


Abstract
Processed pseudogenes result from reverse transcribed mRNAs. In general, because processed pseudogenes lack promoters, they are no longer functional from the moment they are inserted into the genome. Subsequently, they freely accumulate substitutions, insertions and deletions. Moreover, the ancestral structure of processed pseudogenes could be easily inferred using the sequence of their functional homologous genes. Owing to these characteristics, processed pseudogenes represent good neutral markers for studying genome evolution. Recently, there is an increasing interest for these markers, particularly to help gene prediction in the field of genome annotation, functional genomics and genome evolution analysis (patterns of substitution). For these reasons, we have developed a method to annotate processed pseudogenes in complete genomes. To make them useful to different fields of research, we stored them in a nucleic acid database after having annotated them. In this work, we screened both mouse and human complete genomes from ENSEMBL to find processed pseudogenes generated from functional genes with introns. We used a conservative method to detect processed pseudogenes in order to minimize the rate of false positive sequences. Within processed pseudogenes, some are still having a conserved open reading frame and some have overlapping gene locations. We designated as retroelements all reverse transcribed sequences and more strictly, we designated as processed pseudogenes, all retroelements not falling in the two former categories (having a conserved open reading or overlapping gene locations). We annotated 5823 retroelements (5206 processed pseudogenes) in the human genome and 3934 (3428 processed pseudogenes) in the mouse genome. Compared to previous estimations, the total number of processed pseudogenes was underestimated but the aim of this procedure was to generate a high-quality dataset. To facilitate the use of processed pseudogenes in studying genome structure and evolution, DNA sequences from processed pseudogenes, and their functional reverse transcribed homologs, are now stored in a nucleic acid database, HOPPSIGEN. HOPPSIGEN can be browsed on the PBIL (Pôle Bioinformatique Lyonnais) World Wide Web server (http://pbil.univ-lyon1.fr/) or fully downloaded for local installation.
INTRODUCTION
Processed pseudogenes arise by reverse transcription of mRNAs and integration of the resulting cDNAs into the genome (1–4). Hence, they lack introns, possess relics of the poly(A) tail at their 3′ ends. They are flanked by target-site duplications and in particular show a strong similarity to the mRNAs they originate from. Their number was estimated to be 8000–100 000 (4–10) in the human genome and 5000–14 000 in the mouse genome (11,12). Despite the existing interest in studying them, available sequences of processed pseudogenes are poorly annotated in general databases (GenBank or EMBL). Recently, a specialized database for these markers was developed by Zhang et al. (9) (http://pseudogene.org/human/index.php). Therefore, this database contains data only from the human genome whereas processed pseudogenes' sequences from the mouse genome are also available separately (12). In this context, and considering the amount of data available, we developed a method to detect and annotate processed pseudogenes in complete human and mouse genomes. These data are stored in a database HOPPSIGEN (HOmologous Processed PseudoGENes), which contains useful information concerning processed pseudogenes location, potential function, base composition, gene structure and a few other features. HOPPSIGEN is available through WWW-query (13), a WWW-server on the PBIL (http://pbil.univ-lyon1.fr/).
We searched for reverse transcribed sequences similar to known genes and having the characteristics of processed pseudogenes: no introns, a polyadenylation track, frameshifts or internal stop codons, and direct flanking repeats. With this procedure, we can only detect processed pseudogenes generated by genes with introns. Each identified processed pseudogenes was then classified into a family. A family is a group composed of a functional reverse transcribed gene to which we had associated one or more homologous processed pseudogenes. Using our method, we identified 5823 human and 3934 mouse retroelements localized in the human and mouse genomes (ENSEMBL 18.3) (14). A total of 3491 families (1966 specific to human, 1202 specific to mouse and 323 shared between mouse and human) are stored in HOPPSIGEN. More information concerning database browsing are available at http://pbil.univ-lyon1.fr/databases/hoppsigen.html. The main site is linked to several resources. Few examples and a list of ‘How to’ use the database are available at http://pbil.univ-lyon1.fr/databases/hoppsigen_examples.html. A table containing the most abundant families (51 human and 27 mouse only single species family), associated to known proteins, is available at http://pbil.univ-lyon1.fr/databases/hoppsigen_families.html. Finally, a table of the 323 families of processed pseudogenes shared between human and mouse is available at http://pbil.univ-lyon1.fr/databases/hoppsigen_orthologs.html.
