

Abstract
Although discriminating self from nonself is a cardinal animal trait, metazoan allorecognition genes do not appear to be homologous. Here, we characterize the Aggregation Factor (AF) gene family, which encodes putative allorecognition factors in the demosponge Amphimedon queenslandica, and trace its evolution across 24 sponge (Porifera) species. The AF locus in Amphimedon is comprised of a cluster of five similar genes that encode Calx-beta and Von Willebrand domains and a newly defined Wreath domain, and are highly polymorphic. Further AF variance appears to be generated through individualistic patterns of RNA editing. The AF gene family varies between poriferans, with protein sequences and domains diagnostic of the AF family being present in Amphimedon and other demosponges, but absent from other sponge classes. Within the demosponges, AFs vary widely with no two species having the same AF repertoire or domain organization. The evolution of AFs suggests that their diversification occurs via high allelism, and the continual and rapid gain, loss and shuffling of domains over evolutionary time. Given the marked differences in metazoan allorecognition genes, we propose the rapid evolution of AFs in sponges provides a model for understanding the extensive diversification of self–nonself recognition systems in the animal kingdom.
Keywords: aggregation factor, intron phase, allorecognition, polymorphism, Porifera, RNA editing
Introduction
Self–nonself recognition is central to the multicellular condition, allowing individuals to avoid invasion and parasitism from conspecific neighbors and other organisms. Sophisticated allorecognition systems capable of discriminating between single individuals within a species are found in a wide range of metazoans. Given the apparent conservation of this process, the genes underlying allorecognition should share a common origin, as is the case with other “essential” metazoan genes, such as those employed during development (Srivastava et al. 2010). However, allorecognition genes show no evidence of homology or conservation between invertebrate animals in which they have been best studied, the colonial ascidian Botryllus schlosseri (Scofield et al. 1982; Rinkevich et al. 1995; De Tomaso et al. 2005; Nyholm et al. 2006; McKitrick and De Tomaso 2010; Nydam et al. 2013a, 2013b; Voskoboynik et al. 2013) and the cnidarian Hydractinia symbiolongicarpus (Mokady and Buss 1996; Cadavid et al. 2004; Powell et al. 2007, 2011; Nicotra et al. 2009; Rosa et al. 2010; Gloria-Soria et al. 2012; Karadge et al. 2015). The lack of similarity between such systems supports either multiple independent origins or rapid evolution of animal allorecognition genes. However, as the majority of allorecognition research to date has focused on a small number of representative species with apparently non-homologous allorecognition systems, little is known about the evolutionary processes by which allorecognition novelty is produced.
Here, we explore allorecognition in poriferans or sponges, one of the oldest surviving phyletic lineages (Erwin et al. 2011). Sponges discriminate between self and nonself by allorecognition, with tissue graft acceptance in all analysed species restricted to grafts involving pieces from a single sponge (Moscona 1968; Hildemann et al. 1979; Smith and Hildemann 1986; Fernàndez-Busquets and Burger 1997; Gauthier and Degnan 2008). As first demonstrated by Wilson in 1907, sponges can be dissociated to the cellular level and allowed to reaggregate, a process which occurs with species specificity (Wilson 1907; Humphreys et al. 1960). An extracellular product named “aggregation factor” (AF) is responsible for this reaggregation (Moscona 1968; Müller and Zahn 1973). These extracellular proteoglycans (Henkart et al. 1973) have been characterized structurally and biochemically in multiple sponge species. AFs appear to exist in either a linear form, similar to a classical proteoglycan, or a circular “sunburst”-like form that is currently unknown outside the sponges (Fernàndez-Busquets and Burger 2003) (supplementary fig. S1.1, Supplementary Material online).
In addition to their well-characterized species-specific cell adhesion role, the AFs have also been proposed as conspecific allorecognition molecules (Fernàndez-Busquets and Burger 1999). Evidence of AF activity in immunologically challenging contexts provides the first link between these molecules and allorecognition. MAFp3 and MAFp4, contiguously transcribed AF genes in the demosponge Clathria (née Microciona) prolifera, are upregulated in both autografted and allografted tissue, compared with normal tissue (Fernàndez-Busquets and Burger 1997; Fernàndez-Busquets et al. 1998). The deglycosylated form of the MAFp3 protein, present exclusively in archeocytes, is recruited to the site of allogeneic contact (Fernàndez-Busquets et al. 1998, 2002). Furthermore, AFs bear hallmarks found in allorecognition molecules, including the ability to detect, interact with and determine whether a biological entity is derived from self or nonself, and promote a downstream response on the basis of this decision (Grice and Degnan 2015a). AFs allow physical interactions between compatible sponge cells by contributing to a molecular “bridge” between cells, which also includes aggregation receptors and associated glycans and glycoproteins (Jumblatt et al. 1980; Misevic and Burger 1990, 1993). MAFp3 and an associated glycoprotein, p210, are highly polymorphic in C. prolifera and there is a perfect correlation between MAFp3 sequence similarity/dissimilarity and graft fusion/rejection (Fernàndez-Busquets and Burger 1997). Finally, AF-receptor binding is coupled to various downstream signaling and regulatory pathways, which may stimulate an active rejection response upon exposure to nonself (Müller et al. 1976, 1987, 1994; Dunham et al. 1983; Rottmann et al. 1987; Schröder et al. 1988; Pfeifer et al. 1993; Wimmer et al. 1999). Although these observations gathered over many years from a number of sponge species are together consistent with a central role for AFs in sponge allorecognition, experimental demonstration of AFs directly regulating allorecognition is lacking.
