We furthermore compared the Euclidean distance-based measure with a similar measure based on the mean of the absolute logFC values. They are highly correlated (Kendall’s τ = 0.95, P < 2e−16), suggesting that differences are only small. Conceptually, both approaches answer how much a gene’s expression deviates from uniformity, i.e., how strongly the different logFC values deviate from 0. The absolute mean is calculated by summing the absolute values of each logFC. When using the Euclidean distance, we sum the squares of each logFC (Formula 1). Due to the square included in the calculation of Euclidean distances, high logFC are given more weight, i.e., resulting in relatively higher π for genes with higher expression in a single morph. Therefore, π is better suited for our purpose to quantify expression plasticity.
The gene expression data used for this study was generated from 28 individually sequenced larvae of C. obscurior (Schrader et al. 2015). Seven larvae per morph were selected for RNA sequencing at an approximate age of ten to eleven days; i.e., at the beginning of the third and last instar, when morph determination is fixed (Schrempf and Heinze 2006). The use of independent individual biological replicates substantially increases statistical power and allowed us to assess individual-level variation in gene expression as a proxy for neutral rates of gene expression variation. To quantify expression variation within morphs, we calculated the coefficient of variance (CV) for each gene. CV is defined as the ratio of the standard deviation to the mean of normalized read counts across the seven biological replicates per morph. We also calculated the mean CV across all four morphs for each gene.
Among the 17,552 genes annotated in the C. obscurior genome (Schrader et al. 2014), 10,012 were expressed in larvae. The overall distributions of gene expression levels were not significantly different between the four morphs (supplementary fig. S2a, b, Supplementary Material online). Significant differences between morphs occurred in the variation of expression, where genes in EM (median CV = 0.12) and QU (median CV = 0.13) showed higher variation in expression compared with genes in WO and WM (median CV = 0.09 each) (supplementary fig. S2c, d, Supplementary Material online). Whereas some genes showed several fold differences in expression in pairwise comparisons, most genes differed only mildly (supplementary fig. S3, Supplementary Material online). Accordingly, the distribution of expression plasticity as measured by π is skewed towards 0, with only a few genes showing very strong overall plasticity (median π = 0.83, fig. 3A).
Fig. 3.
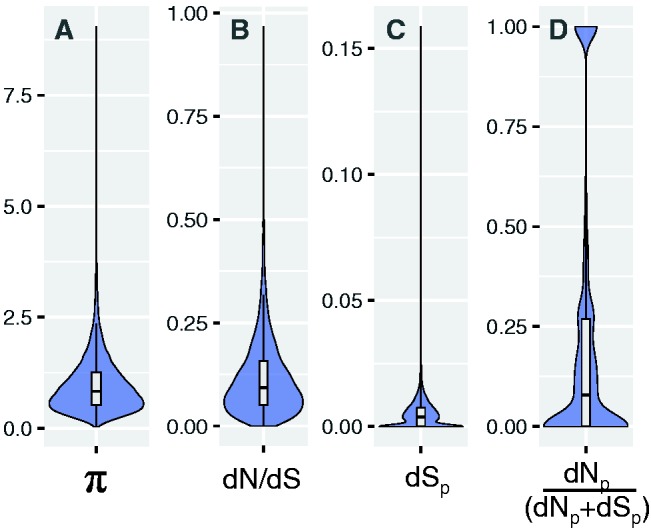
Distribution of expression plasticity (π), species divergence (dN/dS), rates of synonymous substitutions (dSp), and population divergence (dNp/(dNp + dSp)). Violin plots showing the distribution of different quantitative measures of gene expression and sequence evolution for all 10,012 genes expressed in larvae of Cardiocondyla obscurior. (A) Distribution of π as a measure of gene expression plasticity. (B) Distribution of dN/dS ratios for 1-to-1 orthologs between Cardiocondyla obscurior and Monomorium pharaonis. (C) Distribution of the rate of synonymous substitutions (dSp) separating two populations of C. obscurior from Brazil (BR) and Japan (JP). (D) Distribution of the ratio of nonsynonymous substitutions per nonsynonymous site (dNp) and the sum of dNp and nonsynonymous substitutions per nonsynonymous site (dSp) separating the BR and JP populations [dNp/(dNp+dSp)]. Boxplots show the median, inter-quartile ranges (IQR) and 1.5 × IQR.
To quantify levels of molecular evolution of protein-coding genes, we used two different measures. First, we estimated inter-specific dN/dS rates between C. obscurior and M. pharaonis, based on alignments of single-copy orthologs of protein-coding genes. The distribution of dN/dS rates (fig. 3B) suggests that the majority of genes evolved under purifying selection (dN/dS ≪ 1) and that no gene evolved strictly under positive selection (dN/dS > 1). Second, single nucleotide variant (SNV) annotations from a genomic comparison between a Brazilian (BR) and a Japanese (JP) population were used to calculate intra-specific evolutionary rates (Schrader et al. 2014, data available at hymenopteragenome.org/cardiocondyla/). The two populations show significant phenotypic divergence (Schrader et al. 2014) and the gene expression data used in this study was generated from one laboratory colony of the BR strain that was kept under strict inbreeding for several generations. We generated estimates of evolutionary rates between populations as dNp/(dNp + dSp), a measure that allowed us to include genes that are not separated by synonymous substitutions (Stoletzki and Eyre-Walker 2010). The comparison revealed that 33% of the expressed genes contain no synonymous variants (ndSp = 0 = 3,303, fig. 3C). In 6,459 of the 7,452 genes for which rates of population divergence could be calculated, rates of dNp were lower than dSp, (dNp/(dNp + dSp) < 0.5), indicating that purifying selection acts on the majority of protein coding genes between the populations (fig. 3D).