DATABASE CONTENT
Processed pseudogenes identification
Processed pseudogenes were identified in the human and mouse genomes by looking for similarities between nucleic acid sequences of functional genes to introns and the whole genome sequence. The basic principles of the search are illustrated in Figure 1. Our goal was to identify only processed paralogs of a functional gene with introns. From complete genomes annotated by ENSEMBL, we used genomic fragments of size ∼1 Mb. Our annotation procedure was divided into several steps (Figure 1).
Figure 1.
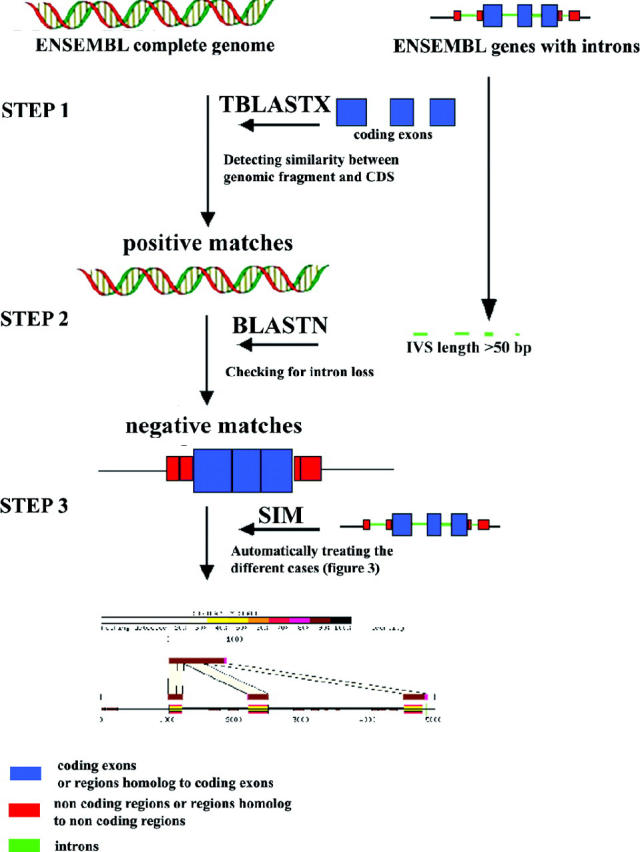
Description of the method used to identify processed pseudogenes.
Step 1. We selected all coding sequences (CDS) of genes with introns (IVS) from ENSEMBL 18.3 (27 156 human and 28 696 mouse CDS). We excluded CDS from intron-less genes because it is difficult to determine if the retroelements of such genes resulted from a real reverse transcription event. Moreover, some intron-less genes could be wrongly annotated retroelements (15). Each dataset of nucleic acid sequences was masked using RepeatMasker (http://ftp.genome.washington.edu/RM/RepeatMasker.html) with the Repbase release of the May 5, 2002 (option -norna for both datasets and option -mus for the mouse dataset).
For each species, we ran TBLASTX (16) between the masked dataset of CDS and ENSEMBL genome fragments to find DNA sequences similar to functional CDS. We retained positive matches >80 bp, with no similarity threshold and an E-value >10−5.
Step 2. The aim of this step was to separate positive matches resulting from a complete or partial gene duplication and those corresponding to the gene itself (Figure 2, case A). For this purpose, the sequences of the positive matches were extracted, including 1 kb on each side. Then, BLASTN was run between each candidate and the introns (IVS) of the homologous gene in ENSEMBL (length >50 bp, no similarity threshold and E-value >10−5). Only cases without significant similarities to introns (Figure 1, Step 2, negative matches) were retained (Figure 2, cases B, C and D) and the others were removed (Figure 2, case A).
Figure 2.

Duplication and reverse transcription. In complete genomes, we can identify four kinds of homologous sequences to a functional gene with introns. (A) A complete duplication of a functional gene leads to the formation of a paralogous gene. If the duplicated gene became non-functional after its insertion, it is classified as an unprocessed pseudogene. However, it may have evolved to give another gene with a new function. If the duplication is recent, we still detect similarities on the introns. (B) Another case of old duplicated gene. In this case, the duplicated gene may be still functional but introns have diverged much faster than exons, therefore we can detect a similarity between the two homologous copies only on the exons. (C) Single exon sequences are generated either by partial or old reverse transcription, or by partial or old gene duplication. (D) A retroelement is generated by reverse transcription. The retroelement lacks introns and is similar only to exons and UTR regions.