Analysis of AF-related gene products in three demosponges, C. prolifera, Geodia cydonium and Suberites domuncula, reveals both similarities and marked differences between species. In C. prolifera, multiple AF isoforms, containing nucleotide, intronic, exonic and length variants, have been identified across individuals (Fernàndez-Busquets et al. 1996, 1998; Fernàndez-Busquets and Burger 1997). MAFp3 does not have any characterized domains, while MAFp4 isoforms possess between 3 and 15 Calx-beta domains (Fernàndez-Busquets and Burger 1997; Fernàndez-Busquets et al. 1998) (fig. 1). The G. cydonium AF, named GEOCY AF, includes two Sushi domains (Müller et al. 1999), while the S. domuncula AF, named SdSLIP, has one Calx-beta domain (Wiens et al. 2005); both also have regions similar to C. prolifera MAFp3 (fig. 1).
Fig. 1.
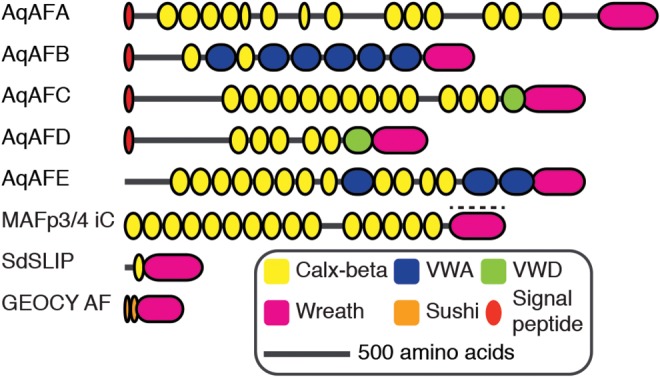
Domain architecture of aggregation factor proteins. Aggregation factor proteins from Amphimedon queenslandica (AqAFA–AqAFE), Clathria prolifera (MAFp3/4 iC), Suberites domuncula (SdSLIP) and Geodia cydonium (GEOCY AF) are shown. Colored shapes represent predicted protein domains and sequence features. Models are approximately to scale. For C. prolifera, MAFp3 (indicated by dashed line) and MAFp4 are represented as a single contiguous sequence; the longest isoform (isoform C) is shown. iC, isoform C; VWA, von Willebrand type A domain; VWD, von Willebrand type D domain.
Here, we present a comparative analysis of the AF genes from multiple sponge species, permitting for the first time, to our knowledge, a systematic investigation of the origin and evolution of a putative allorecognition gene family across an animal phylum. We first characterize the genomic structure and organization of the AF locus in the demosponge Amphimedon queenslandica, as this genomic perspective has been lacking in previous studies. We demonstrate that A. queenslandica AF genes bear remarkable similarities to those from other animal allorecognition loci, in that they are highly polymorphic, clustered, encode large extracellular proteins with repeated protein domains, and have structural properties consistent with loci that can generate a high number of variants. Using our new insights from A. queenslandica, combined with previously published observations, we then survey the transcriptomes or genomes of 24 representative species spanning all four poriferan classes—Calcarea, Demospongiae, Hexactinellida and Homoscleromorpha—for AF candidate genes. This phylum-wide analysis reveals that the AF gene family evolved in demosponges after they diverged from other sponge lineages, and included the evolution of a novel domain that we have coined the “Wreath” domain. Differences in extant AFs between and within species are consistent with the continual evolution of this gene family, and provide an explanation as to how other allorecognition genes present in the animal kingdom obtain their unique molecular structure and organization.
Results
Domain Architecture of the Amphimedonqueenslandica Aggregation Factor Gene Family
Using BLAST similarity searches, we identified five genomically clustered Amphimedon queenslandica genes (AqAFA–AqAFE; fig. 2A) that exhibit significant sequence similarities to aggregation factor (AF) or AF-like sequences from Clathria prolifera, Geodia cydonium and Suberites domuncula (table 1). Overall amino acid sequence similarities between pairs of AqAF predicted proteins or with other known AFs are relatively low (i.e., <40% identity between matching amino acid regions). Membrane topology predictions from translated peptide sequences indicate that all AqAF proteins are secreted, except perhaps AqAFE, which is predicted to occur extracellularly yet lacks a discernable signal peptide (fig. 2B). The AqAFs are predicted to encode three characterized domains from the Pfam protein family database (fig. 1). As in C. prolifera MAFp4 and S. domuncula SdSLIP, all AqAFs include Calx-beta domains in varying numbers from two in AqAFB to 14 in AqAFA. The Calx-beta domains of the AqAFs share very little sequence identity with each other (average 23% amino acid identity), the Calx-beta domains from MAFp3 (average 30% identity within MAFp3 isoform C) or Calx-beta domains elsewhere in the A. queenslandica genome (average 25% identity). AqAFB and AqAFE also encode von Willebrand type A (VWA) domains (average 31% identity), and AqAFC and AqAFD each have one von Willebrand type D (VWD) domain (23% pairwise identity).
