

Abstract
A major topic of interest in human prehistory is how the large-scale genetic structure of modern populations outside of Africa was established. Demographic models have been developed that capture the relationships among small numbers of populations or within particular geographical regions, but constructing a phylogenetic tree with gene flow events for a wide diversity of non-Africans remains a difficult problem. Here, we report a model that provides a good statistical fit to allele-frequency correlation patterns among East Asians, Australasians, Native Americans, and ancient western and northern Eurasians, together with archaic human groups. The model features a primary eastern/western bifurcation dating to at least 45,000 years ago, with Australasians nested inside the eastern clade, and a parsimonious set of admixture events. While our results still represent a simplified picture, they provide a useful summary of deep Eurasian population history that can serve as a null model for future studies and a baseline for further discoveries.
Keywords: population genetics, human history, admixture graph
Introduction
Modern humans are widely believed to have evolved first in sub-Saharan Africa and then to have spread at least once, and possibly several times, into Asia and Europe (Groucutt et al. 2015). The earliest strong archaeological evidence of modern human occupation outside of Africa comes from the Levant, approximately 100 thousand years ago (kya), but these and other Middle Paleolithic remains from Southwest (Armitage et al. 2011) and South Asia (Blinkhorn et al. 2013) may or may not be from groups related to present-day Eurasians. By contrast, the modern human remains that appear across most of the broad areas of Eurasia during the Late Paleolithic (∼40–50 kya) can plausibly be interpreted as continuous with present-day populations (Groucutt et al. 2015).
Numerous studies have addressed the genetic history of the modern human expansion out of Africa. Many of the first insights were provided by single-locus systems, and while we generally adopt an autosomal perspective here, active research continues with mitochondrial DNA (Posth et al. 2016), Y chromosomes (Hallast et al. 2015; Karmin et al. 2015), and microbial pathogens (e.g., H. pylori; Montano et al. 2015). Multi-locus analyses have shown that, to a first approximation, modern humans in Eurasia can be divided into what we will refer to as eastern and western clades. The former includes present-day East Asians and had differentiated as early as the ∼40 kya Tianyuan individual (Fu et al. 2013), while early members of the latter include ancient European hunter-gatherers (Lazaridis et al. 2014; Seguin-Orlando et al. 2014; Fu et al. 2016) and the ancient northern Eurasian Mal’ta 1 (MA1, a ∼24 kya Upper Paleolithic individual from south-central Siberia) (Raghavan et al. 2014). More recent (Neolithic and later) western Eurasians, such as Europeans, are mostly descended from the western clade but with an additional component of “Basal Eurasian” ancestry (via the Near East) splitting more deeply than any other known non-African lineage (Lazaridis et al. 2014, 2016). The timing of the eastern/western split is uncertain, but several papers (Gutenkunst et al. 2009; Laval et al. 2010; Gravel et al. 2011) have used present-day European and East Asian populations to infer dates of initial separation of 40–45 kya (adjusted for a mutation rate of per year; Scally 2016). Interestingly, two early modern Eurasians (Ust’-Ishim (Fu et al. 2014), from ∼45 kya in western Siberia, and Oase 1 (Fu et al. 2015), from ∼40 kya in Romania) have been found that share little or no ancestry with either clade, unlike any known present-day population. After the initial modern human colonization of Eurasia, later migrations led to the formation of major populations with mixed ancestry from both clades, notably including South Asians (Reich et al. 2009) and Native Americans (Raghavan et al. 2014). It has also been proposed that ancestors of Europeans and East Asians experienced continuing gene flow after the initial eastern/western separation (Gutenkunst et al. 2009; Laval et al. 2010; Gravel et al. 2011; Fu et al. 2016).
It has been argued by some authors that this model of a primary split between eastern and western Eurasians is incorrect for certain present-day populations from Oceania and Southeast Asia. Under the “southern route hypothesis,” Australians, New Guineans, and perhaps Southeast Asian “Negrito” populations are descended in part from an early out-of-Africa dispersal through southern Asia, with this component of ancestry splitting prior to the common ancestor of other Eurasians (Lahr and Foley 1994; Rasmussen et al. 2011; Reyes-Centeno et al. 2014). Recently, several studies based on whole-genome sequence data have presented more refined models of Australasian ancestry (Mondal et al. 2016; Mallick et al. 2016; Malaspinas et al. 2016; Pagani et al. 2016); in particular, we proposed a historical model that fit better without any such deep-source ancestry than with it (Mallick et al. 2016).
Here, we study the deep relationships of most non-African continental groups by building a unified historical model based on patterns of allele frequency correlations due to genetic drift. Where available, we incorporate relevant ancient individuals alongside present-day populations. We also account for gene flow from archaic humans (Green et al. 2010; Reich et al. 2010), preventing potential confounding in the relationships among modern human lineages. While we acknowledge that our final model will not represent the complete truth, it represents (to our knowledge) the largest and most detailed such effort to date and can provide a baseline for future work.
Results
Overview of Best-Fitting Admixture Graph
As a starting point for our model, we used the set of populations (minus Dai) from an admixture graph formulated in Mallick et al. (2016): Chimpanzee, Altai Neanderthal (Prüfer et al. 2014), Denisova (Meyer et al. 2012), Dinka, Kostenki 14 (K14, a ∼37 kya Upper Paleolithic individual from Russia belonging to the western Eurasian clade) (Seguin-Orlando et al. 2014), New Guinea, Australia, Onge (an indigenous population from the Andaman Islands), and Ami (aboriginal Taiwanese, representing East Asians). The elements of the model in Mallick et al. (2016) were mostly relatively straightforward, with no admixture events aside from those involving archaic humans. The primary finding of interest was that the Australasians (plus Onge) fit best as a clade with East Asians; incorporating a deeper “southern route” ancestry component did not improve the fit.
Here, for our primary results, we used single nucleotide polymorphisms (SNPs) genotyped on the Affymetrix Human Origins array, which gave us access to larger sample sizes and additional populations beyond those that are currently available with whole-genome sequencing data. With the nine populations listed above (here New Guinea Highlanders {Reich et al. 2011} rather than the SGDP Papuan {Mallick et al. 2016}), we replicated the earlier results: The graph fit well with the same topology, correctly predicting all f-statistic relationships to within (standard errors estimated by block jackknife; see Materials and Methods).
To this preliminary model, we added four additional populations: MA1, Ust’-Ishim, Mamanwa (a “Negrito” population from the Philippines), and Suruí (an indigenous population from Brazil). Mamanwa (with one component related to Australasians and the other to East Asians; Reich et al. 2011; Lipson et al. 2014) and Suruí (with one component related to MA1 and the other to East Asians; Raghavan et al. 2014) immediately had clear signatures of admixture, while Ust’-Ishim required excess Neanderthal ancestry (Fu et al. 2014, 2016). After adding these admixtures and optimizing the topology, the resulting model had 42 statistics that differed by at least two standard errors from their fitted values (max Z = 2.68). Many of these were highly correlated, for example with New Guinea substituted for Australia or with either Dinka or Chimp as an outgroup. We identified four residuals that were independent and reflected quartets of populations forming approximately unadmixed subtrees in the fitted model (to aid in interpretation; absolute value < 0.1 in 1000 times drift units): f4(MA1, Ami; Denisova, Dinka) (residual Z = 2.03), f4(Suruí, Altai; New Guinea, Australia) (Z = 2.07), f4(Ust’-Ishim, Onge; Australia, Ami) (Z = 2.17), and f4(MA1, K14; Ami, Ust’-Ishim) (Z = 2.31). The first and last of these motivated us to add additional admixture events to the model (see “Archaic humans” and “Western and northern Eurasians”); the third is related to a residual signal we discuss in more detail below (“Replication with SGDP data”); and the second could be connected to deeply splitting ancestry in Amazonian populations (Skoglund et al. 2015) (see “Native Americans”) but was not addressed here.