Step 3. In this step, we checked if the observed pattern of similarity presented evidence of splicing (Figure 2, cases B and C): the alignment between the CDS and its homolog contained some large gaps (>50 bp) located near splice-site junctions.
We used SIM (17) to make an alignment between each selected case (extended by 2 kb on each side) and the complete gene corresponding to the homologous CDS (extended by 1 kb on each side). Outputs were automatically treated to distinguish between the three cases and selected only retroelements (Figure 2, case D). We used a simple algorithm (Figure 3), based on the position of the splicing sites to determine if a selected region was truly processed or not.
Figure 3.

- ← If (SIM score > 20 and PR similar to at least two exons) ↠ PR is retained;
- else if (SIM score > 20 and PR similar to one exon) ↠ PR is a single exon (case C);
- else if (SIM score < 20) ↠ PR is discarded;
- ↑ for each splicing site: test if ai or ai±1 is near the splicing site Sj on the gene (Sj ± 10 bp);
- If test succeeded: then if d(ai, ai+1) > 50 bp ↠ PR is an unprocessed paralog (case A and B);
- if test failed ↠ PR is a retroelement (case D);
These homologs might correspond to distantly related unprocessed paralogs and have been discarded (Figure 2, case B). When the similarity encompassed only one exon (Figure 2, case C), it was not possible to determine whether it resulted from a retrotransposition or not. Such cases were also discarded. With this approach, our procedure could not be used to detect retrosequences from intron-less genes. The other sequences were classified as retroelements (Figure 2, case D).
Processed pseudogenes annotation
All retroelement sequences were annotated as ppgene. We annotated several features in previously detected retroelements as described in Figure 4. Three homologous regions were distinguished on a retroelement: 5′-flanking regions (5′-FL), 3′-FL and CDE (CoDing Exons), respectively, and homologous to the 5′-untranslated regions (5′-UTRs), 3′-UTRs and CDS of the functional gene. Some retroelements lack 5′- and 3′-FL regions. In such cases, retroelements were annotated using the keyword ‘truncated’ (5FL_truncated or 3FL_truncated).
Figure 4.

Structure of a retroelement in HOPPSIGEN. After being annotated, retroelements were classified into three classes: (i) putative ORF: retroelements having a conserved_ORF; (ii) putative retroelements, overlapping an ENSEMBL gene position; and (iii) other cases: all retroelements that are processed pseudogenes.
We extracted regions similar to CDS (CDE regions) from all retroelements and we scanned them to detect conserved open reading frame (ORF). A conserved ORF indicates either that the retroelement is non-functional but did not accumulate enough changes to destroy its original ORF, or that it is still functional and a selective pressure maintains the ORF. Therefore, very few retroelements were described as functional. In order to distinguish retroelements with a conserved ORF, we added the keyword ‘putative_orf’ to their description (Figure 4).
Retroelements were located on the assembled human (NCBI Human v34) and mouse genomes (NCBI Mouse build 3). We compared retroelement locations with all known-gene locations to detect if a retroelement is overlapping a gene. Only the retroelements inside exons were retained. We annotated such cases as ‘putative_retroelement’ (Figure 4).
The polyadenylation track (Figure 4) is in general very hard to identify correctly in retroelements because it is often degenerated. Usual methods cannot be applied. Therefore, we used a method based on a sliding window. We calculated the composition in adenine for a sliding window of size 30 bp, with a step of 10 bp, in a region of 1 kb starting from the 3′ end of the retroelement. In the window showing the significantly highest composition in adenine (Student's t-test, P-value < 0.05), we calculated the exact probability of the longest adenine repetition. Only windows containing a significant repetition (exact probability <0.05) of at least five adenines were selected. In order to have a control, we used the same protocol to identify the window having the significantly highest composition in thymine (Student's t-test, P-value < 0.05) and a significant repetition of at least five thymines (exact probability <0.05). Subsequently, for each retroelement, we concluded that there was a polyadenylation track only if we found a significant adenine window and no significant thymine window. This method is not totally efficient because there are some ambiguous cases. When a transposable element (SINE or LINE) is inserted in the 3′-FL region of a retroelement, we can wrongly attribute the polyadenylation track to the retroelement.