Based on this data set, we tested for correlations between plasticity of gene expression (π) and rates of molecular evolution. Overall, π and average gene expression levels (ExprAve) are negatively correlated (τKendall = −0.116, P < 1e−16, fig. 4A, supplementary table S1, Supplementary Material online); i.e., highly plastic genes tend to be expressed at a lower level. Note however, that the LOWESS function applied to the data suggests that the correlation is non-monotonic and that the correlation is in fact weak positive for genes with high expression plasticity. To control for the known effect of expression level on sequence evolutionary rates (Pál et al. 2006), we conducted all subsequent correlation analyses using semi-partial correlations, with ExprAve as the excluded factor. The semi-partial correlation for neutral evolutionary rates (dSp) with π was positive (τKendall = 0.022, P < 0.0012, fig. 4B, supplementary table S1, Supplementary Material online), suggesting that plastic genes accumulate synonymous substitutions faster than unbiased genes. The semi-partial correlation between expression plasticity (π) and dNp/(dNp + dSp) was significant with P < 5.54e−11 (τKendall = 0.051). Similarly, dN/dS rates between M. pharaonis and C. obscurior (Figure 4c, d, supplementary table S1, Supplementary Material online) were positive correlated with π (τKendall = 0.057, P < 2.51e−11). To confirm that the significant correlations of π and evolutionary rates are independent of gene length, we divided genes in five groups based on size and repeated the analyses for each bin separately. These analyses yielded similar results, indicative of only a minor effect of gene length (supplementary fig. S4, Supplementary Material online).
Fig. 4.
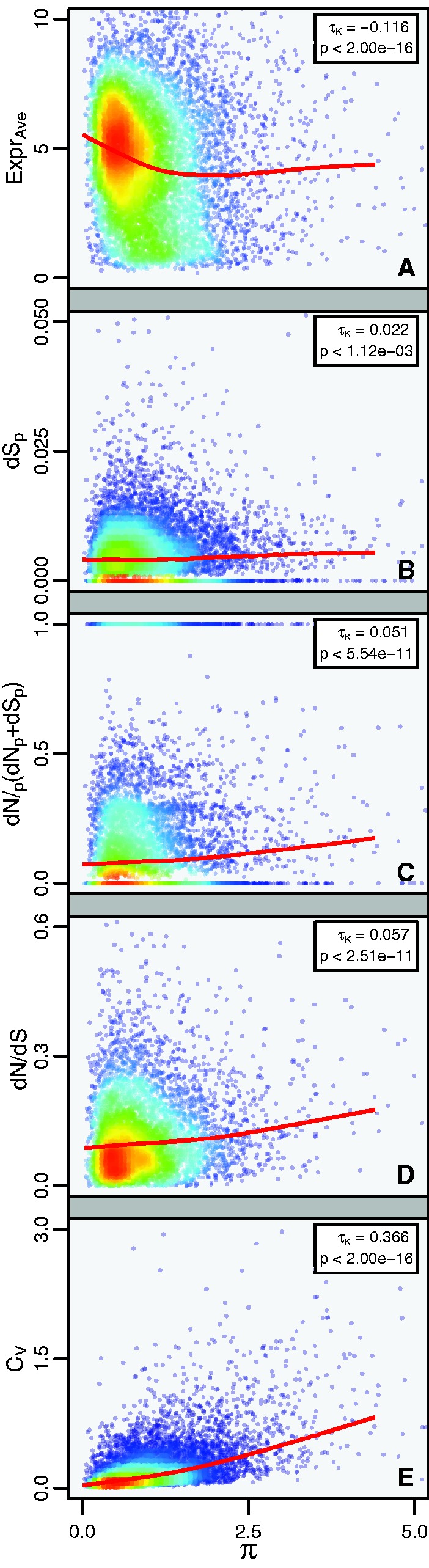
Correlation of gene expression plasticity during development with average gene expression, gene expression variability, and gene sequence evolution in Cardiocondyla obscurior. (A) Negative correlation between plasticity index π and expression level (ExprAve). (B) Positive (semi-partial) correlation between π and intraspecific rates of nearly neutral synonymous substitutions (dSp). (C) Positive (semi-partial) correlation between π and dNp/(dNp+dSp) as a measure of divergence between a Japanese (JP) and Brazilian (BR) population of C. obscurior. (D) Positive (semi-partial) correlation between π and gene-wise dN/dS rates for C. obscurior and M. pharaonis. (E) Positive (semi-partial) correlation between π and average expression variation (CV). Each dot represents one gene. The red–blue gradient shows the density of data points at the respective position in the plot. A LOWESS-smoothed line illustrates a generalized trend of the relationship (red line, smoother span = 0.9). Correlation coefficients (τK) and P-values for Kendall’s correlation between π with ExprAve (A) and for Kendall’s semi-partial correlations between π with dSp, dNp/(dNp+dSp), dN/dS, and CV, respectively (B–E), are given in the top right corner. Additional statistics are available in supplementary table S1, Supplementary Material online.