The full best-fitting admixture graph is shown in figure 1. It is based on a total of ∼123k SNPs and includes 13 leaf nodes (i.e., directly sampled groups): Seven present-day populations, three ancient modern humans, two archaic humans, and Chimp. There are nine admixture events, of which six are from archaic humans (although these likely do not all represent separate historical events, as discussed below). All f-statistics fit to within , including all pairwise f2-statistics to within (fig. 2); inferred mixture proportions are indicated in figure 1 and can also be found in table 1. In what follows, we describe the features of the graph in more detail.
Fig. 1.

Final best-fitting graph model. Colors of filled nodes (sampled populations and selected internal split points) and edge arrows correspond to subsets of the graph: green, Chimp and archaic; yellow, African and basal non-African; dark blue, eastern clade; light blue, Australasian sub-clade; red, western clade; purple, northern sub-clade. Tree edges (solid lines) are labeled with branch lengths in 1000 times drift units (rounded to the nearest integer value), while admixtures (dotted lines) are shown with their inferred proportions. The three drift lengths surrounding an admixture event (immediately preceding each mixing population and immediately following the admixed population) cannot be solved for individually in our framework and instead form a single compound parameter (Lipson et al. 2013); we omit the first two and report the total drift on the edge following the admixture. The terminal drifts leading to ancient individuals are inflated as a result of a combination of single-individual populations, lower coverage, and/or haploid genotype calls.
Fig. 2.

Pairwise f2 residuals in the final model, in units of Z-score (fitted minus observed divided by standard error).
Table 1.
Inferred Mixture Proportions with Alternative Data Sets.
| Admixture event | HO (%) | SGDP (%) | SGDP (tv) (%) |
|---|---|---|---|
| Neanderthal to non-Africans | 2.6 | 4.3 | 4.2 |
| Neanderthal to Ust’-Ishim | 1.5 | 1.9 | 1.6 |
| Neanderthal to K14 | 1.5 | 1.2 | 0.9 |
| Denisova to Australasians | 3.5 | 3.0 | 3.1 |
| Denisova to MA1 | 1.2 | 0.5 | 0.6 |
| Western Eurasian to Suruí | 26.6 | 28.2 | 25.2 |
| East Asian to Mamanwa | 68.5 | – | – |
| Eastern Eurasian to MA1 | 17.4 | 10.8 | 16.2 |
NOTE.—Inferred mixture proportions in the admixture graph. Final ancestry proportions in leaf-node populations may be lower if diluted by a second admixture event. The three sets of values are for the primary (Human Origins) data, SGDP, and SGDP restricted to transversions. Exact archaic admixture parameters should be treated with a degree of caution (see Materials and Methods, “Possible methodological caveats”).
Archaic Humans
The top portion of the graph contains Altai, Denisova, and their ancestors. We included one of two previously inferred admixtures into Denisova (Prüfer et al. 2014): “unknown archaic” ancestry from a source splitting deeper than the common ancestor of the Neanderthal/Denisova clade with modern humans. We do not have enough constraint to solve for the precise mixture proportion (Materials and Methods) and thus prespecified it at 3%, within the range of the initial estimate. Allowing this proportion to vary freely only slightly improved the log-likelihood score of the model, whereas removing the admixture decreased the log-likelihood by about 5.9 (P < 0.005 by likelihood ratio test {LRT}; see Materials and Methods). We also considered the previously reported Neanderthal admixture into Denisova, but our model does not provide sufficient constraint to observe this signal, so for the sake of parsimony we omitted it from the final graph.
Our model also includes several instances of gene flow from archaic to modern humans. All present-day non-African populations in the graph fit well with a single, shared Neanderthal introgression event. Consistent with previous results (Fu et al. 2014, 2016; Seguin-Orlando et al. 2014), Ust’-Ishim and K14 require extra Neanderthal ancestry, with inferred proportions of 1.5% each; we use the same mixing Neanderthal for all events. We note that while there is evidence that present-day Europeans and related groups have less Neanderthal ancestry than East Asians (Wall et al. 2013; Sankararaman et al. 2014; Vernot and Akey 2014; Lazaridis et al. 2016), no such populations are present in our model (although see “Western and northern Eurasians” below). As an overall trend, we recapitulate the finding of increased archaic ancestry in ancient individuals (Fu et al. 2016), which could be evidence of purifying selection against introgressed segments over time (Harris and Nielsen 2016; Juric et al. 2016). Thus, while the graph contains separate Neanderthal gene flow events for Ust’-Ishim and K14, these do not necessarily reflect additional historical episodes of admixture.
In addition to the previously documented Denisova-related introgression into Australasians (here 3.5% into the common ancestor of New Guinea, Australia, and Mamanwa), we find suggestive new evidence for Denisova-related ancestry in MA1, which we believe may explain the preliminary residual statistic f4(MA1, Ami; Denisova, Dinka) mentioned above. A consistent signal of excess allele sharing between MA1 and archaic humans can be observed when using any of Denisova, Altai Neanderthal, or the Vindija and Mezmaiskaya Neanderthals (Green et al. 2010) (table 2). We also used an ancient ingroup in place of Ami to ensure that this pattern does not reflect an ancient DNA artifact (table 2, bottom half). While the differences between the rows in table 2 are not statistically significant, MA1 appears to share the most drift with Denisova; the excess shared drift with Neanderthals would also be expected in a scenario of Denisova-related introgression on the basis of the sister relationship between Neanderthals and Denisova.
Table 2.
Relationship between MA1 and Archaic Humans.
| Pop X | Pop Y | f4(MA1, X; Y, Dinka) | Z-score |
|---|---|---|---|
| Ami | Denisova | 1.15 | 2.19 |
| Ami | Altai | 0.84 | 1.48 |
| Ami | Vindija | 0.86 | 1.60 |
| Ami | Mezmaiskaya | 1.76 | 1.39 |
| WHG | Denisova | 1.52 | 2.60 |
| WHG | Altai | 0.85 | 1.42 |
NOTE.—Statistics of the form f4(MA1, X; Y, Dinka) for a comparison population X and archaic humans Y, along with Z-scores for difference from zero, computed on all available SNPs from panels 4 and 5 of the Human Origins array. WHG is defined as a combined Mesolithic western hunter-gatherer population consisting of the ∼8 kya Loschbour (Lazaridis et al. 2014) and La Braña 1 (Olalde et al. 2014) individuals. A larger positive value indicates that MA1 shares more alleles with population Y than does X. Because Ami and WHG have very similar levels of archaic ancestry (f4(Ami, WHG; Altai, Dinka) ), we would expect similar values with X = Ami or WHG.
Motivated by these results, we tested the effects of including extra archaic ancestry in MA1 in our full graph model. Adding Denisova-related admixture resulted in a significant log-likelihood score improvement of 6.3 (P < 0.002), whereas instead allowing additional Neanderthal gene flow improved the score by 2.3 (P = 0.10). (We note that while the inferred best-fitting source for the Denisova-related introgression was closer to the Denisova sample than for Australasians, the difference in fit quality was negligible, so for the sake of parsimony we used the same source for both events.) Given the consistent pattern of greater Neanderthal ancestry in ancient samples, however, a model with excess Neanderthal ancestry would perhaps be a more reasonable null hypothesis. Using such a model as a starting point, adding Denisova-related admixture improved the score by a marginally significant 4.0 (P < 0.02; when including Denisova-related admixture, the graph fit best without any extra Neanderthal gene flow). In our final model, we therefore (tentatively) included Denisova-related (but not excess Neanderthal) gene flow into MA1, with an inferred mixture proportion of 1.2%, or 1.0% Denisova-related ancestry (95% confidence interval 0.4–1.6%; see Materials and Methods) in MA1 after dilution by eastern Eurasian gene flow (while we specified the Denisova-related admixture to be older, exchanging the order did not affect the quality of fit). Placing the Denisova-related admixture in the deeper northern Eurasian lineage shared with Native Americans made the score slightly worse, so in the absence of any evidence for shared Denisova-related ancestry, we retained the mixture only into MA1. We also experimented with allowing Denisova-related ancestry in East Asians but did not find any improvement in the fit, although we would not have power to detect a very small contribution as previously inferred (Prüfer et al. 2014; Sankararaman et al. 2016).