We have also identified and annotated direct flanking repeats around retroelements (Figure 4). For that purpose, we extracted 200 bp on each side of the retroelement and made an alignment between them using SIM. We selected the best local alignment showing at least a score of nine (default parameters) and a length of 10 bp. We annotated the corresponding regions as direct flanking repeats. We also identified and annotated other repeat boundaries around the retroelement using RepeatMasker (Repbase May 5, 2002, option mus to detect mouse repeats).
Finally, we computed miscellaneous features related to the level of similarity between a retroelement and its functional homolog, and the GC-content of the retroelement.
In order to make all these annotations easily available, we built HOPPSIGEN, a nucleic acid database of retroelements.
HOPPSIGEN database
HOPPSIGEN (release 4.0, January 2004) nucleotide sequences (retroelements and functional paralogs) are structured in an ACNUC database (18). HOPPSIGEN is available in the form of EMBL flat files (19) and divided into 3491 families. A family contains a functional (human or mouse) gene and all its homologous retroelements. Each family is pointed out by a keyword: HOP003477 for example corresponds to a family of 86 sequences, 84 retroelements shared by the human and mouse, and their two functional genes: human and mouse genes coding for 40S ribosomal protein SA (RPS4). The human gene and its homologous retroelements contain in their annotations the ENSEMBL gene name ‘ENSG00000168028’ and the Swiss-Prot-TrEMBL (20) accession number Q96RS2 (RSP4_HUMAN). Both can also be used as keywords to retrieve them. The mouse gene and its homologous retroelements are associated with the ENSEMBL gene name ‘ENSMUSG00000032518’ and the Swiss-Prot-TrEMBL (20) accession number Q91V31 (RSP4_MOUSE). HOPPSIGEN annotations are divided into several categories. A useful description of these annotations is available at http://pbil.univ-lyon1.fr/databases/hoppsigen_desc.html.
For each HOPPSIGEN family we have associated two files containing:
A nucleotide alignment. We built automatically this alignment with CLUSTAL W (21), using for each species in the database a functional reverse transcribed gene and its homologous retroelements. Some functional reverse transcribed genes in the mouse or human genomes could also be orthologs. In such a case, alignments included both species. To test whether two reverse transcribed mouse and human genes were orthologs or not, we used reference datasets of orthologous genes extracted from HOVERGEN (22). Each alignment in CLUSTAL format is available in a file called HOPxxxxx.aln.
For families with more than three sequences, a phylogenetic tree was built with the nucleotide alignment using the Neighbor-Joining method (23) implemented in CLUSTAL W with K2 distances (24). Each tree was rooted automatically using NJPLOT (24) and stored in NEWICK format in a file called HOPxxxxx.phb. The root was chosen to minimize the branch length variance between this point and each leaf in the tree.
DATABASE ACCESS
Following our protocol, we identified 5823 human and 3934 mouse retroelements associated with 2289 human CDS and 1525 mouse CDS (Table 1). Whereas, the size of initial CDS datasets were approximately the same for human (27 156 CDS) and mouse (28 696 CDS), we found less retroelements in mouse genome. This difference can be explained in two ways: (i) the quality of the annotations and assembly are better for human genome compared to mouse genome. (ii) There was a possible burst of retroelements formation in primate lineage 40–50 Myr ago (25). Within our dataset, nearly 3% of the human and mouse retroelements were overlapping a gene position as described in ENSEMBL (matches overlapping exons). We also found that 8.1% of human and 9.6% of the mouse retroelements had a conserved ORF. After excluding retroelements belonging to these two categories, 5206 human and 3428 mouse retroelements were strictly identified as non-functional processed pseudogenes. Several features concerning our dataset and comparisons between the two species are described briefly (Table 1).