Fig. 2.

Genomic and domain organization of the Amphimedon queenslandica aggregation factor genes. Five aggregation factor (AF) genes are encoded in the Amphimedon queenslandica genome. (A) AqAFA–AqAFE are clustered in an ∼80-kb region. Two non-AF genes are also nested within this cluster: Autophagy-related protein 13-like (1) and sn1-specific diacylglycerol lipase beta-like (2). Non-AF genes were identified based on the best BLASTp or BLASTx hit obtained from NCBI. AFs are shown in orange and non-AFs are shown in gray. Chromosomal gene orientation is indicated by arrowheads representing the 3′ end of each gene. Genes are drawn to scale. (B) The gene model prediction for each A. queenslandica AF gene is shown, with boxes representing exons and lines representing introns. Genomic DNA regions encoding protein domains are colored accordingly. Numbers above introns indicate the phase of each intron. AqAFC R1–R3 and AqAFE R1–R2: Location of repeated sequences encoded within the genomic DNA of each gene. (C) The majority of A. queenslandica AF protein domains are encoded by exons that are organized in a consistent way between domains within the AFs. Domain boundaries and average sizes are shown for each domain type in bright colors. Paler colors represent other domains that typically begin or end within the same exon. Exons are not to scale within or between models. VWA, von Willebrand type A domain; VWD, von Willebrand type D domain.
Table 1.
General Properties of Amphimedon queenslandica Aggregation Factor Genes.
| Gene | Accession | gDNA Size (kb) | cDNA Size (kb) | Exon No. | Ave. Intron Size (bp) | Domain Architecture | Intergenic Distances | Hotspot Size (gDNA/cDNA) |
|---|---|---|---|---|---|---|---|---|
| AqAFA | Aqu2.1.38623_001 | 15.44 | 9.09 | 48 | 108 | SP–14 x Calx-beta–1x Wreath | Overlap | 120 bp | n/a |
| AqAFB | Aqu2.1.38624_001 | 9.46 | 5.96 | 19 | 181 | SP–2x Calx-beta–6x VWA–1x Wreath | 120 bp | 543 bp | 1,872/1,198 bp |
| AqAFC | Aqu2.1.38625_001 | 11.83 | 7.85 | 41 | 96 | SP–13x Calx-beta–1x VWD–1x Wreath | 543 bp | 110 bp | 1,447/617 bp |
| AqAFD | Aqu2.1.38627_001 | 13.34 | 5.16 | 18 | 383 | SP–5x Calx-beta–1x VWD–1x Wreath | 96 bp | 29 bp | n/a |
| AqAFE | Aqu2.1.38629_001 | 17.03 | 8.42 | 34 | 250 | 12x Calx–3x VWA–1x Wreath | 704 bp | 1,489 bp | 3,075/1,443 bp (hotspot); 2,122/ 1,423 bp (cntl) |
Note.—gDNA, genomic DNA; cDNA, complementary DNA; downstream | upstream; SP, signal peptide.
In C. prolifera, MAFp3 self-adheres to form the central ring of the core AF sunburst structure (Jarchow et al. 2000). BLASTp searches revealed that regions exhibiting MAFp3 sequence similarity also exist in SdSLIP, GEOCY AF, and in all AqAFs. Considering the demonstrated functional importance, structural independence, and multi-species distribution of this region, we propose that MAFp3 and homologous sequences be considered to possess a novel protein domain (Richardson 1981). We suggest the name “Wreath domain” due to this protein region’s role in C. prolifera central AF ring formation (Jarchow et al. 2000). A multiple sequence alignment of the Wreath regions from MAFp3, SdSLIP and AqAFC (supplementary fig. S1.5, Supplementary Material online) was used to generate a profile hidden Markov Model (HMM) (supplementary file S5, Supplementary Material online) for this putative novel domain. HMM searches with this new model identified a single Wreath domain in AqAFA to AqAFE. The Wreath domain was not found in non-AF predicted proteins encoded in the A. queenslandica genome, nor in any non-demosponge species.
Modular Exon Structure of A.queenslandica AF Protein Domains
To investigate the relationship between AqAF domain architecture and genomic structure, we mapped the positions of all AqAF Calx-beta, VWA, VWD and Wreath domains back to the genome (fig. 2B). Each domain type displays remarkably similar exonic coverage patterns across all AqAFs (fig. 2C andsupplementary tables S1.1–S1.4, Supplementary Material online). With the exception of AqAFA Calx-beta domain 10, all AqAF Calx-beta domains are encoded by three exons, which on average span the final 27 base pairs (bp) of exon i, the entirety of exon ii (average 145 bp), and the first 126 bp of exon iii. This pattern repeats starting in the final ∼27 bp of exon iii. All VWA domains in AqAFB and AqAFE localize to single exons and are flanked by short spacer regions at both ends of the exon (average 22 bp at either end). Similarly, the single VWD domains in AqAFC and AqAFD both map to single exons, with a short spacer sequence at the beginning of the exon, but with the adjacent Wreath domains beginning immediately after the domains’ end. Finally, all AqAF Wreath domains are encoded by the final three coding exons of each gene, commencing partway through the antepenultimate exons of each gene and running to the end of the sequence. The lengths of the first Wreath domain-containing exons vary between sequences, while the other two exons are more consistently sized between genes (average 523 and 336 bp, respectively).