Overall, the strongest positive correlation in our analysis was found between gene expression plasticity (π) and gene expression variation (CV) (fig. 4E, supplementary table S1, Supplementary Material online, τKendall = 0.366, P < 2e−16), showing that the strongest expression differences between morphs occur predominantly in genes that also show high expression variation between individuals of the same morph. At low expression levels, technical noise can be a significant component of CV (Law et al. 2014). To rule out that the correlation of π and CV is predominately a result of technical noise rather than biological variation, we tested whether the relationship persists if we restrict the analysis to genes with high expression. For genes with ExprAve > 10, π and CV are positively correlated at τKendall = 0.463 (supplementary fig. S5, Supplementary Material online, P < 3.1e−12).
To verify our findings, we performed permutation tests. To this end, we compared genes with high and low π and found stronger differences in evolutionary rates between those genes than between randomly drawn genes (N = 500, 105 permutations, dN/dS: P < 0.003, dNp/(dNp + dSp): P < 0.018). Similarly, permutation tests also confirmed the correlation between gene expression levels and π (P < 1e−5) and the correlation between CV and π (P < 1e−5). The permutation test for dSp was not significant (P > 0.05). Note however that these permutation tests are sensitive to the number of genes N sampled in each permutation (supplementary fig. S6, Supplementary Material online), most likely due to large variances of the data.
Following a previously established approach to disentangle effects of expression bias and expression level on sequence evolutionary rates (Hunt et al. 2011), we performed a principal component analysis on average expression levels (ExprAve) and the six different gene expression contrasts. PC1, which explains 43.3% of the variance in the data, is dominated by the six different gene expression contrasts that contribute to similar extent (fig. 5A, supplementary fig. S7a, Supplementary Material online). ExprAve loads heavily on PC2. We tested for significant (Kendall’s rank) correlations between evolutionary rates and principal components. dSp was significantly correlated with PC2 but not PC1 (fig. 5B; supplementary fig. S7b and supplementary tables S2–S3, Supplementary Material online). dNp/(dNp + dSp), dN/dS, and CV were each significantly correlated with both PC1 and PC2 (fig. 5C–E; supplementary fig. S7b–e and supplementary tables S2–S3, Supplementary Material online). Correlations with PC1 were negative and correlations with PC2 were positive. These results, together with the weak contribution of ExprAve to PC1 thus confirm an independent effect of expression bias on evolutionary rates.
Fig. 5.

Principal component analysis of gene expression contrasts and average expression level in Cardiocondyla obscurior. (A) Loadings of gene expression contrasts (blue dots) and average expression level (ExprAve, red dot) on PC 1 and PC 2. (B–E) Kendall’s rank correlation of the first two PCs with dSp, dNp/(dNp+dSp), dN/dS, and CV. Stacking of the barplots shows the relative contributions of the different gene expression contrasts and ExprAve to the two different PCs. Asterisks indicate significant correlations with P < 1e−5, ns = not significant (P > 0.05). Additional information is provided in the Supplementary Material online. QU = queens, WO = workers, EM = ergatoid males, WM = winged males.
Discussion
This study on larval gene expression in the ant C. obscurior reveals correlations between developmental expression patterns and gene evolution. Our results corroborate that relaxed selection and neutral processes are important contributors to the evolution of adaptive plasticity. In accordance with previous studies on polymorphic species, we find a positive correlation between evolutionary rates and expression plasticity. Albeit providing only a single snapshot into development, here we show that this correlation extends to gene expression plasticity early after the critical stage of morph determination.
As any organismal trait, phenotypic plasticity is subject to selection and evolutionary change. It is important to assess how plasticity of a trait has been shaped by selection and whether the plasticity itself is adaptive (Borenstein et al. 2006; Fitzpatrick 2012). For example, the queen-worker polymorphism found in ants is highly adaptive, as it underlies the reproductive division of labor that has been key to the ants’ evolutionary success (Smith and Szathmary 1995). Such adaptive forms of phenotypic plasticity are referred to as active plasticity. They involve anticipatory changes in physiological and highly integrated developmental networks (Forsman 2014). Caste determination in social insects for example involves many conserved developmental and physiological pathways (Corona et al. 2016; Klein et al. 2016). In contrast, passive plasticity as an unspecific physical response to environmental conditions can often be considered neutral and nonadaptive (Forsman 2014). Given that both active and passive plasticity are driven by changes in gene expression, these changes in expression will accordingly be shaped by different evolutionary constraints: whereas gene expression variation underlying passive plasticity should evolve under relaxed selection, variation associated with active plasticity should involve positive or purifying selection.