Table 1. General statistics extracted from the database HOPPSIGEN.
| Homo sapiens | Mus musculus | Comparisons (P-value for χ2-test) | |
|---|---|---|---|
| Reverse transcribed genes | |||
| Total number of retroelements | 5823 | 3934 | |
| Number of processed pseudogenes | 5206 (89.4%) | 3428 (87.1%) | NS |
| Maximum number of retroelements for a family (known genes and genes with unknown functions) | 101 | 61 | Not calculated |
| Retroelements features | |||
| Number of truncated retroelements (%) | 3809 (65.4%) | 2519 (64.0%) | NS |
| Number of retroelements with a 5′-flanking region (%) | 342 (5.9%) | 386 (9.8%) | *** |
| Number of retroelements with a 3′-flanking region (%) | 2262 (38.8%) | 1392 (35.4%) | NS |
| Number of retroelements overlapping a known gene (‘putative_retroelement’) (%) | 166 (2.9%) | 141 (3.6%) | NS |
| Number of retroelements with a conserved ORF. (‘putative_orf’) (%) | 472 (8.1%) | 378 (9.6%) | * |
| Homology distribution | |||
| Number of recent CDE (<5% divergent with their functional homolog) (%) | 516 (8.9%) | 620 (15.8%) | *** |
We used the χ2-statistics to compare the number of sequences in each category for the two species: ***P ≤ 10−4; **P ≤ 0.001; *P ≤ 0.01; P > 0.01 (NS). The reference values for the test were the total number of retroelements for each species.
HOPPSIGEN can be accessed through the PBIL World Wide Web server (13). Thanks to the retrieval system set up on this server, it is possible to query the HOPPSIGEN database either by sequence name or by family name (http://pbil.univ-lyon1.fr/databases/hoppsigen.html) (Figure 5). With a local installation, the HOPPSIGEN database can also be accessed with the Query (18) or Query_win (26) programs allowing complex queries.
Figure 5.

Browsing HOPPSIGEN database. HOPPSIGEN help pages (http://pbil.univ-lyon1.fr/databases/hoppsigen.html) contain information on how to query the database. The WWW-Query tools (http://pbil.univ-lyon1.fr/search/query_fam.php) are a powerful way to make queries to HOPPSIGEN.
The WWW-Query system offers many possibilities to browse the database (Figure 5). Through the WWW-Query system, HOPPSIGEN offers direct links with several databases like ENSEMBL or Swiss-Prot-TrEMBL. If the database is browsed for families, among interesting possibilities, the user can extract all retroelements associated with a Swiss-Prot-TrEMBL protein (knowing the accession number), or an ENSEMBL gene (knowing the gene name), or simply with a family name. If the database is browsed for all sequences, it is possible to extract a group of retroelements following the criteria defined by the user. For example, to select only human retroelements following the true definition of processed pseudogenes (i.e. no putative_orf and no putative_retroelements), we made a query based on the sequences level. First, the species homo has been specified, second, the type ppgene was also specified and, finally, we excluded all sequences containing the keywords putative_orf and putative_retroelements (by choosing the ‘AND/NOT’ option from the left-hand side menu comprising those keywords). This query returns 5206 human retroelements considered as non-functional processed pseudogenes. The same query made by replacing homo by mus returns 3428 mouse processed pseudogenes. Another interesting query concerns the similarity between retroelements and functional genes. It is possible to select retroelements (or CDE regions) having a similarity (or GC-content <50%) <80% to their functional genes by using the keyword i < 80% (CG < 50%). The insertion pattern of retroelements generated by reverse transcribed genes may follow the same pattern as human and mouse LINEs and SINEs (27,11). In particular, we should find more recently inserted retroelements in the mouse genome because there was a burst of recent reverse transcription specific to the mouse lineage and not observed in the human lineage (25). A similar, but more important, burst was observed in human genome but 40–50 Myr ago. To test this hypothesis, we assume that the age of insertion is correlated with the observed divergence between retroelements and their functional homologous genes, and we extracted recently inserted retroelements (more than 95% similar to their functional genes). We found a higher frequency of recently inserted retroelements in the mouse lineage (χ2-test, P < 10−16)—15.8% compared to 8.9% in the human lineage (Table 1).
Retroelements could be studied considering their genome location. Whereas the exact chromosomal location is available in the general description, WWW-Query does not offer the possibility to use this feature to browse the database. Therefore, by using the keywords HSn for human chromosomes and MMn for mouse chromosomes, where n is the chromosome number, chromosome names can be used to browse HOPPSIGEN.