Genomic Organization of the A.queenslandica AF Locus
The five AqAF genes (AqAFA–AqAFE) cluster together within an 80-kilobase (kb) genomic region (fig. 2A). The AqAF cluster is tightly packed even in comparison to other regions in the highly compacted A. queenslandica genome. The median intergenic distance in the AF region is 103 bp (table 1), which is considerably smaller than that observed genome-wide (589 bp) (Fernandez-Valverde et al. 2015). All AqAFs encode a single contiguous sequence equivalent to C. prolifera MAFp4 + MAFp3. The AqAFs are large genes (between 9.5 and 17.0 kb in length) with many exons (between 18 and 48 exons per gene; table 1). The average intron lengths of AqAFA (108 bp), AqAFB (181 bp), AqAFC (96 bp) and AqAFE (250 bp) are shorter than the genome-wide average of 327 bp (Fernandez-Valverde et al. 2015), while those from AqAFD (383 bp) are slightly longer (table 1). The AqAF introns are also generally shorter than those observed in C. prolifera MAFp3 (300–600 bp; Fernàndez-Busquets and Burger 2003).
Two sets of highly similar repeats are present in the genomic regions encoding AqAFC and AqAFE (fig. 2B). In AqAFC, three repeat units span intron 10 to exon 14, intron 14 to exon 18, and intron 26 to exon 30. These AqAFC repeats cover regions encoding two Calx-beta domains each, include both introns and exons, and share ∼85% total pairwise nucleotide sequence identity to one another. Two repeats are present in AqAFE, in exons 30 and 31 (96% pairwise identity), and each encode a single VWA domain. These repeats do not cover any intronic sequences and do not bear any particular sequence similarity to the AqAFC repeats.
Intron Phase Distribution in the AqAFs and Other Calx-Beta Domain-Encoding Sequences
We compared intron phase frequencies across the AqAFs and other A. queenslandica Calx-beta domain-containing genes to those observed genome-wide for this species. The AqAFs show an extreme bias in intron phase distribution; all AqAF introns except one are in phase 1 (n = 154 of 155; fig. 2B and table 2). This distribution differs significantly from both the genome-wide (44% phase 0, 36% phase 1, 20% phase 2) and non-AF Calx-beta domain-containing (16% phase 0, 78% phase 1, 6% phase 2) gene sets. For comparison, all introns of MAFp3 and MAFp4 are equipped with phase 0 introns only (Fernàndez-Busquets and Burger 1999). The distribution of intron phases across all A. queenslandica non-AF Calx-beta domain-containing genes also differs significantly from the genome-wide distribution (table 2), however not to the extreme extent as in the AqAFs.
Table 2.
Intron Phase Frequencies across Metazoan Calx-Beta Domain-Containing Genes.
| Data Set | Phase 0 | Phase 1 | Phase 2 |
|---|---|---|---|
| Aq genome | 44% | 36% | 20% |
| Aq Calx-beta genes | 109 (13%) σ = 0.012a | 683 (82%) σ = 0.013a | 44 (5%) σ = 0.008a |
| AqAFs | 1 (0.6%) σ = 0.006a,b | 154 (99%) σ = 0.006a,b | 0 (0%) σ = 0a,b |
Note.—σ, standard deviation of the mean.
Statistically significant difference between genome-wide and Calx-beta phase frequency.
(for AqAFs) Statistically significant difference between total Calx-beta and AqAF phase frequency. Statistical significance calculated as per Fedorov et al. (1998) (see “Materials and Methods” section); Aq—Amphimedon queenslandica.
Polymorphism and RNA–DNA Differences in the AqAFs
As the C. proliferaMAFp3 and MAFp4 genes are highly polymorphic (Fernàndez-Busquets and Burger 1997; Fernàndez-Busquets et al. 1998), we determined whether the A. queenslandica AF genes also vary between alleles and individuals. First, we compared Illumina RNA-Seq reads from three sponge adults (supplementary methods, Supplementary Material online). This analysis revealed that polymorphism in the AqAFs is extensive (supplementary tables S1.5–S1.7 and file S3, Supplementary Material online) but variably distributed across the locus (supplementary fig. S1.2, Supplementary Material online). Non-synonymous variants (average 45.9%) are statistically significantly over-represented within the AqAF polymorphic sites relative to the A. queenslandica transcriptome as a whole (average 28.4%; P < 0.0001 for all individuals) (supplementary tables S1.5–S1.7, Supplementary Material online).