Asian and Australasian Populations
Consistent with previous results obtained with a simpler admixture graph in Mallick et al. (2016), New Guinea and Australia fit well as sister groups, with their majority ancestry component forming a clade with East Asians (with respect to western Eurasians). Onge fit as a near-trifurcation with the Australasian and East Asian lineages, while Mamanwa are inferred to have three ancestry components: One branching deeply (but unambiguously) from the Australasian lineage (prior to the split between New Guinea and Australia); one East Asian-related (interpreted as Austronesian admixture); and one from Denisova. The Denisova-related introgression in Mamanwa is shared with New Guinea and Australia and then diluted ∼3× by the Austronesian admixture (here 68.5%, when compared with 73% in Reich et al. (2011) and 50–60% in a simpler model in Lipson et al. (2014)). In a previous study (Reich et al. 2011), Australia and New Guinea were modeled as having about half of their ancestry from each of two components: One forming a trifurcation with Onge and East Asians, and the other splitting more recently from the Onge lineage. Here, we obtain a satisfactory fit without this admixture, and while we cannot rule it out entirely, we do not have strong evidence for rejecting our simpler model. We also note that the previous model, by virtue of its different topology, included relatively more Denisova-related ancestry in Mamanwa (∼50% as much as in Australia), although both versions appear to fit the data satisfactorily.
We also performed two additional analyses involving minimal modeling assumptions to test for possible southern route ancestry in Australasians. First, we used a method that leverages a large set of f4-statistics from different outgroup populations in elucidating admixture in a population of interest (Haak et al. 2015). Given a population Test, we plot f4-statistics f4(Test, Ref1; Oi, Oj) against f4(Test, Ref2; Oi, Oj) for references Ref1 and Ref2 and all pairs of outgroups from a set O1, O2,…, Ok. If Test is well modeled as a two-way admixture of populations related to Ref1 and Ref2 in proportions α and , then f4(Test, Ref1; Oi, Oj) = (Ref2, Ref1; Oi, Oj), and f4(Test, Ref2; Oi, Oj) = (Ref1, Ref2; Oi, Oj) = (Ref2, Ref1; Oi, Oj). Thus, the points should show a negative correlation, where the slope is informative about the mixture proportions (Haak et al. 2015).
We tested New Guinea as a two-way mixture between an East Asian-related population (Ref1 = Ami) and Denisova (Ref2), using outgroups Chimp, Altai, Dinka, Ust’-Ishim, and K14. The negative correlation is very strong (fig. 3), with no points in the plot indicating a significant violation of a New Guinea/East Asian clade. We note that if a deep-lineage component were present in New Guinea, we would expect to detect it via this set of outgroups, as it would push the points defined by (Oi, Oj) for Oi = Dinka and Oj = Ust’-Ishim or K14 off the line.
Fig. 3.

Plot of f4-statistics f4(New Guinea, Ami; Oi, Oj) against f4(New Guinea, Denisova; Oi, Oj) for all pairs of outgroup populations O from the set consisting of Chimp, Altai, Dinka, Ust’-Ishim, and K14. The R2 value for the best-fitting line through the origin is shown. The negative correlation implies that New Guinea can be modeled as a mixture of populations related to Ami and Denisova, with the slope informative about the relative proportions (as shown). Standard errors are approximately 0.0005 along the x-axis and 0.001 along the y-axis.
We then applied qpWave (Reich et al. 2012) to a larger set of test populations simultaneously to test formally for evidence of multiple waves of admixture. Using the same outgroups plus Denisova (right pop list: Chimp, Altai, Denisova, Dinka, Ust’-Ishim, K14), we computed how many ancestry components are necessary to relate the following set of (left) test populations: Ami, Dai, Kinh, Han, Bougainville, New Guinea Highlanders, HGDP Papuan, Australian, Onge, and Mamanwa. We find that this set is consistent with just two ancestral components (rank 1 tail P = 0.27), where the loadings appear to be driven primarily by the gradient of Denisova-related ancestry, as expected. As above, this test can only distinguish components that differ relative to the outgroups, but we would expect to see a signal of any substantial ancestry from a source diverging prior to the eastern/western Eurasian split.
Western and Northern Eurasians
We used the K14 and MA1 individuals to capture the roots of two major western Eurasian lineages (a western clade and a northern/eastern clade, respectively; Raghavan et al. 2014; Seguin-Orlando et al. 2014; Haak et al. 2015). Recent focused studies of later European prehistory have developed detailed models involving numerous admixture events (Lazaridis et al. 2014; Seguin-Orlando et al. 2014; Haak et al. 2015; Fu et al. 2016); as a result of this complexity, we deemed it beyond the scope of the present work to include present-day western Eurasians (although we address Europeans below).
In our model, K14 fits well as unadmixed (aside from archaic introgression), but MA1 receives, in addition to its archaic admixture, a component of eastern Eurasian ancestry. The latter gene flow explains the preliminary residual f4(MA1, K14; Ami, Ust’-Ishim), which is of a similar form to several other relatively poorly fitting statistics from our initial graph, for example f4(MA1, K14; Ami, New Guinea) = 2.00 (fitted 0.08; Z = 2.68) and f4(MA1, Ust’-Ishim; Ami, New Guinea) = 1.73 (fitted 0.08; Z = 2.49). We added this admixture into our model with its best-fitting source position (near the root of the East Asian lineage) and mixture proportion (17.4% East Asian-related ancestry, 95% CI 7.7–27.4%). The graph score improved by 7.0 (P < 0.001), indicating a significant improvement in the fit. We also attempted to fit the full graph with west-to-east gene flow instead, and the overall score was significantly worse (log-likelihood difference of 4.6 with the same number of free parameters).
Similar statistics (D(MA1, K14; Han, Mbuti) with Z = 5.4 and D(Loschbour, K14; Han, Mbuti) with Z = 5.3) were reported in the initial genetic analysis of K14 (Seguin-Orlando et al. 2014) and were interpreted there as evidence that K14 harbored Basal Eurasian ancestry. However, it has been shown in an analysis of a larger set of pre-Neolithic Europeans (Fu et al. 2016) that these signals in fact appear to reflect shared drift between a subset of western Eurasian hunter-gatherers (including MA1 and Mesolithic Europeans such as Loschbour) and East Asians. This is particularly evident from our primary statistic f4(MA1, K14; Ami, Ust’-Ishim): Even if K14 did have a component of deeply diverging ancestry, it would not share extra drift with Ust’-Ishim (likewise, this statistic is essentially unaffected by excess archaic ancestry in K14).
To support our inference of the directionality of gene flow between eastern Eurasians and MA1 (which was not addressed in Fu et al. 2016), we compared the two statistics: (1) f4(MA1, K14; Ami, Ust’-Ishim) = 1.89 (Z = 2.76) and (2) f4(Ust’-Ishim, MA1; Onge, Ami) = 0.23 (Z = 0.52) (computed on all available Human Origins SNPs). In figure 4, we show alternative models in which the flow is east to west, with MA1 admixed (A), or west to east, with Ami admixed (B). The statistics (1) and (2) have expected values equal to a branch length (red for (1) and black for (2)) times the mixture proportion α. As can be seen, the models are distinguishable by which statistic has the greater magnitude (as observed independently in more generality by Pease and Hahn (2015)). In truth, the admixture may have been complex and bidirectional, but the fact that the observed value of (1) is significantly larger in magnitude than (2) (Z = 2.29 for the difference via block jackknife) argues for east-to-west (fig. 4A) as the primary direction, with MA1 admixed. The same pattern is observed with other outgroups in place of Ust’-Ishim (, 2.74, 1.84, 2.90 for the analogous difference using Dinka, Altai, Denisova, and Chimp, respectively). We also repeated the computation with other western Eurasian populations in place of MA1 and found the same signal of eastern Eurasian relatedness, including the same preferred directionality, in WHG (defined as in table 2; Z = 2.04 for the difference), Caucasus hunter-gatherers (CHG; Jones et al. 2015; Z = 1.77), and Afontova Gora 3 (AG3, a ∼17 kya individual from Siberia closely related to MA1; Fu et al. 2016; Z = 2.17). We note that a recent study (Lazaridis et al. 2016) found a cline of MA1-relatedness among a large number of present-day eastern Eurasian populations and argued for admixture from west to east instead; while the present analysis supports the other direction, an important subject for future work will be to reconcile these results.