Another interesting feature of HOPPSIGEN is the possibility to screen the database using various keywords like biological function or gene names. For example, the query using the keyword ‘*ribosom*’ and the type ‘ppgene’ returns 1763 retroelements containing this keyword. The same query, using a search for families, returns 155 families.
This is just an overview of the database. It could be used for other purposes. For example, in genome annotation, by selecting retroelements overlapping a known gene position (keyword: putative_retroelement), the database may be used to re-examine these regions.
COMPARISON WITH EXISTING RESOURCES AND OVERVIEW
Processed pseudogenes identification in large genomic sequences is difficult because it is not easy to detect highly divergent sequences. We developed a mostly automatic method to detect and to annotate them in complete genomes. This method could be generalized to many genomes, and we expect soon to apply it to the rat and chimpanzee genomes. We found 5823 retroelements having several or all of the characteristics of processed pseudogenes in the human genome (for a dataset of 27 156 CDS of genes with introns) and 3934 in the mouse genome (for a dataset of 28 696 CDS of genes with introns). Compared to other recent estimations, we underestimated the total number of processed pseudogenes (12). However, the goal of this database of retroelements was not to be exhaustive but to minimize the false positive rate (high specificity) and to detect retroelements of genes with introns (85% of all genes in ENSEMBL 18.3).
HOPPSIGEN was compared to other available resources about mouse and human processed pseudogenes (9,12). Zhang's human dataset (http://pseudogene.org/human/index.php) contains 7868 processed pseudogenes. In order to make the comparison, we selected only processed pseudogenes homologous to genes with introns extracted from ENSEMBL 18.3. We therefore retained 5348 processed pseudogenes from Zhang's dataset, which is very close to the value of 5206 processed pseudogenes in HOPPSIGEN. We identified 2903 (54.3%) human processed pseudogenes shared between HOPPSIGEN and Zhang's human dataset. We also compared HOPPSIGEN to a recently published dataset of 4476 mouse processed pseudogenes (12). After applying the same criteria as before, 2956 processed pseudogenes were retained from Zhang's dataset. We identified 1266 mouse processed pseudogenes (42.8%) shared between HOPPSIGEN and Zhang's mouse dataset. Whereas the two methods follow the same approach to identify retroelements, they do not give exactly the same results. We noticed that the length of the human genes homologous to processed pseudogenes was higher in HOPPSIGEN than in Zhang's dataset (mean ± SD: 1532 ± 1241 bp compared to 1195 ± 953 bp; Wilcoxon-test: P < 10−16). The same result was found for the mouse genes (1283 ± 1053 bp compared to 826 ± 655 bp; Wilcoxon-test: P < 10−16). It seems that our method performs better for long genes but Zhang's method is more efficient for short genes. Moreover, we found no relationship between GC-content and the efficiency of both methods. We also used a third dataset of human processed pseudogenes (Torrent's dataset) (10) to compare HOPPSIGEN and Zhang's dataset. The method used to build this dataset is different from the two previous methods. It was less conservative to annotate processed pseudogenes, so it found many more sequences: 17 760. We identified 4666 (80.1% for 8823 initial sequences) human processed pseudogenes shared between HOPPSIGEN and Torrents's dataset. We also identified 4625 (59.2% for 7819 initial sequences) human processed pseudogenes shared between Zhang's dataset and Torrent's dataset. Whereas we were highly specific, it seems that our method performs better than Zhang's method: more processed pseudogenes were found in Torrent's dataset using our method than that in Zhang's method.
HOPPSIGEN offers many possibilities, which makes it very useful. It offers the possibility to extract complete retroelement sequences or to extract part of them (5′-FL, CDE, 3′-FL, direct repeats, polyadenylation tracks). The users can, for example, focus on retroelements containing a conserved ORF, or extract retroelements similar to a specific Swiss-Prot-TrEMBL protein. Another interesting aspect of HOPPSIGEN is the possibility to extract processed pseudogenes of different GC-content or similarity percent (according to their functional paralogous genes). Thus, this should be very helpful for evolutionary studies. Moreover, HOPPSIGEN not only provides information about sequence structure but it also provides alignments and phylogenetic trees.