We selected three “hotspots” of higher variability for closer analysis—one from each of AqAFB, AqAFC and AqAFE—with each hotspot spanning between ∼1,500 and ∼3,000 bp of genomic DNA. We also included a ∼2,000-bp region of AqAFE (including the two-exon repeat region) that exhibited lower sequence variability in this initial screen (supplementary fig. S1.2, Supplementary Material online). Genomic DNA and complementary DNA (cDNA) of these regions from four unrelated larval individuals (Larvae 3.1, 6, 7 and 9) were amplified and directly Sanger sequenced, which generated individual sequencing reads potentially capturing multiple alleles. These were compared with the A. queenslandica reference genome to identify variable sites in DNA and messenger RNA (mRNA) in each individual (supplementary file S3, Supplementary Material online). Levels of polymorphism were found to differ both spatially (i.e., in different exons, introns and genes) and between individuals within the hotspots (figs. 3 and4A and B), such that each individual has its own unique AF sequence pattern. All three hotspot regions exhibit higher frequencies of non-synonymous nucleotide substitutions (average 44.9%, P≤ 0.0001–0.002 per individual) than is observed genome-wide (e.g., 25.4% in Sponge A). The putatively less variable AqAFE region also showed this trend (average 68%; P = 0.0001–0.0167 per individual), although the number of variants observed here is lower than in the hotspot regions (fig. 4B). One non-synonymous difference in a single individual (“Larva 9”) is predicted to introduce a termination codon halfway through AqAFB exon 17, truncating the final VWA domain (supplementary file S3, Supplementary Material online); no other variants are predicted to introduce frameshifts, termination codons or signal peptides.
Fig. 3.

Locations of somatic polymorphism and putative RNA editing sites. Introns, exons and domain architecture of sequenced regions are shown as in figure 2; arrows above gene model fragments represent primer binding sites. Gray boxes highlight intronic regions. Each horizontal line represents the sequenced region of a larval (L6–L3.6) or adult (A1–A10) individual. Two regions of AqAFE are shown: one initially predicted to be highly polymorphic (1) and the other predicted to be less polymorphic (2). Dashes above the line (s) are identified somatically encoded polymorphisms, while dashes below the line (e) are putative RNA editing sites. Dash color represents the predicted effect of each variant: red—non-synonymous, non-conservative amino acid change; orange—non-synonymous, conservative amino acid change; green—synonymous nucleotide change; black—intronic change.
Fig. 4.

Quantification of AqAF polymorphism and RNA editing. (A) Proportion of variant sites per exon, e, and intron, i. Percentages are shown relative to the size of each exon or intron. Two regions of AqAFE are shown: one initially predicted to be highly polymorphic (1) and the other predicted by be less polymorphic (2). (B) Variability in polymorphism levels between larval, L, and adult, A, individuals and between gene regions. Percentages are shown relative to the size of each sequenced region. (C) Percentage of AqAFB RNA-edited sites per individual, relative to the size of the sequenced AqAFB region. (D) Distribution of unique RNA-edited sites across the AqAFB hotspot region. (E) Frequency of genomic DNA-to-cDNA nucleotide substitutions. Percentages in A, D and E refer to total sites across all sequenced individuals. Bar color represents the predicted effect of each variant: red—non-synonymous non-conservative amino acid change; orange—non-synonymous conservative amino acid change; green—synonymous nucleotide change; black—intronic change.
In one of the four individual larvae surveyed (Larva 3.1), we observed 42 AqAFB sites (3.4% exonic region) with nucleotide differences between the genomic and cDNA sequences, which is a potential hallmark of RNA editing (fig. 3 and supplementary file S3, Supplementary Material online). Such RNA–DNA differences (RDDs) were not observed in the other three sequenced regions of Larva 3.1, or in any sequenced regions of Larvae 6, 7 or 9 (fig. 4C). To explore this phenomenon further, we amplified the AqAFB hotspot from an additional 13 individuals: five larvae that were half-sibs of each other and of Larva 3.1 (Larvae 3.2–3.6); and somatic cells isolated from eight unrelated adults (Adults 1–6, 9 and 10). In total, eight individuals (Larvae 3.1, 3.5, 3.6 and Adults 3, 4, 5, 6, 10) possessed between one and 42 AqAFB RDDs (total 56 unique sites; 4.8% of the sequenced exonic area of AqAFB) (figs. 3and 4). Only those sites for which single, different base calls were made between the genomic and cDNA sequences within a given individual are included in this total. All individuals show unique RDD patterns. Each RDD site displayed one of seven genomic DNA-to-mRNA nucleotide substitutions (A-to-G, C-to-U, G-to-A, U-to-C, A-to-U, G-to-C or G-to-U) (fig. 4E). The majority of RDDs are transitions (i.e., purine–purine or pyrimidine–pyrimidine substitutions), with a slightly elevated proportion of these being A-to-G differences and the three other transitions occurring at equal rates (fig. 4E). A small number of transversions (purine–pyrimidine or pyrimidine–purine substitutions) were also observed (fig. 4E). About 43% of RDD sites are predicted to change the encoded amino acid sequence (fig. 4); no frameshifts, stop codons or signal peptide-inducing methionine residues are introduced by RDDs (supplementary file S3, Supplementary Material online). If a given RDD site was changed in two or more individuals, these individuals all displayed the same specific nucleotide change at that site, as did any other individuals that were otherwise polymorphic at that site. These observations, plus the existence of multiple sequencing replicates per individual, convinced us that the variable sites represent genuine biological variability and not artifacts of the PCR or sequencing processes.