The strong correlation between expression variation (CV) and π suggests that expression variation between and the variation within morphs largely follow the same dynamics, suggesting a close relationship of active and passive plasticity with regard to gene expression. Under the assumption that within-morph variation is largely neutral, the acceleration of sequence evolution will be the result of relaxed selection. Only relatively few genes show a strong variation between morphs and low variation within morphs (fig. 4E). Genes showing this pattern might be particularly interesting, because this expression pattern suggests involvement in active plasticity. Thus, these genes should be the ones where adaptive processes underlie the changes in evolutionary rates (Helanterä and Uller 2014). Intriguingly, one such gene is doublesex (CV = 0.39, π = 4.54), which has been co-opted for male and female morph differentiation in C. obscurior (Klein et al. 2016). Similarly, only few genes show the inverse pattern, with high variation within morphs but low variation between. This is in accordance with the assumption that similar sets of genes are involved in generating actively and passively plastic phenotypes and also that genes showing fluctuating expression are more likely to be co-opted to the expression of an actively plastic trait (Helanterä and Uller 2014).
CV is composed of technical and biological variation, which could compromise this argument. To detect and control for technical variation in gene expression data, synthetic transcript-mimicking RNA molecules at known concentrations (spike-ins) can be added to the RNA samples (e.g., ERCCs (Jiang et al. 2011) or the recently developed sequins (Hardwick et al. 2016)). However, in datasets lacking spike-ins levels of biological variation can be reliably quantified at high expression levels, where the contribution of technical variation to CV is small (Law et al. 2014). Replicating the analysis using highly expressed genes confirmed the significant correlation between π and CV (supplementary fig. S5, Supplementary Material online).
The analysis of expression variation (CV) also revealed significantly higher variation in the QU and EM morphs. Compared with the sterile WO and the rarely produced WM there is higher potential for evolutionary change in the reproducing morphs, potentially driven by a combination of passive plasticity and alternative strategies within sexes. This increased expression variation in QU and EM larvae might also be linked to passive plasticity of adult morphs. However, it remains to be tested whether this higher CV also translates into higher variation of larval or adult traits in the different morphs of C. obscurior.
The correlation between expression plasticity π and population divergence demonstrates that plastically expressed genes continue to evolve faster even after their recruitment to the expression of a plastic trait. Assuming similar selection pressures on larvae in the two populations, the continuing divergence of genes can be attributed largely to relaxed selection. In general however, whereas relaxed selection appears to be ancestral to the evolution of plastic expression and phenotypic plasticity (Hunt et al. 2011; Leichty et al. 2012), subsequent changes in the selection regime can accelerate the evolution of plastic genes once plasticity has been established, and caste-antagonistic pleiotropy alleviated.
Positive selection can explain the observed divergence between populations to some extent, if differences between habitats in Brazil and Japan invoked genetic adaptations. However, whereas the data presented in this study provides evidence for evolutionary divergence of the populations from Brazil and Japan, it does not allow for specifically dissecting contributions of relaxed and positive selection and genetic drift. To do so, future analyses of inter-population divergence should include several colonies per population for assessing standing levels of polymorphism and genetic diversity, which would allow for intraspecific tests of selection (Vitti et al. 2013).
We furthermore found that expression plasticity and rates of synonymous mutations are weakly correlated. It is likely that this correlation reflects the fact that synonymous changes are not fully neutral but can also be targeted by positive or purifying selection (Chamary et al. 2006). This effect is likely to be even stronger in eusocial insects for their low effective population size (Ne) (Chamary et al. 2006; Romiguier et al. 2014). Alternatively, assuming nearly neutral evolution of synonymous sites, the observed pattern could indicate that biased expression patterns are more likely to evolve at genomic locations with increased mutational activity. Intriguingly, such mutational rate differences in the genome are remarkably prominent in C. obscurior (Schrader et al. 2014).
In general, small Ne might also lead to stronger neutral (or nearly neutral) variation in gene expression (i.e., passive plasticity) in social insect than in nonsocial insects. Such neutral variation in expression could then be targeted by positive selection under changing environments, leading to phenotypic innovation and adaptation (Ghalambor et al. 2007). It is tempting to speculate that higher regulatory drift due to small Ne is an important driver in the remarkable phenotypic diversification within and across eusocial insect species.
Complex phenotypes such as the elaborate polymorphisms found in extant eusocial insects are unlikely to evolve de novo, independent from ancestral traits. It is thus likely that, prior to the evolution of reproductive division of labor in eusocial insects, nonadaptive passively plastic traits in the solitary ancestors preceded the evolution of the actively plastic caste system (Nijhout 2003; Linksvayer and Wade 2005; Amdam et al. 2006). Consequently, it seems feasible that the decisive step towards the transition from passive to active plasticity lies in establishing morph-specific expression profiles from genes that are under relaxed selection and already show an increased level of expression variation (Ghalambor et al. 2007; Hunt et al. 2011; Ruden et al. 2015). The correlation of within and between morph variation provides further support for this hypothesis, by showing that genes involved in passive plasticity are likely to produce active plasticity. In accordance, environmentally and in particular stress-induced variation in gene expression (i.e. passive plasticity) is considered to be positively correlated with the evolvability of discrete expression profiles of genes (López-Maury et al. 2008).