Fig. 4.
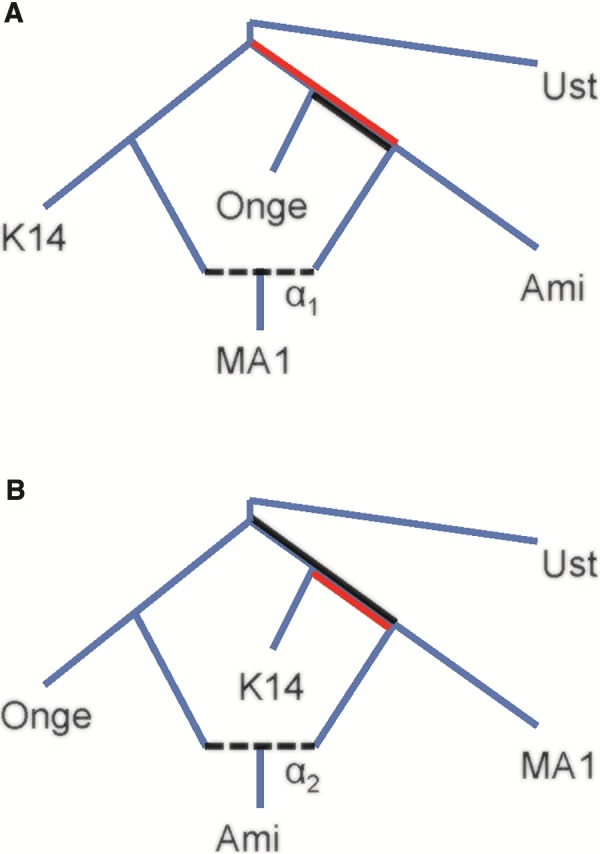
Admixture graph schematics representing alternative historical scenarios to explain shared drift between MA1 and East Asians. (A) MA1 is admixed, with East Asian-related ancestry. (B) Ami is admixed, with MA1-related ancestry. Other relationships are assumed as shown (the position of the root is arbitrary). The expected values of the statistics (1) f4(MA1, K14; Ami, Ust’-Ishim) and (2) f4(Ust’-Ishim, MA1; Onge, Ami) are equal to a branch length times a mixture proportion: red for (1) and black for (2) [times α1 in (A) and α2 in (B)].
While we did not carefully model present-day Europeans in our main admixture graph, we did build an extended graph with French added (25 individuals). A good fit was obtained with four ancestry components, related to western (K14), northern (near the base of the MA1 lineage), and eastern (specified as the same source as for MA1) Eurasians, plus Basal Eurasian (specified without Neanderthal introgression; Lazaridis et al. 2016). The inferred proportions were 27.7%, 34.9%, 23.2%, and 14.2%, respectively, with essentially no change in the list of residuals. We note that these sources do not represent the proximal ancestral populations of present-day Europeans (Lazaridis et al. 2014; Haak et al. 2015), and this fit also may not be the optimal one, but it does provide a sense of the relationships of Europeans to the major lineages defined in our model.
Lastly, we also briefly studied two other ancient European lineages. First, we built a version of our model with WHG in place of MA1 and found that it fit in a similar fashion (majority component of WHG’s ancestry as a sister group to K14, plus eastern Eurasian gene flow). Second, we fit an expanded graph with MA1, K14, and the early modern human Oase 1 (Fu et al. 2015). Because of possible contamination, we used the published damage-restricted data, which reduced the set of SNPs with coverage in all populations to ∼28k. The inferred graph was similar overall, with modest changes due to the smaller set of SNPs. Oase 1 was inferred to diverge from the western Eurasian (K14) lineage, slightly later than Ust’-Ishim (shared drift 1.6), but still close to the split of the eastern and western clades. As shown in Fu et al. (2015), Oase 1 has a significant excess of Neanderthal ancestry, which we inferred at 8.3% in the extended model.
Native Americans
We included Suruí, from Brazil, a Native American population without recent European admixture. As previously demonstrated for Native Americans generally (Raghavan et al. 2014), we found that they fit well in the model as a mixture of components related to East Asians (73%) and MA1 (27%). This proportion of western Eurasian ancestry is lower than previously inferred (∼40% in Raghavan et al. 2014), which may be because we are separately modeling East Asian-related gene flow into MA1. It has also been shown that Suruí harbor a few percent ancestry from a “Population Y” related to Onge and Australasians (Skoglund et al. 2015). In the context of our model, with only one Native American population present, this admixture should only have a minor effect, although we do see hints of such a signal, as mentioned above.
Early Out-of-Africa Split Points
After the divergence of Dinka from non-Africans, the next split point on the modern human lineage in our model is that between the major eastern and western clades (the node labeled “Non-African”—although we note that the split point of Basal Eurasian would be deeper.) This split is soon followed on the western Eurasian branch by the split between K14 and Ust’-Ishim (i.e., their respective modern-human ancestry components). The original Ust’-Ishim analysis (Fu et al. 2014) inferred a near-trifurcation at this point, and we wished to test whether K14 (and other western Eurasians) and Ust’-Ishim form a statistically supported clade. In fact, while the best-fitting position for Ust’-Ishim is on the western lineage (0.6 shared drift), the inferred 95% confidence interval for this point overlaps the eastern/western split (standard error 0.4 for the Ust’-Ishim split position), so that we cannot confidently resolve the branching order. We therefore continue to regard this cluster as approximately a trifurcation; while we show Ust’-Ishim at its best-fitting split point in figure 1, we color-code it as a basal non-African rather than a member of the western clade.
We also investigated another near-trifurcation, near the top of the eastern Eurasian clade, where the East Asian, Onge, and Australasian lineages are inferred to diverge in a short span. Here, the best-fitting arrangement features Onge and East Asians as a weak clade (), but the model reaches a second, only slightly inferior local optimum with Onge and Australasians as sister groups instead, possibly suggesting admixture between two of the three lineages. An admixture event in either Onge (between the Australasian and East Asian lineages) or Australasians (between the Onge and deep eastern Eurasian lineages) is likewise weakly significant (), but with no discriminatory power between these two scenarios. Ultimately, we chose to present the model with a trifurcation at this point because we felt it best conveyed our uncertainty: No pair of lineages clearly shares more drift, and it is likely that some admixture took place, but we cannot accurately determine which lineage or lineages were involved or constrain the exact proportions or sources.
Power to Detect Admixture Events
The set of admixture events we have included is limited by our power to detect statistically significant deviations from the proposed model. Suppose that we specify a quartet of populations as unadmixed with the topology ((A, B),(C, D)). If population A is in fact admixed with a component of ancestry related to C (in an unrooted sense), then we will observe a residual statistic f4(A, B; C, D) with expected value , where γ is the C-related mixture proportion and L is the branch length in the graph that is shared uniquely between this component and population C (Reich et al. 2009; Patterson et al. 2012). For our data set, in 1000 times drift units, standard errors on observed f4-statistics are on the order of 0.5. Thus, in order to observe a residual at Z = 2 (for example), an admixture event would have to satisfy (approximately) . We note that while γ is a fixed parameter, L depends on how close a reference C is available (and one must also have access to populations B and D with the proper topology).