Although we did not observe more than two nucleotide types at any given position in our sequence alignments, our dataset was generated by directly Sanger sequencing PCR products, meaning that each read represented all captured alleles from a given sample. To reduce the possibility that the mRNA variants were from a previously undetected, duplicated AqAFB gene, we amplified this region again from the genomic and cDNA from six individuals (Larvae 3.1, 3.5, 6 and 7; Adults 5 and 9), and Sanger sequenced 10 cloned sequences of each (120 sequences in total). In no case did we find evidence of a second AqAFB locus and indeed five of the six individuals surveyed appear to be homozygous in their genomic DNA sequences at this locus. Comparison of RDDs detected in individual cloned sequences matched those from the original direct-sequencing sequences, except in some positions in Adult 9 (where a previously undetected cDNA allele was identified) (supplementary file S2, Supplementary Material online).
Identification of AF Candidates in Other Poriferans
We compared the features of the A. queenslandica AFs and other known sponge AF-like sequences, and used this information to design a sequence filtering workflow to allow more efficient identification of novel AF candidates from large transcriptome or genome datasets (fig. 5). We then applied these criteria to identify candidate AFs from the transcriptomes (or genomes, where specified) of 24 sponge species: A. queenslandica (developmental transcriptomes), Aphrocallistes vastus, Chondrilla nucula, C. prolifera, Cliona varians, Corticium candelabrum, Crella elegans, Ephydatia muelleri, Haliclona amboinensis, Hyalonema populiferum, Ircinia fasciculata, Kirkpatrickia variolosa, Latrunculia apicalis, Niphatidae indet., Oscarella carmela (genome), Petrosia ficiformis, Pseudospongosorites suberitoides, Rossella fibulata, Spongilla lacustris, Sycon ciliatum (genome), Sycon coactum, Sympagella nux, Tethya wilhelma, and Xestospongia testudinaria (genome and transcriptome).
Fig. 5.

Methodology for aggregation factor candidate sequence identification. Flowchart depicting the filtering process to isolate aggregation factor (AF)-like and candidate AF sequences from genome or transcriptome datasets. Sequences possessing Wreath, Calx-beta, von Willebrand type A (VWA) or D (VWD) domains were identified by searching sequence datasets with hidden Markov model (HMM) profiles. Sequences were eliminated (X) if they encoded only a Wreath domain and this domain did not cover at least 60% of the HMM model. Short or redundant sequences were also removed. The resulting list was divided into three groups, based on domain architecture and sequence similarity. Group 1 sequences possess a Wreath domain, with or without other domain types. Group 2 sequences have a top BLAST hit to a known AF sequence from Amphimedon queenslandica, Clathria prolifera or Suberites domuncula, but do not possess a Wreath domain. Group 3 sequences represent all other filtered sequences, and were not considered AF candidates for the purposes of this study.
Filtered sequences were considered candidate AFs if they possessed a Wreath domain (Group 1) or showed AF-like domain architecture (fig. 1) and a top BLAST hit to an AqAF, GEOCY AF, MAFp3, MAFp4 or SdSLIP sequence (Group 2). All remaining sequences (Group 3) were not considered to be candidate AFs for the purposes of this study, although they encoded at least one domain found in the well-characterized AF-like proteins. Sequence counts per species are shown in figure 6 and sequences are available in supplementary file S4, Supplementary Material online.
Fig. 6.
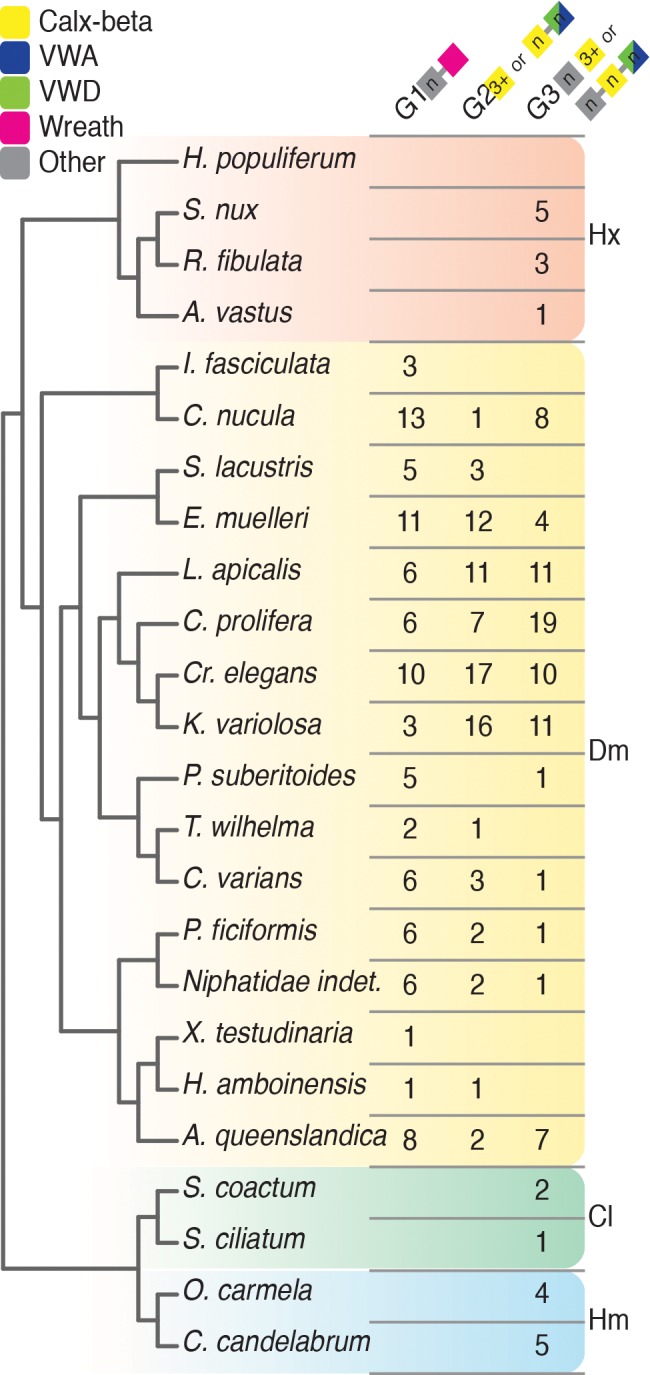