Several theoretical and empirical studies suggest that passive plasticity producing a phenotype close enough to a new fitness optimum can form the basis for the evolution of adaptive and eventually active plasticity (Denver 1997; Nijhout 2003; Ghalambor et al. 2007; Gomez-Mestre et al. 2008; Leichty et al. 2012; Ghalambor et al. 2015). This process involves genetic accommodation of plastic traits, release from pleiotropic constraint, and an increase in directional positive selection of genes co-opted for morph-specific function (Levis and Pfennig 2016). Accordingly, release from pleiotropic constraint and increased positive selection most likely contribute to the observed acceleration of sequence evolution in our study as well. A study on adaptive evolution in honey bees revealed that genes with worker-biased brain expression evolve under strong positive selection (Harpur et al. 2014), challenging the conception of relaxed selection as the main driver of molecular evolution of biased genes (Hunt et al. 2011). Similarly, studies in Drosophila showed signatures of strong positive selection in sex-biased genes (Ellegren and Parsch 2007). The results from these studies show that relaxed selection is not the only driver in the molecular evolution of plastic genes and that positive selection can be particularly strong during the evolution of novel traits involving biased genes (Jasper et al. 2015). It is thus important to recognize that genes evolving under relaxed selection might become both subject to directional and purifying selection, following their recruitment to the expression of plastic traits, either through selective co-option or through regulatory drift (Helanterä and Uller 2014). However, the general route to active phenotypic plasticity most likely lies in the co-option of genes under relaxed selection that are ancestrally involved in the expression of passively plastic traits. Whereas an increase in relaxed selection appears to be a precursor to the evolution of actively plastic gene expression patterns, positive selection likely only follows once such plastic patterns have evolved. By combining population data of monomorphic and polymorphic species with phylogeny-based ancestral reconstructions of expression patterns, it should be possible to more specifically tease apart neutral and adaptive contributions to the evolution of plastic genes in future studies.
Methods
Sampling and Gene Expression Analysis
Samples for RNA sequencing were collected from a strain of C. obscurior from Brazil (BR) (Heinze et al. 2006) that has been kept under strict inbreeding over several generations in the lab. Detailed protocols for sample collection, sample preparation and sequencing as well as the sequencing data have been published previously (Schrader et al. 2015). In brief, four sets of experimental colonies were set up under different conditions, allowing for the production of only QU, WO, WM, or EM individuals, respectively. Reads were mapped using bowtie2 and tophat2 (v2.0.8, v.2.1.0, –b2-sensitive, default settings) against the C. obscurior reference genome (version Cobs1.4) (Schrader et al. 2014). Genes with 20 or fewer reads per million reads were removed from the subsequent gene expression analysis with limma (Ritchie et al. 2015), which allows for appropriate modeling of multi-sample comparisons (Rapaport et al. 2013). We performed TMM normalization and subsequent variance stabilization with the voom function implemented in limma (Law et al. 2014). Gene expression values produced by limma are reported as normalized logarithmic read counts, with average expression (ExprAve) being the mean of all 28 samples, and gene expression differences between treatment pairs as logarithmic fold-changes (logFC) as calculated by limma.
Estimating Rates of Expression Regulation and Sequence Evolution
We used the GATK’s FastaAlternateReferenceMaker (McKenna et al. 2010) to generate a JP genome sequence including the homozygous SNV annotations from a genomic comparison between BR and JP. Genes where SNVs introduced premature stop-codons in the JP population were removed from the analysis. We then extracted coding sequences of each gene for both the BR and JP genome and used PAML’s yn00 algorithm (Yang and Nielsen 2000) to calculate the number of nonsynonymous substitutions per nonsynonymous site (cross-population dN = dNp) and the number of synonymous substitutions per synonymous site (cross-population dS = dSp) values for each gene. dSp values were used as an estimate of neutral evolutionary rates in C. obscurior protein-coding genes. We subsequently calculated a relative measure of non-neutral population divergence between the JP and BR populations as dNp/(dNp + dSp) (the equivalent to the first term in the “Direction of Selection” test (Stoletzki and Eyre-Walker 2010)), allowing us to calculate evolutionary rates for genes without synonymous differences as well.
To assess divergence between species, we calculated pairwise dN/dS ratios for single-copy orthologs between C. obscurior and the most closely related ant for which genome sequences were available, M. pharaonis (Mikheyev and Linksvayer 2015). Genomic sequences, protein sequences, and gene annotations were downloaded from NCBI for M. pharaonis (genome assembly id 231934) and from hymenopteragenome.org for C. obscurior. For each gene, we extracted the longest predicted protein isoform and corresponding CDS sequence using bioperl (Stajich 2002). Single copy orthologs were inferred with the orthoDB software (v1.6, http://www.orthodb.org/?page=software; last accessed November 8, 2016), which uses a best-reciprocal-hit clustering algorithm based on Smith-Waterman protein alignments. For each of the 7,802 single-copy ortholog pairs, we produced consensus pairwise protein alignments with MCoffee, combining results from mafft, muscle and clustalw. Protein alignments were subsequently back-translated with TCoffee (Notredame et al. 2000). About 1,226 alignments were removed from downstream analyses, either because their CDS sequence translation differed from the corresponding predicted protein, CDS sequences did not start with a start codon, did not end in a stop codon, or were not a multiple of three. After removing poorly aligned positions and divergent regions of the alignments with Gblocks (Castresana 2000), we calculated pairwise and gene-wide dN/dS rates with yn00 (PAML 4.8, Yang and Nielsen 2000). We excluded genes from downstream analyses with too short alignments (less than 100 bases), dS = 0 and very high dS rates (larger than the 1.5 × IQR of all calculated dS values) potentially indicating mutational saturation. This resulted in a final set of 6,076 genes.