We were particularly interested in the question of power to detect possible southern route ancestry in Australasians. In this case, population A would be Australasians, population B would be an East or Southeast Asian group without a southern route component, population C would be an outgroup (African, archaic, or Chimp), and population D would be a western Eurasian. The length L would measure the distance from the “Non-African” node up to the split point of the southern route source. A more thorough empirical analysis of this question in Mallick et al. (2016) concluded that any southern route component in Australasians is unlikely to comprise more than several percent. If, for example, the deeper ancestry were from a population that split halfway between the “Non-African” node and the African/non-African ancestor (), then via the inequality above, we would have power to find roughly 6% admixture or more, similar to the previous results. For a split closer to the “Non-African” node, our power would be reduced, while for a deeper split (e.g., the ∼120 kya proposed by Pagani et al. (2016)), it would be enhanced.
We also carried out two tests using our admixture graph to examine our power empirically in cases of well-known admixture. First, we studied the ∼27% MA1-related ancestry in Suruí. In order to make our available constraints similar to the hypothetical southern route case, we removed MA1, K14, and Ust’-Ishim from the model. With this reduced graph, if we model Suruí as unadmixed, we observe six residuals with Z > 3 (max 3.6), all of the form f4(Suruí, Asian; outgroup, Australasian). Thus, even without any western Eurasian references, we can easily locate the admixture signal (the relevant length L is the branch between the “East1” and “Non-African” nodes, or approximately 6 units).
Second, we conducted a similar analysis for the Denisova-related ancestry in Australasians, having removed Onge, Chimp, Altai, and Denisova from the model. In this case, we observe a residual f4(Australian, Suruí; Dinka, K14) at Z = 2.01, roughly as expected given the known parameters of a few percent introgression and (the full distance between the “Non-African” node and the African/non-African ancestor). This demonstrates our ability to detect a very small proportion of deep ancestry in Australasians without a close surrogate available, as would also be true for southern route admixture. While in this example the residual was only weakly significant, it was obtained after relaxing a number of key constraints from the full model, and even 1–2% more deep ancestry would have made for an unambiguous signal.
Replication with SGDP Data
To gain further perspective, we repeated our admixture graph analysis with full sequence data from the SGDP (Mallick et al. 2016). The set of populations we used was similar (although generally with smaller sample sizes; see Materials and Methods), with only two changes: We substituted HGDP Papuans instead of New Guinea Highlanders (which should not be substantively different) and no longer had access to data for Mamanwa. We ascertained SNPs as polymorphic among the four SGDP Mbuti individuals and merged with Chimp, Altai, Denisova, Ust’-Ishim, K14, and MA1. The resulting data set contained 1.99M SNPs overlapping all populations.
The best-fitting graph is shown in figure 5. Overall, it is very similar to our primary model, with only one change in topology (the eastern Eurasian admixture source for MA1 now splits closer to Ami) and small differences in inferred mixture proportions (table 1). The SGDP model does have two residual statistics falling more than 3 standard errors from their fitted values: f4(Ust’-Ishim, Ami; Australia, Dinka) = −4.34 (fitted value −6.69; Z = 3.52) and f4(Ust’-Ishim, Papuan; Australia, Dinka) = −24.64 (fitted value −26.83; Z = 3.02). A number of other residuals are also present with Z > 2 showing the same apparent signal of shared drift between Ust’-Ishim and Australasians (for example, with Altai or Denisova in place of Dinka); these statistics are not independent, but the overlap argues that it is indeed this pair of populations that drives the two most significant residuals.
Fig. 5.

Model fit with SGDP data. Notation is the same as in figure 1.
While this signal is intriguing, we ultimately decided not to add a new admixture event to the model to account for it. We considered the possibility either of gene flow from a population related to Ust’-Ishim into the ancestors of Australasians or vice versa. Both would create a pattern of shared drift as reflected in the residuals, but the two directions would make distinct predictions in relation to other populations. In the first scenario, we would expect the genetic affinity to Ust’-Ishim to be unique to Australasians, and this affinity should be detectable in our previous analyses. However, neither the f4 correlation nor qpWave methods showed evidence of Ust’-Ishim-related admixture in Australasians, and the signal of shared drift between Australasians and Ust’-Ishim was only marginally significant in our main graph model; while based on fewer SNPs, the corresponding statistics in fact had lower standard errors than those from the SGDP model, so the difference was not due to a lack of power. In the second scenario, with Ust’-Ishim admixed, we would expect a slightly different signature, since Ust’-Ishim would then possess a component of ancestry that, in addition to being related to Australasians, also shares excess drift with other eastern Eurasians. We do in fact observe two such (weaker) residual statistics: f4(Ust’-Ishim, K14; Onge, Dinka) = 1.37 (fitted value −0.21; Z = 2.24) and f4(Ust’-Ishim, K14; Ami, Dinka) = 1.30 (fitted value −0.21; Z = 2.07). However, when we replicated these statistics with our main data set, using all available SNPs, they were only weakly positive (f4(Ust’-Ishim, K14; X, Dinka) = 0.31, 0.62, 0.72, 1.10 for X = Onge, Ami, New Guinea, and Australia, respectively; , 1.18, 1.23, 1.18).
We also compared the quality of fit of full graph models (using the SGDP data) in which we added an extra admixture event between Ust’-Ishim and Australasians in either direction, with the hypothesis that if one of these models is correct, it should score better than the other. In both cases, the score improved by more than 11, but the two models had similar residual lists, and their log-likelihood scores only differed by ∼0.3. This would seem to indicate either that 1) the shared drift signal does not reflect a true admixture, 2) there was very evenly bidirectional gene flow, or 3) our model is not sufficiently powered to detect the true mixture source. Moreover, both graphs still had numerous other residuals involving Ust’-Ishim with Z-scores above 2 (up to Z = 2.48 with Australasians admixed and Z = 2.89 with Ust’-Ishim admixed). For the sake of comparison, using our main graph model, we applied a similar analysis to the inferred admixture event from eastern Eurasians into MA1. In that case, while the signal was relatively weak, and there could be reason to believe that the gene flow was not strictly east-to-west, we were able to show that the admixture had a preferred directionality; as noted above, a model with gene flow in the reverse direction scored 4.6 worse. (For an example of a strongly constrained admixture event, if we fit a model with gene flow from Mamanwa to Ami rather than in reverse, the score is more than 400 worse.) To draw a more precise analogy, we repeated the model-fitting without K14 in the graph, so that MA1 was the only western Eurasian population present (in the same way that Ust’-Ishim is the only representative of its lineage). Not surprisingly, this caused the signal of directionality to be attenuated, but it was still present: A model with east-to-west gene flow scored 1.2 better than the reverse.
Finally, we fit a version of our model on SGDP data but restricted to transversion polymorphisms (a total of ∼622k SNPs) to test whether there could be any effects of ancient DNA damage patterns either on these residual signals or in the model-fitting as a whole. The graph fit well overall, with the same optimized topology as in our main results (restoring the one difference present in the full-SGDP graph) and similar mixture proportions (table 1). The list of significant residuals was slightly shorter than with all SNPs, but the Ust’-Ishim-related signal was still present (although now stronger with Papuan than with Australia), with the most significant residual, f4(Ust’-Ishim, Ami; Papuan, Dinka), now at Z = 2.98. This leads us to conclude that neither this signal nor other results obtained in the full models are driven by ancient DNA damage. However, in light of our other analyses, we do not feel that we have sufficient evidence at this time to assign the Ust’-Ishim-related signal as a true admixture.
Discussion
Our proposed admixture graph provides both an integrated summary of many population relationships among diverse non-African modern human groups and a framework for testing additional hypothesis. As an example of the model’s utility, it can help to evaluate a signal previously used to argue for deeply diverged ancestry in Aboriginal Australians (Rasmussen et al. 2011): f4(Australia, Han; Yoruba, French) = 2.02 (Z = 8.23), or with related populations in our admixture graph, f4(Australia, Ami; Dinka, MA1) = 1.43 (Z = 3.01). While southern route ancestry would indeed cause these statistics to be positive, the admixture events specified in our model provide two (partial) alternative explanations, namely Denisova-related introgression into Australasians and gene flow between eastern and western Eurasians. In fact, the predicted value of f4(Australia, Ami; Dinka, MA1) in our final graph is 1.72, slightly larger than the observed value, without any southern route ancestry in Australasians. Thus, this example illustrates how fitting a large number of groups simultaneously can add context to the interpretation of observed patterns in population genetic data.