Phylogenetic distribution of Group 1, 2 and 3 aggregation factor candidates and related sequences. The phylogenetic relationships between analysed sponge species is depicted on the left (Kocot KM, personal communication) (Thacker et al. 2013; Whelan et al. 2015). The table gives the number of sequences per species in Groups 1 (i.e., possessing a Wreath domain), 2 (i.e., having homology to known aggregation factors (AFs) or AF-related sequences from Amphimedon queenslandica, Clathria prolifera, Geodia cydonium or Suberites domuncula) and 3 (i.e., sequences equipped with Calx-beta and von Willebrand types A (VWA) or D (VWD) domains only, without top sequence homology to known AFs). The counts given for A. queenslandica refer to the AF transcripts encoded in the developmental transcriptome dataset. Colored box diagrams represent stylized domain architectures of group members. Hx—Hexactinellida, Dm—Demospongiae, Cl—Calcarea, Hm—Homoscleromorpha.
We identified at least one AF candidate in each demosponge transcriptome, but none in any non-demosponge (i.e., calcarean, hexactinellid and homoscleromorph) species surveyed (figs. 6 and7; supplementary fig. S1.3–S1.4 and supplementary file S4, Supplementary Material online). The majority of identified AF transcripts have the same overall domain composition as seen in the AqAFs: Calx-beta, VWA, VWD and Wreath domains occur in different numbers and combinations. In a small number of species—C. nucula, Niphatidae indet. and P. ficiformis—Calx-beta, VWA and/or VWD, and Wreath domains all co-occur as in AqAFB–AqAFE.
Fig. 7.

Summary and phylogenetic distribution of aggregation factor candidate domain architectures. The domain architectures of all identified Group 1 and 2 aggregation factor (AF) candidates from each analysed demosponge species are shown. Phylogenetic relationships are as shown in figure 6. Colored circles represent protein domains; symbols above the table represent different domain combinations but do not show numbers of each domain. Gray circles represent the presence of one or more proteins encoding each domain combination in each demosponge species. Shaded columns highlight domain architectures present in Amphimedon queenslandica, Clathria prolifera, Geodia cydonium or Suberites domuncula AFs. Note that the A. queenslandica distribution was here derived from a transcriptome not the genome, and thus displays slightly different results to those seen in the genome-wide data (i.e. no Calx-beta + VWA + Wreath domain-containing sequence was recovered). VWA—von Willebrand type A domain; VWD—von Willebrand type D domain; PSI—Plexin, Semaphorin and Integrin domain; EGF—Epidermal Growth Factor domain; GPS—GPCR (G-protein coupled receptor) Proteolysis Site domain.
When present, Wreath domains are always found at the 3′-most end of the coding sequence. Many (n = 31) demosponge transcripts encode a Wreath domain only. In most cases, it is unknown whether these represent full-length or truncated sequences. However, four such sequences (C. nucula Cn_13331.30; C. varians Cv_16635, Cv_16636; T. wilhelma Tw_1404) include signal peptides, suggesting these do represent full-length transcripts.
Several AF transcripts encode additional domain types not observed in the A. queenslandica or C. prolifera AFs. Two closely related freshwater haploscleromorph demosponges E. muelleri (Em_90236) and S. lacustris (Sl_2436.75) both encode proteins equipped with one copy each of Sema (PF01403), PSI (PF01437) and Wreath domains. Immunoglobulin superfamily (IgSF; Ig-2 [PF13895], V-set [PF07686], I-set [PF07679], Ig_3 [PF13927]), fibronectin type III (fn3; PF00041) and EGF-related (calcium-binding EGF domain, PF07645; human growth factor-like EGF domain, PF12661) domains, and a GPS motif (GPCR (G-protein coupled receptor) proteolysis site; PF01825) were found in various AF candidates. Finally, Sushi domains (PF00084), as previously documented in the possible G. cydonium AF GEOCY AF, are present in one sequence each from I. fasciculata (IF_3013.75, three domain copies) and P. suberitoides (Ps_6648.67, one copy).