Statistical Analysis
To analyze correlations between expression bias and the different measures of molecular evolution and expression regulation, we used Kendall’s rank correlation coefficient τ, testing for positive or negative correlations. As expression levels are known to be negatively correlated with evolutionary rates (Drummond et al. 2005; Zhang and Yang 2015), we used semi-partial correlations with Kendall’s coefficients (R package “ppcor”) to exclude effects of expression level from correlations of expression bias and sequence evolution and expression regulation (Kim and Yi 2006). To further test whether median levels of gene sequence evolution [i.e., dSp, dN/dS, dNp/(dNp + dSp)] and expression regulation (i.e., ExprAve, CV) differ significantly more strongly between highly and weakly biased genes than between randomly selected genes, we performed permutation tests (see Supplementary Material online). For this, we calculated medians (dSp, dN/dS, dNp/(dNp + dSp), ExprAve and CV) for two gene sets of size N sampled from the top 25% of genes with the highest and lowest π. We then calculated the medians for two groups of N randomly selected genes. These calculations were performed for 105 permutations. If expression bias (as measured by π) correlates with rates of evolution and regulation, we expect that the distance (i.e., the absolute differences) between medians are larger for the high-vs.-weak bias gene sets than for random gene sets. We calculated the probability P as the proportion of iterations, where differences of medians for random gene sets were equal to or larger than those observed in the high-vs.-weak bias gene sets. Statistical tests were considered significant at P < 0.05.
Data Accessibility
Additional data and scripts used for this study have been uploaded as part of the Supplementary Material online. Raw reads are available at the NCBI short read archive under the accession numbers SRX879674, SRX879676, SRX879678 and SRX692538.
Supplementary Material
Supplementary tables S1–S3 and figures S1–S7 are available at Molecular Biology and Evolution online.
Author Contributions
L.S. and J.O. designed the study. L.S. performed the analyses and L.S., H.H., and J.O. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Julia C. Engelmann for valuable input on the statistical analysis of the data. We furthermore thank the anonymous reviewers whose comments and suggestions helped improve and clarify this manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (He1623/31 to JO), the Academy of Finland (135970 to HH and 252411 (decision number 284666) to the Centre of Excellence in Biological Interactions) and the Kone Foundation (HH).
Footnotes
Associate editor: Hideki Innan
References
- Amdam GV, Csondes A, Fondrk MK, Page RE. 2006. Complex social behaviour derived from maternal reproductive traits. Nature 439:76–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borenstein E, Meilijson I, Ruppin E. 2006. The effect of phenotypic plasticity on evolution in multipeaked fitness landscapes. J Evol Biol. 19:1555–1570. [DOI] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552. [DOI] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. 2006. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 7:98–108. [DOI] [PubMed] [Google Scholar]
- Corona M, Libbrecht R, Wheeler DE. 2016. Molecular mechanisms of phenotypic plasticity in social insects. Curr Opin Insect Sci. 13:55–60. [DOI] [PubMed] [Google Scholar]
- Denver RJ. 1997. Environmental stress as a developmental cue: Corticotropin-releasing hormone is a proximate mediator of adaptive phenotypic plasticity in amphibian metamorphosis. Horm Behav. 31:169–179. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. 2005. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 102:14338–14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. 2007. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 8:689–698. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick BM. 2012. Underappreciated Consequences of Phenotypic Plasticity for Ecological Speciation. Int J Ecol. 2012:1–12. [Google Scholar]
- Forsman A. 2014. Rethinking phenotypic plasticity and its consequences for individuals, populations and species. Heredity 115:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghalambor CK, Hoke KL, Ruell EW, Fischer EK, Reznick DN, Hughes KA. 2015. Non-adaptive plasticity potentiates rapid adaptive evolution of gene expression in nature. Nature 525:372–375. [DOI] [PubMed] [Google Scholar]
- Ghalambor CK, McKay JK, Carroll SP, Reznick DN. 2007. Adaptive versus non-adaptive phenotypic plasticity and the potential for contemporary adaptation in new environments. Funct Ecol. 21:394–407. [Google Scholar]
- Gilad Y, Oshlack A, Rifkin SA. 2006. Natural selection on gene expression. Trends Genet. 22:456–461. [DOI] [PubMed] [Google Scholar]
- Gomez-Mestre I, Wiens JJ, Warkentin KM. 2008. Evolution of adaptive plasticity: risk-sensitive hatching in neotropical leaf-breeding treefrogs. Ecol Monogr. 78:205–224. [Google Scholar]
- Grath S, Parsch J. 2012. Rate of amino acid substitution is influenced by the degree and conservation of male-biased transcription over 50 Myr of Drosophila evolution. Genome Biol Evol. 4:346–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DW, Yi SV, Goodisman MAD. 2013. Kin selection, genomics and caste-antagonistic pleiotropy. Biol Lett. 9:20130309.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick SA, Chen WY, Wong T, Deveson IW, Blackburn J, Andersen SB, Nielsen LK, Mattick JS, Mercer TR. 2016. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat Methods 13:792–798. [DOI] [PubMed] [Google Scholar]