In addition to providing a synthesis of previous results, we have also proposed two new admixture events in MA1, both of which seem plausible on geographical grounds but are not overwhelmingly statistically significant and would be interesting topics for further study. One event, consisting of gene flow from an eastern Eurasian population, appears to be present as well in other later western Eurasians, which makes it unlikely that the signal could be due to contamination in MA1. We note though that this event could have involved one or more (unknown) intermediate populations rather than being direct, and we also cannot rule out a small amount of admixture in the reverse direction. The second, consisting of Denisova-related gene flow, provides intriguing evidence of a novel instance of archaic introgression, but it should be subject to additional scrutiny with more sensitive methods to confirm whether the source has been accurately inferred (as opposed to excess introgression from Neanderthal or a different archaic group).
Furthermore, we show that we can obtain a good fit to the data with no further admixture events beyond those specified in our model. In other words, even though our graph is in some ways relatively simple, with only three admixtures among the 10 modern human populations, we do not find any large residuals (to the extent that we have statistical power). This does not mean that we have identified all admixture events in the ancestry of these populations or that our graph represents the exact historical truth; rather, we propose that our model be viewed as a reasonable and relatively comprehensive starting point given currently available data, in the spirit of previous demographic null models (Schaffner et al. 2005; Gravel et al. 2011). We also have not included certain groups with known complicated histories, including present-day European and Indian populations. We attempted to add Indians to the graph but failed to obtain a satisfactory fit, which we believe was primarily due to difficulty in modeling the western Eurasian (ANI) ancestry found in all Indian groups today (in addition to eastern, “ASI” ancestry) (Reich et al. 2009).
Overall, our model supports a rapid radiation of Eurasian populations following an out-of-Africa dispersal, in line with results from uniparental markers (Karmin et al. 2015; Posth et al. 2016). Here, this pattern is reflected in the near-trifurcations among the main eastern and western Eurasian clades plus Ust’-Ishim (with Oase 1 splitting very close as well) and at the base of the eastern clade among Andamanese, Australasians, and East Asians. We note that archaeological evidence increasingly points to early eastward modern human dispersals, with suggestive remains from East and Southeast Asia (Mijares et al. 2010; Demeter et al. 2015; Liu et al. 2015) in addition to the relatively early sites confidently assigned to modern humans in Australasia (O’Connell and Allen 2015; Clarkson et al. 2015). If these finds do all represent true modern human occupation, this would change our understanding of the timing of out-of-Africa migrations, but it would not necessarily be the case that present-day Asians and Australasians are related to these first inhabitants. It is also possible that Australasians possess a few percent ancestry from an early-dispersal population which could be detectable with more sensitive genetic analyses but not with our allele-frequency-based methods (Mallick et al. 2016; Pagani et al. 2016). Finally, we caution that we are limited in our ability to infer the geographical locations and calendar dates of events in the admixture graph; our most powerful temporal constraint comes from Ust’-Ishim, whose date of ∼45 kya places the eastern/western Eurasian split no later than this time. Further analysis of ancient DNA in the context of present-day genetic variation promises to provide additional data points to refine and add detail to our understanding.
Materials and Methods
Admixture Graph Fitting Methodology
Our central approach is to summarize information about population relationships (in the form of f-statistics) within the framework of an admixture graph, a phylogenetic tree augmented with admixture events. A full description of the mathematical underpinnings of our methods can be found in Patterson et al. (2012) and Lipson et al. (2013). Briefly, a proposed admixture graph implies f2, f3, and f4-statistic values for all pairs, triples, and quadruples of populations, where we use the notation f4(A, B; C, D) interchangeably as (the expected allele frequency correlation of populations A, B, C, and D) and as the estimator of this quantity (the empirical sum of the correlation over many loci). (The other statistics can be written as f3(A; B, C) = f4(A, B; A, C) and f2(A, B) = f4(A, B; A, B).) The parameters in the graph consist of mixture proportions and branch lengths, the latter in units of genetic drift (where populations that share descent from a common ancestor will covary in their allele frequencies, measured at SNPs polymorphic across populations, as a result of shared drift). A graph model can be optimized by solving a system of equations (which are linear in terms of branch lengths) for a linearly independent set of f-statistics (e.g., all pairwise f2 values).
The results presented here are obtained via the ADMIXTUREGRAPH software (Patterson et al. 2012). ADMIXTUREGRAPH takes as input a user-defined admixture graph topology and solves for the best-fitting branch lengths and mixture proportions given the observed f-statistics. Our usual strategy for building a model in ADMIXTUREGRAPH is to start with a small, well-understood subgraph and then add populations (either unadmixed or admixed) one at a time in their best-fitting positions. This involves trying different branch points for the new population and comparing the results. If a population is unadmixed, then if it is placed in the wrong position, the fit of the model will be poorer, and the inferred split point will move as far as it can in the correct direction, constrained only by the specified topology. Thus, searching over possible branching orders allows us to find a (locally) optimal topology. If no placement provides a good fit (in the sense that the residual errors are large), then we infer the presence of an admixture event, in which case we test for the best-fitting split points of the two ancestry components. After a new population is added, the topology relating the existing populations can change, so we examine the full model fit and any inferred zero-length internal branches for possible local optimizations. When compared with an automated method such as TreeMix (Pickrell and Pritchard 2012), this procedure is more laborious, but for this application we preferred to build the graph in a more careful, supervised way.
We note that in order to constrain all the parameters of an admixture event, it is necessary to have four reference populations in the graph splitting at different points along the path connecting the mixing populations (for example, for the shared Neanderthal introgression into non-Africans, the references are Altai, Denisova, Chimp, and Dinka). With three distinct references, it is possible to detect the presence of an admixture, but the mixture proportion and one of the relevant branch lengths are confounded as a compound parameter. Also, even with four available references, if two of them have distinct but very close phylogenetic positions, or if one or more are themselves admixed, then it may be possible to infer the admixture proportions but only with relatively large uncertainty.
Our ADMIXTUREGRAPH settings are slightly different from the default. First, we use the option “outpop: NULL” rather than specifying an outgroup population in the graph in which SNPs must be polymorphic, and we also set “lambdascale: 1” in order to preserve the standard scaling of the f-statistics without an extra denominator. Second, we use the full matrix form of the objective function (with diag = 0.0001) to avoid the basis dependence of the least-squares version of the computation. There could be some concern that the empirical covariance matrix Q is unstable, but we did not see any evidence of such behavior. We also note that while we show graphs with Chimp as the root, the fitting should not depend on the root position (including whether Chimp itself or a common ancestral node is used). We tested versions of the graph with Dinka as the root, and the results were essentially identical.
Measures of Fit Quality
When evaluating the quality of fit of a graph model, we use two metrics provided by the ADMIXTUREGRAPH program. First, the program returns a list of residuals above a specified Z-score threshold. These are f-statistics for which the fitted value in the model differs from the actual value measured from the data by a significant amount (in terms of the Z-score, i.e., fitted minus observed divided by standard error) according to a block jackknife (block size 5 cM). It is difficult to assess the exact false-positive rate corresponding to a given threshold; the Z-score for an individual statistic is approximately distributed as a standard normal under the null, but we are testing many statistics simultaneously. Moreover, while the total number of linearly independent f-statistics in the graph is known (, where n is the number of populations), as is the number of free parameters (, where a is the number of admixture events, assuming no parameters are locked), the residual errors in the statistics are correlated because of shared history (for example, f4(A, B; C, Australia) and f4(A, B; C, New Guinea) are always similar). Thus, rather than conducting hypothesis tests with the residuals, we use them as a heuristic: Better-fitting models should generally have fewer significant residuals and lower Z-scores, and the most significant residuals in a given model generally point to the populations that are being the most poorly fit.