We expect HOPPSIGEN to be an important tool for all evolutionary and genetic studies using processed pseudogenes. Some authors highlighted the existence of mutational biases in processed pseudogenes correlated to base composition in DNA regions where these sequences are located (28). We have already demonstrated a link between recombination and the evolution of processed pseudogenes in the human and mouse genomes. We found that the GC-content expected at equilibrium in processed pseudogenes is correlated with the recombination pattern (data not shown).
HOPPSIGEN contains retroelements associated to known genes and to predicted genes. Therefore, the association of retroelements with predicted genes could be used as a supplementary proof to classify them as functional genes.
More generally, HOPPSIGEN should be useful in comparative and functional genomics. Some processed pseudogenes are still functional after translation or only after transcription (29,30). The processed pseudogene identified is homologous to the human Makorin1 gene (Swiss-Prot-TrEMBL accession number: Q9UHC7), the nucleic acid sequence is 700 bp long. We also found in HOPPSIGEN two human processed pseudogenes homologous to the Makorin1 human gene, and one of them corresponds to the processed pseudogene detected by Hirotsune et al. (29). What is however interesting is that we also found two mouse processed pseudogenes homologous to the mouse Makorin1 gene (Swiss-Prot-TrEMBL name: MKR1_MOUSE). It would be therefore very interesting to test if one of these retroelements is still functional. Knowing conserved processed pseudogenes between the two species should help us to detect such cases. Using global alignments between human and mouse genomes (31), we have already identified such cases.
Acknowledgments
ACKNOWLEDGEMENTS
Thanks to Julien Grassot and Marie-France Sagot for discussion and comments on the manuscript. We thank the Centre de Calcul de l'IN2P3 for providing computer resources. This work was supported by the Centre National de la Recherche Scientique and the Claude Bernard University (Lyon, France).
REFERENCES
- 1.Vanin E.F. (1985) Processed pseudogenes: characteristics and evolution. Annu. Rev. Genet., 19, 253–272. [DOI] [PubMed] [Google Scholar]
- 2.Esnault C., Maestre,J. and Heidmann,T. (2000) Human LINE retrotransposons generate processed pseudogenes. Nature Genet., 24, 363–367. [DOI] [PubMed] [Google Scholar]
- 3.Mighell A.J., Smith,N.R., Robinson,P.A. and Markham,A.F. (2000) Vertebrate pseudogenes. FEBS Lett., 468, 109–114. [DOI] [PubMed] [Google Scholar]
- 4.Pavlicek A., Paces,J., Zika,R. and Hejnar,J. (2002) Length distribution of long interspersed nucleotide elements (LINEs) and processed pseudogenes of human endogenous retroviruses: implications for retrotransposition and pseudogene detection. Gene, 300, 189–194. [DOI] [PubMed] [Google Scholar]
- 5.Crollius H.R., Jaillon,O., Dasilva,C., Ozouf-Costaz,C., Fizames,C., Fischer,C., Bouneau,L., Billault,A., Quetier,F., Saurin,W., Bernot,A. and Weissenbach,J. (2000) Characterization and repeat analysis of the compact genome of the freshwater pufferfish Tetraodon nigroviridis. Genome Res., 10, 939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ewing B. and Green,P. (2000) Analysis of expressed sequence tags indicates 35,000 human genes. Nature Genet., 25, 232–234. [DOI] [PubMed] [Google Scholar]
- 7.Gonçalves I., Duret,L. and Mouchiroud,D. (2000) Nature and structure of human genes that generate retropseudogenes. Genome Res., 10, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrison P.M., Hegyi,H., Balasubramanian,S., Luscombe,N.M., Bertone,P., Echols,N., Johnson,T. and Gerstein,M. (2002) Molecular fossils in the human genome: identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res., 12, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z., Harrison,P.M., Liu,Y. and Gerstein,M. (2003) Millions of years of evolution preserved: a comprehensive catalog of the processed pseudogenes in the human genome. Genome Res., 13, 2541–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torrents D., Suyama,M., Zdobnov,E. and Bork,P. (2003) A genome-wide survey of human pseudogenes. Genome Res., 13, 2559–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mouse Genome Sequencing Consortium (2002) Initial sequencing and comparative analysis of the mouse genome. Nature, 420, 520–562. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z., Carriero,N. and Gerstein,M. (2004) Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends Genet., 20, 62–67. [DOI] [PubMed] [Google Scholar]