- Harpur BA, Kent CF, Molodtsova D, Lebon JMD, Alqarni AS, Owayss AA, Zayed A. 2014. Population genomics of the honey bee reveals strong signatures of positive selection on worker traits. Proc Natl Acad Sci U S A. 111:2614–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PW, Wright AE, Zimmer F, Dean R, Montgomery SH, Pointer MA, Mank JE. 2015. Sexual selection drives evolution and rapid turnover of male gene expression. Proc Natl Acad Sci U S A. 112:4393–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinze J, Cremer S, Eckl N, Schrempf A. 2006. Stealthy invaders: the biology of Cardiocondyla tramp ants. Insectes Soc. 53:1–7. [Google Scholar]
- Heinze J, Hölldobler B. 1993. Fighting for a harem of queens: physiology of reproduction in Cardiocondyla male ants. Proc Natl Acad Sci U S A. 90:8412–8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helanterä H, Uller T. 2014. Neutral and adaptive explanations for an association between caste-biased gene expression and rate of sequence evolution. Front Genet. 5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt BG, Ometto L, Keller L, Goodisman MAD. 2013. Evolution at two levels in fire ants: the relationship between patterns of gene expression and protein sequence evolution. Mol Biol Evol. 30:263–271. [DOI] [PubMed] [Google Scholar]
- Hunt BG, Ometto L, Wurm Y, Shoemaker D, Yi SV, Keller L, Goodisman MAD. 2011. Relaxed selection is a precursor to the evolution of phenotypic plasticity. Proc Natl Acad Sci U S A. 108:15936–15941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt BG, Wyder S, Elango N, Werren JH, Zdobnov EM, Yi SV, Goodisman MAD. 2010. Sociality is linked to rates of protein evolution in a highly social insect. Mol Biol Evol. 27:497–500. [DOI] [PubMed] [Google Scholar]
- Innocenti P, Morrow EH. 2010. The sexually antagonistic genes of Drosophila melanogaster. PLoS Biol. 8:e1000335.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasper WC, Linksvayer TA, Atallah J, Friedman D, Chiu JC, Johnson BR. 2015. Large-scale coding sequence change underlies the evolution of postdevelopmental novelty in honey bees. Mol Biol Evol. 32:334–346. [DOI] [PubMed] [Google Scholar]
- Jiang L, Schlesinger F, Davis CA, Zhang Y, Li R, Salit M, Gingeras TR, Oliver B. 2011. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 21:1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-H, Yi SV. 2006. Understanding relationship between sequence and functional evolution in yeast proteins. Genetica 131:151–156. [DOI] [PubMed] [Google Scholar]
- Klein A, Schultner E, Lowak H, Schrader L, Heinze J, Holman L, Oettler J. 2016. Evolution of social insect polyphenism facilitated by the sex differentiation cascade. PLoS Genet. 12:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryuchkova-Mostacci N, Robinson-Rechavi M. 2016. A benchmark of gene expression tissue-specificity metrics. Brief Bioinform bbw008:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15:R29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leichty AR, Pfennig DW, Jones CD, Pfennig KS. 2012. Relaxed genetic constraint is ancestral to the evolution of phenotypic plasticity. Integr Comp Biol. 52:16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levis NA, Pfennig DW. 2016. Evaluating “plasticity-first” evolution in nature: key criteria and empirical approaches. Trends Ecol Evol. 31:563–574. [DOI] [PubMed] [Google Scholar]
- Linksvayer TA, Wade MJ. 2005. The evolutionary origin and elaboration of sociality in the aculeate Hymenoptera: maternal effects, sib-social effects, and heterochrony. Q Rev Biol. 80:317–336. [DOI] [PubMed] [Google Scholar]
- López-Maury L, Marguerat S, Bähler J. 2008. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat Rev Genet. 9:583–593. [DOI] [PubMed] [Google Scholar]
- Mank JE, Hultin-Rosenberg L, Zwahlen M, Ellegren H. 2008. Pleiotropic constraint hampers the resolution of sexual antagonism in vertebrate gene expression. Am Nat. 171:35–43. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheyev AS, Linksvayer TA. 2015. Genes associated with ant social behavior show distinct transcriptional and evolutionary patterns. eLife 4:e04775–e04775.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhout HF. 2003. Development and evolution of adaptive polyphenisms. Evol Dev. 5:9–18. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. 2000. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 302:205–217. [DOI] [PubMed] [Google Scholar]
- Nuzhdin SV. 2004. Common pattern of evolution of gene expression level and protein sequence in Drosophila. Mol Biol Evol. 21:1308–1317. [DOI] [PubMed] [Google Scholar]
- Oettler J, Schrempf A. 2016. Fitness and aging in Cardiocondyla obscurior ant queens. Curr Opin Insect Sci. 16:58–63. [DOI] [PubMed] [Google Scholar]
- Oettler J, Suefuji M, Heinze J. 2010. The evolution of alternative reproductive tactics in male Cardiocondyla ants. Evolution 64:3310–3317. [DOI] [PubMed] [Google Scholar]
- Parsch J, Ellegren H. 2013. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 14:83–87. [DOI] [PubMed] [Google Scholar]
- Pál C, Papp B, Lercher MJ. 2006. An integrated view of protein evolution. Nat Rev Genet. 7:337–348. [DOI] [PubMed] [Google Scholar]