The second metric is an overall model fit score, , where f is the vector of observed f-statistics (the full linearly independent set according to the chosen basis), g is the corresponding vector of statistics according to the model, and Q is an estimated covariance matrix. Under an approximation that noise is multivariate normal distributed under the null, this score is a log-likelihood for the model. We do not use the score directly to gauge whether a given model is an adequate fit for the data, but we can compare alternative models on the basis of their scores, favoring the one with the higher log-likelihood. Specifically, we believe the score is suitable to use for LRTs. In our context, we are typically interested in the question of whether adding a new admixture event provides a significantly better fit: The more complex model will always have at least as good a score, but it will also contain two additional free parameters (the split point of the second mixing population and the mixture proportion). We perform LRTs by taking the difference in scores; twice this difference is approximately chi-squared distributed with two degrees of freedom under the null. Thus, the score must improve by ∼3 for an admixture to be significant at P = 0.05.
Lastly, we also use relative scores to generate confidence intervals for parameters in the graph. To do so, we create a one-dimensional grid of values for a given parameter (either a branch length or mixture proportion) and run the program for each value, where all parameters but that one are optimized. For example, to determine the confidence interval for a certain mixture proportion, we might compute the log-likelihood for the model where this proportion is fixed at 1%, 2%, etc. Because of the multivariate normal score approximation and the linearity of f-statistics, this procedure in general produces a normal likelihood function for the parameter of interest, from which we compute confidence intervals.
Possible Methodological Caveats
One potential weakness of our graph-fitting approach is that with such a large space of possible models, it is difficult to say for certain that our proposed graph is optimal. However, between previous knowledge, our careful stepwise construction of the graph, and the (local) optimization of the final model, we believe that our graph is a reasonable representation. Also, while we have used a principled approach to choosing the admixture events present in the model, it is certain that we have missed some that truly occurred. Partly this is a matter of power, as some residuals resulting from incorrect model specification might be too small to separate from statistical noise (and because we are attempting to fit many relationships simultaneously, it would be expected that we would find some modest residuals by chance). Finally, the results we obtain could also depend to some extent on which reference populations are included in the graph.
Another possible concern is ascertainment bias. Ideally, admixture graphs should be built with SNPs that are (a) polymorphic at the root, and (b) not subject to any ascertainment involving the populations in the graph. Since we included archaic humans, it was not strictly possible for us to satisfy these conditions. However, we used what should be almost bias-free ascertainment schemes, particularly with regard to relationships among modern humans, as all SNPs were ascertained in outgroup or near-outgroup African populations. The one set of parameters we would recommend treating with caution are the exact proportions of archaic admixture; for example, the inferred Neanderthal admixture proportion in non-Africans in the graph based on full sequence data is large (4.3%), which we suspect could be caused by condensed branch lengths in the archaic section of the tree (a combination of ascertainment effects and large drifts).
Data
For the main admixture graph, as well as other analyses (unless otherwise specified), we use autosomal data for present-day populations (Lazaridis et al. 2014) from panels 4 and 5 of the Human Origins array (SNPs ascertained as heterozygous in single San and Yoruba individuals), a total of approximately 259k SNPs. The present-day human populations have sample sizes of 3 Australian, 7 Dinka, 8 Suruí, 11 Onge, 16 Mamanwa, 19 Ami, and 19 New Guinea. For ancient individuals, we use diploid genotype calls from full genome sequences for the high-coverage samples (Altai, Denisova, and Ust’-Ishim, plus Loschbour) and majority-allele calls for most of the lower-coverage samples (K14 and MA1, plus Oase 1 and La Braña 1). The only exceptions (both of which are only used in follow-up analyses and not in the admixture graph models) are the Caucasus hunter-gatherer individuals (Jones et al. 2015) and Afontova Gora 3 (Fu et al. 2016), which have random haploid allele calls. We only use SNPs for which no populations in the graph have missing data, resulting in a total of approximately 123k SNPs for the final model. For f-statistics presented as part of additional analyses to support the admixture graph results, we use all SNPs covered by the populations in question.
We also conduct analyses on full sequence data from the SGDP (Mallick et al. 2016), for which we ascertain SNPs as polymorphic among the four Mbuti individuals. We then merge data from K14 and MA1, here utilizing single random allele calls at each locus. This genotyping approach can reduce some biases but can also potentially be more susceptible to damage or contamination; empirically, we do not see any major differences in the inferred parameters for K14 and MA1 in the two models. The final data set contains approximately 1.99M SNPs. Present-day human populations are represented by 2 individuals for Onge, Ami, and Suruí; 3 for Dinka; 4 for Australia; and 16 for Papuan.
Acknowledgments
We would like to thank Qiaomei Fu, Iosif Lazaridis, Nick Patterson, and Pontus Skoglund for technical assistance and helpful comments and suggestions. This work was supported by the National Science Foundation (HOMINID grant BCS-1032255), National Institutes of Health (NIGMS grant GM100233), and the Howard Hughes Medical Institute.
References
- Armitage SJ, Jasim SA, Marks AE, Parker AG, Usik VI, Uerpmann HP.. 2011. The southern route “out of Africa”: Evidence for an early expansion of modern humans into Arabia. Science 331(6016): 453–456. [DOI] [PubMed] [Google Scholar]
- Blinkhorn J, Achyuthan H, Petraglia M, Ditchfield P.. 2013. Middle Palaeolithic occupation in the Thar Desert during the Upper Pleistocene: The signature of a modern human exit out of Africa? Quaternary Sci Rev. 77:233–238. [Google Scholar]
- Clarkson C, Smith M, Marwick B, Fullagar R, Wallis LA, Faulkner P, Manne T, Hayes E, Roberts RG, Jacobs Z, et al. 2015. The archaeology, chronology and stratigraphy of Madjedbebe (Malakunanja II): A site in northern Australia with early occupation. J Hum Evol. 83:46–64. [DOI] [PubMed] [Google Scholar]
- Demeter F, Shackelford L, Westaway K, Duringer P, Bacon AM, Ponche JL, Wu X, Sayavongkhamdy T, Zhao JX, Barnes L, et al. 2015. Early modern humans and morphological variation in Southeast Asia: Fossil evidence from Tam Pa Ling, Laos. PLoS ONE 10(4): e0121193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Meyer M, Gao X, Stenzel U, Burbano HA, Kelso J, Pääbo S.. 2013. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci U S A. 110(6): 2223–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Li H, Moorjani P, Jay F, Slepchenko SM, Bondarev AA, Johnson PL, Aximu-Petri A, Prüfer K, de Filippo C, et al. 2014. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514(7523): 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Hajdinjak M, Moldovan O, Constantin S, Mallick S, Skoglund P, Patterson N, Rohland N, Lazaridis I, Nickel B, et al. 2015. An early modern human from Romania with a recent Neanderthal ancestor. Nature 524:216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Posth C, Hajdinjak M, Petr M, Mallick S, Fernandes D, Furtwängler A, Haak W, Meyer M, Mittnik A, et al. 2016. The genetic history of Ice Age Europe. Nature 534:200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Henn B, Gutenkunst R, Indap A, Marth G, Clark A, Yu F, Gibbs R, Bustamante C, Altshuler D, et al. 2011. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A. 108(29): 11983–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R, Krause J, Briggs A, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz M, et al. 2010. A draft sequence of the Neandertal genome. Science 328(5979): 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groucutt HS, Petraglia MD, Bailey G, Scerri EM, Parton A, Clark-Balzan L, Jennings RP, Lewis L, Blinkhorn J, Drake NA, et al. 2015. Rethinking the dispersal of Homo sapiens out of Africa. Evol Anthropol. 24(4): 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD.. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5(10): e1000695.