- 13.Perriere G., Combet,C., Penel,S., Blanchet,C., Thioulouse,J., Geourjon,C., Grassot,J., Charavay,C., Gouy,M., Duret,L. and Deleage,G. (2003) Integrated databanks access and sequence/structure analysis services at the PBIL. Nucleic Acids Res., 31, 3393–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birney E., Andrews,D., Bevan,P., Caccamo,M., Cameron,G., Chen,Y., Clarke,L., Coates,G., Cox,T., Cuff,J., Curwen,V., Cutts,T., Down,T., Durbin,R., Eyras,E., Fernandez-Suarez,X.M., Gane,P., Gibbins,B., Gilbert,J., Hammond,M., Hotz,H., Iyer,V., Kahari,A., Jekosch,K., Kasprzyk,A., Keefe,D., Keenan,S., Lehvaslaiho,H., McVicker,G., Melsopp,C., Meidl,P., Mongin,E., Pettett,R., Potter,S., Proctor,G., Rae,M., Searle,S., Slater,G., Smedley,D., Smith,J., Spooner,W., Stabenau,A., Stalker,J., Storey,R., Ureta-Vidal,A., Woodwark,C., Clamp,M. and Hubbard,T. (2004) Ensembl 2004. Nucleic Acids Res., 32, D468–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z., Harrison,P. and Gerstein,M. (2002) Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome. Genome Res., 12, 1466–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X. and Miller,W. (1991) A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math., 12, 337–357. [Google Scholar]
- 18.Gouy M., Gautier,C., Attimonelli,M., Lanave,C. and Di Paolo,G. (1985) ACNUC—a portable retrievial system for nucleic acid sequence databases: logical and physical designs and usage. Comput. Appl. Biosci., 1, 167–172. [DOI] [PubMed] [Google Scholar]
- 19.Kulikova T., Aldebert,P., Althorpe,N., Baker,W., Bates,K., Browne,P., Van Den Broek,A., Cochrane,G., Duggan,K., Eberhardt,R., Faruque,N., Garcia-Pastor,M., Harte,N., Kanz,C., Leinonen,R., Lin,Q., Lombard,V., Lopez,R., Mancuso,R., McHale,M., Nardone,F., Silventoinen,V., Stoehr,P., Stoesser,G., Tuli,M.A., Tzouvara,K., Vaughan,R., Wu,D., Zhu,W. and Apweiler,R. (2004) The EMBL Nucleotide Sequence Database. Nucleic Acids Res., 32, D27–D30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boeckmann B., Bairoch,A., Apweiler,R., Blatter,M.C., Estreicher,A., Gasteiger,E., Martin,M.J., Michoud,K., O'Donovan,C., Phan,I., Pilbout,S. and Schneider,M. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res., 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duret L., Mouchiroud,D. and Gouy,M. (1994) HOVERGEN: a database of homologous vertebrate genes. Nucleic Acids Res., 22, 2360–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saitou N. and Nei,M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4, 4406–4425. [DOI] [PubMed] [Google Scholar]
- 24.Kimura M. (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol., 16, 111–120. [DOI] [PubMed] [Google Scholar]
- 25.Ohshima K., Hattori,M., Yada,T., Gojobori,T., Sakaki,Y. and Okada,N. (2003) Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol., 4, R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perriere G. and Thioulouse,J. (1996) On-line tools for sequence retrieval and multivariate statistics in molecular biology. Comput. Appl. Biosci., 12, 63–69. [DOI] [PubMed] [Google Scholar]
- 27.International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 28.Francino M.P. and Ochman,H. (1999) Isochores result from mutation, not selection. Nature, 400, 30–31. [DOI] [PubMed] [Google Scholar]
- 29.Hirotsune S., Yoshida,N., Chen,A., Garrett,L., Sugiyama,F., Takahashi,S., Yagami,K., Wynshaw-Boris,A. and Yoshiki,A. (2003) An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature, 423, 91–96. [DOI] [PubMed] [Google Scholar]
- 30.Lee J.T. (2003) Molecular biology: complicity of gene and pseudogene. Nature, 423, 26–28. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz S., Kent,W.J., Smit,A., Zhang,Z., Baertsch,R., Hardison,R.C., Haussler,D. and Miller,W. (2003) Human–mouse alignments with BLASTZ. Genome Res., 13, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]