- Perry JC, Harrison PW, Mank JE. 2014. The ontogeny and evolution of sex-biased gene expression in Drosophila melanogaster. Mol Biol Evol. 31:1206–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purandare SR, Bickel RD, Jaquiery J, Rispe C, Brisson JA. 2014. Accelerated evolution of morph-biased genes in pea aphids. Mol Biol Evol. 31:2073–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, Mason CE, Socci ND, Betel D. 2013. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 14:R95.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlfs RV, Harrigan P, Nielsen R. 2014. Modeling gene expression evolution with an extended Ornstein-Uhlenbeck process accounting for within-species variation. Mol Biol Evol. 31:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y. 2012. Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet. 13:505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romiguier J, Lourenco J, Gayral P, Faivre N, Weinert LA, Ravel S, Ballenghien M, Cahais V, Bernard A, Loire E, Wurm Y, Smith CD, Yandell M, Heinze J, Gadau J, Oettler J. 2014. Population genomics of eusocial insects: the costs of a vertebrate-like effective population size. J Evol Biol. 27:593–603. [DOI] [PubMed] [Google Scholar]
- Ruden DM, Cingolani PE, Sen A, Qu W, Wang L, Senut M-C, Garfinkel MD, Sollars VE, Lu X. 2015. Epigenetics as an answer to Darwin's ‘special difficulty,’ Part 2: natural selection of metastable epialleles in honeybee castes. Front Genet. 6:60.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichting CD, Pigliucci M. 1998. Phenotypic evolution: a reaction norm perspective. Sunderland (MA: ): Sinauer Associates [Google Scholar]
- Schlichting CD, Wund MA. 2014. Phenotypic plasticity and epigenetic marking: an assessment of evidence for genetic accommodation. Evolution 68:656–672. [DOI] [PubMed] [Google Scholar]
- Schrader L, Kim JW, Ence D, Zimin A, Klein A, Wyschetzki K, Weichselgartner T, Kemena C, Stökl J, Schultner E, et al. 2014. Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat Commun. 5:5495.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader L, Simola DF, Heinze J, Oettler J. 2015. Sphingolipids, transcription factors, and conserved toolkit genes: developmental plasticity in the ant Cardiocondyla obscurior. Mol Biol Evol. 32:1474–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrempf A, Heinze J. 2006. Proximate mechanisms of male morph determination in the ant Cardiocondyla obscurior. Evol Dev. 8:266–272. [DOI] [PubMed] [Google Scholar]
- Smith JM, Szathmary E. 1995. The major transitions in evolution. Oxford: Oxford University; Press. [Google Scholar]
- Snell-Rood EC, Cash A, Han MV, Kijimoto T, Andrews J, Moczek AP. 2011. Developmental decoupling of alternative phenotypes: insights from the transcriptomes of horn-polyphenic beetles. Evolution 65:231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell-Rood EC, Van Dyken JD, Cruickshank T, Wade MJ, Moczek AP. 2010. Toward a population genetic framework of developmental evolution: the costs, limits, and consequences of phenotypic plasticity. Bioessays 32:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE. 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 12:1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. 2010. Estimation of the neutrality index. Mol Biol Evol. 28:63–70. [DOI] [PubMed] [Google Scholar]
- Van Dyken JD, Wade MJ. 2010. The genetic signature of conditional expression. Genetics 184:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitti JJ, Grossman SR, Sabeti PC. 2013. Detecting Natural Selection in Genomic Data. Annu Rev Genet. 47:97–120. [DOI] [PubMed] [Google Scholar]
- Vojvodic S, Johnson BR, Harpur BA, Kent CF, Zayed A, Anderson KE, Linksvayer TA. 2015. The transcriptomic and evolutionary signature of social interactions regulating honey bee caste development. Ecol Evol. 5:4795–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnefors M, Kaessmann H. 2013. Evolution of the correlation between expression divergence and protein divergence in mammals. Genome Biol Evol. 5:1324–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West-Eberhard MJ. 1998. Evolution in the light of developmental and cell biology, and vice versa. Proc Natl Acad Sci U S A. 95:8417–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West-Eberhard MJ. 2003. Developmental plasticity and evolution. New York: Oxford University Press. [Google Scholar]
- West-Eberhard MJ. 2005a. Developmental plasticity and the origin of species differences. Proc Natl Acad Sci U S A. 102:6543–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West-Eberhard MJ. 2005b. Phenotypic accommodation: adaptive innovation due to developmental plasticity. J Exp Zool B Mol Dev Evol. 304B:610–618. [DOI] [PubMed] [Google Scholar]
- Wheeler DE. 1986. Developmental and physiological determinants of caste in social hymenoptera—evolutionary implications. Am Nat 128:13–34. [Google Scholar]
- Whitehead A, Crawford DL. 2006. Neutral and adaptive variation in gene expression. Proc Natl Acad Sci U S A. 103:5425–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. 2005. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21:650–659. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 17:32–43. [DOI] [PubMed] [Google Scholar]
- Zhang J, Yang J-R. 2015. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 16:409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional data and scripts used for this study have been uploaded as part of the Supplementary Material online. Raw reads are available at the NCBI short read archive under the accession numbers SRX879674, SRX879676, SRX879678 and SRX692538.