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak W, Lazaridis I, Patterson N, Rohland N, Mallick S, Llamas B, Brandt G, Nordenfelt S, Harney E, Stewardson K, et al. 2015. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallast P, Batini C, Zadik D, Delser PM, Wetton JH, Arroyo-Pardo E, Cavalleri GL, de Knijff P, Bisol GD, Dupuy BM, et al. 2015. The Y-chromosome tree bursts into leaf: 13,000 high-confidence SNPs covering the majority of known clades. Mol Biol Evol. 32(3): 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K, Nielsen R.. 2016. The genetic cost of Neanderthal introgression. Genetics 203(2): 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ER, Gonzalez-Fortes G, Connell S, Siska V, Eriksson A, Martiniano R, McLaughlin RL, Llorente MG, Cassidy LM, Gamba C, et al. 2015. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat Commun. 6:8912.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juric I, Aeschbacher S, Coop G.. 2016. The strength of selection against Neanderthal introgression. PLoS Genet. 12(11): e1006340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmin M, Saag L, Vicente M, Sayres MAW, Järve M, Talas UG, Rootsi S, Ilumäe AM, Mägi R, Mitt M, et al. 2015. A recent bottleneck of Y chromosome diversity coincides with a global change in culture. Genome Res. 25(4): 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahr MM, Foley R.. 1994. Multiple dispersals and modern human origins. Evol Anthropol. 3(2): 48–60. [Google Scholar]
- Laval G, Patin E, Barreiro L, Quintana-Murci L.. 2010. Formulating a historical and demographic model of recent human evolution based on resequencing data from noncoding regions. PLoS ONE 5(4): e10284.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis I, Patterson N, Mittnik A, Renaud G, Mallick S, Sudmant PH, Schraiber JG, Castellano S, Kirsanow K, Economou C, et al. 2014. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513(7518): 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis I, Nadel D, Rollefson G, Merrett DC, Rohland N, Mallick S, Fernandes D, Novak M, Gamarra B, Sirak K, et al. 2016. Genomic insights into the origin of farming in the ancient Near East. Nature 536(7617): 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson M, Loh PR, Levin A, Reich D, Patterson N, Berger B.. 2013. Efficient moment-based inference of admixture parameters and sources of gene flow. Mol Biol Evol. 30(8): 1788–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson M, Loh PR, Patterson N, Moorjani P, Ko YC, Stoneking M, Berger B, Reich D.. 2014. Reconstructing Austronesian population history in Island Southeast Asia. Nat Commun. 5:4689.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Martinón-Torres M, Cai Yj, Xing S, Tong Hw, Pei Sw, Sier MJ, Wu Xh, Edwards RL, Cheng H, et al. 2015. The earliest unequivocally modern humans in southern China. Nature 334(6052): 94–98. [DOI] [PubMed] [Google Scholar]
- Malaspinas AS, Westaway MC, Muller C, Sousa VC, Lao O, Alves I, Bergström A, Athanasiadis G, Cheng JY, Crawford JE, et al. 2016. A genomic history of Aboriginal Australia. Nature 538(7624): 207–214. [DOI] [PubMed] [Google Scholar]
- Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, et al. 2016. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538(7624): 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, et al. 2012. A high-coverage genome sequence from an archaic Denisovan individual. Science 338(6104): 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijares AS, Détroit F, Piper P, Grün R, Bellwood P, Aubert M, Champion G, Cuevas N, De Leon A, Dizon E.. 2010. New evidence for a 67,000-year-old human presence at Callao Cave, Luzon, Philippines. J Hum Evol. 59(1): 123–132. [DOI] [PubMed] [Google Scholar]
- Mondal M, Casals F, Xu T, Dall’Olio GM, Pybus M, Netea MG, Comas D, Laayouni H, Li Q, Majumder PP, et al. 2016. Genomic analysis of Andamanese provides insights into ancient human migration into Asia and adaptation. Nat Genet. 48(9): 1066–1070. [DOI] [PubMed] [Google Scholar]
- Montano V, Didelot X, Foll M, Linz B, Reinhardt R, Suerbaum S, Moodley Y, Jensen JD.. 2015. Worldwide population structure, long term demography, and local adaptation of Helicobacter pylori. Genetics 200:947–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J, Allen J.. 2015. The process, biotic impact, and global implications of the human colonization of Sahul about 47,000 years ago. J Archaeol Sci. 56:73–84. [Google Scholar]
- Olalde I, Allentoft ME, Sánchez-Quinto F, Santpere G, Chiang CW, DeGiorgio M, Prado-Martinez J, Rodríguez JA, Rasmussen S, Quilez J, et al. 2014. Derived immune and ancestral pigmentation alleles in a 7,000-year-old Mesolithic European. Nature 507(7491): 225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani L, Lawson DJ, Jagoda E, Mörseburg A, Eriksson A, Mitt M, Clemente F, Hudjashov G, DeGiorgio M, Saag L, et al. 2016. Genomic analyses inform on migration events during the peopling of Eurasia. Nature 538(7624): 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D.. 2012. Ancient admixture in human history. Genetics 192(3): 1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease JB, Hahn MW.. 2015. Detection and polarization of introgression in a five-taxon phylogeny. Syst Biol. 64(4): 651–662. [DOI] [PubMed] [Google Scholar]
- Pickrell J, Pritchard J.. 2012. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8(11): e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posth C, Renaud G, Mittnik A, Drucker DG, Rougier H, Cupillard C, Valentin F, Thevenet C, Furtwängler A, Wißing C, et al. 2016. Pleistocene mitochondrial genomes suggest a single major dispersal of non-Africans and a Late Glacial population turnover in Europe. Curr Biol. 26(6): 827–833. [DOI] [PubMed] [Google Scholar]
- Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505(7481): 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan M, Skoglund P, Graf KE, Metspalu M, Albrechtsen A, Moltke I, Rasmussen S, Stafford TW Jr, Orlando L, Metspalu E, et al. 2014. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature 505(7481): 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Guo X, Wang Y, Lohmueller KE, Rasmussen S, Albrechtsen A, Skotte L, Lindgreen S, Metspalu M, Jombart T, et al. 2011. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science 334(6052): 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Thangaraj K, Patterson N, Price A, Singh L.. 2009. Reconstructing Indian population history. Nature 461(7263): 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Green R, Kircher M, Krause J, Patterson N, Durand E, Viola B, Briggs A, Stenzel U, Johnson P, et al. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468(7327): 1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Patterson N, Kircher M, Delfin F, Nandineni M, Pugach I, Ko A, Ko Y, Jinam T, Phipps M, et al. 2011. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet. 89(4): 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Patterson N, Campbell D, Tandon A, Mazieres S, Ray N, Parra MV, Rojas W, Duque C, Mesa N, et al. 2012. Reconstructing Native American population history. Nature 488(7411): 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Centeno H, Ghirotto S, Détroit F, Grimaud-Hervé D, Barbujani G, Harvati K.. 2014. Genomic and cranial phenotype data support multiple modern human dispersals from Africa and a southern route into Asia. Proc Natl Acad Sci U S A. 111(20): 7248–7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, Patterson N, Reich D.. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507(7492): 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Patterson N, Reich D.. 2016. The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr Biol. 26(9): 1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A. 2016. The mutation rate in human evolution and demographic inference. Curr Opin Gen Dev. 41:36–43. [DOI] [PubMed] [Google Scholar]
- Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D.. 2005. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 15(11): 1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguin-Orlando A, Korneliussen TS, Sikora M, Malaspinas AS, Manica A, Moltke I, Albrechtsen A, Ko A, Margaryan A, Moiseyev V, et al. 2014. Genomic structure in Europeans dating back at least 36,200 years. Science 346(6213): 1113–1118. [DOI] [PubMed] [Google Scholar]
- Skoglund P, Mallick S, Bortolini MC, Chennagiri N, Hünemeier T, Petzl-Erler ML, Salzano FM, Patterson N, Reich D.. 2015. Genetic evidence for two founding populations of the Americas. Nature 525(7567): 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernot B, Akey JM.. 2014. Resurrecting surviving Neandertal lineages from modern human genomes. Science 343(6174): 1017–1021. [DOI] [PubMed] [Google Scholar]
- Wall JD, Yang MA, Jay F, Kim SK, Durand EY, Stevison LS, Gignoux C, Woerner A, Hammer MF, Slatkin M.. 2013. Higher levels of Neanderthal ancestry in East Asians than in Europeans. Genetics 194(1): 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]