Xestospongia testudinaria is the only analysed non-Amphimedon demosponge for which both transcriptome and genomic data are available; analysis of intronic properties is therefore possible for the single candidate AF from this species. The introns of the single X. testudinaria candidate AF Xt_88826 (average size 62 bp) are smaller than those of both the AqAFs (average size 172 bp) and of MAFp3 (350–600 bp) (Fernàndez-Busquets and Burger 1997). As in the AqAFs, all introns of Xt_88826 are in phase 1; MAFp3 and MAFp4 have phase 0 introns only (Fernàndez-Busquets and Burger 1999).
Discussion
The disparate nature of animal allorecognition means that little is known about how such systems evolve. With the goal of understanding both sponge allorecognition specifically, and the processes driving evolution of animal allorecognition more generally, we undertook a detailed investigation of the aggregation factor (AF) gene family in sponges. We performed a broad-ranging survey for AFs across Porifera, and identified the sponge lineages in which the AFs were present. We then characterized the various domain organizations that candidate AFs took in each species. This allowed us to assess the evolution of a putative allorecognition system in a phylum. We also scrutinized the AFs at a finer scale, exploring the AF locus in the genome of the demosponge Amphimedon queenslandica. This provided insight into the genomic background underlying broader-scale evolutionary changes. In addition to revealing high levels of polymorphisms between AqAF alleles, we identified one region of AqAFB that displays extensive RNA–DNA differences (RDDs), suggesting that further AqAF diversification can occur by RNA editing. Overall, our study reveals that the AF gene family is a demosponge-specific evolutionary novelty, and has undergone continual modification since the demosponge last common ancestor.
Amphimedon q ueenslandica AFs Appear to Be Diversified by High Allelism and RNA Editing
Amphimedon queenslandica AF genes exhibit a high degree of between-individual polymorphism, including a greater proportion of non-synonymous nucleotide substitutions than are observed across the genome as a whole. Intriguingly, we also identified a large number of RDDs across the AqAFB sequences of some individuals. We performed additional testing to exclude the possibility that these RDDs could be attributed to the presence of a cryptic AqAFB duplicate gene elsewhere in the A. queenslandica genome. While it is impossible to ensure that any sequencing approach targets the full suite of alleles in any given sample, our consistent observation of no more than two genomic DNA alleles across the six tested individuals suggests that gene duplication is an unlikely explanation for our results.
If the observed RDDs are indeed derived from the same genomic locus, this implies that AqAFB RNAs are modified post-transcriptionally, and that RNA editing is another possible contributor to AF diversification. RNA editing appears to be possible in A. queenslandica because ADARs, proteins responsible for A-to-I RNA editing, are present in sponge genomes and are likely to be a metazoan-specific innovation (Grice and Degnan 2015b). However, AqAFB RDDs do not seem to favor any particular combination of nucleotide substitutions, with both transitions and transversions observed across 56 unique editing sites; multiple RNA editing profiles were detected between some A. queenslandica individuals. While similar patterns of non-specific substitution have been observed, e.g., in certain dinoflagellate mitochondrial and plastid genes (Lin et al. 2002; Mungpakdee et al. 2014), its occurrence in a metazoan nuclear gene appears to be unusual. Some unknown targeted or motif-guided mode of RNA nucleotide substitution may be at play, although currently it is unclear if the variety of RDD events in AqAFB may be explained by the activity of one or more classes of editing molecules.
The finding of elevated, individual-specific, and often non-synonymous AF polymorphism and editing is consistent with the requirement of an allorecognition gene to display high levels of between-individual variability to in turn allow rejection of nonself contact (Grice and Degnan 2015a). Such an effect has been observed in the sea urchin Strongylocentrotus purpuratus Sp185/333 immune gene family, where polymorphism and RNA editing appears to both diversify encoded Sp185/333 messages and regulate their expression by the introduction of premature stop codons (reviewed by Smith et al. 2010). In A. queenslandica, one edited region of AqAFB encodes a Wreath domain, which in Clathria prolifera contributes to the AF central ring that interacts with other AF rings to form intercellular bridges (Jumblatt et al. 1980; Misevic and Burger 1990, 1993). Sequence variation in this region may affect AF–AF binding and therefore passive “self” recognition, either by directly affecting protein conformation, or altering glycan attachment sites. In addition to these diversifying changes, a large proportion of nucleotide edits are synonymous. It is unclear whether these are a result of non-specific modification or instead reflect functionally important changes that, e.g., introduce or remove nucleotide motifs or affect the regulation of gene expression in some way.
The scope and distribution of RNA editing across the AqAF locus, and the A. queenslandica genome at large, is presently unknown. Expanded surveys for RNA editing to target larger regions of this locus and more individuals from a range of environmental, developmental and immunological contexts, would help resolve this question.
The AFs Are a Demosponge-Specific Evolutionary Novelty
To reconstruct the evolutionary origins of the AF gene family, we surveyed for their presence across 24 sponge transcriptomes or genomes. Candidate AFs are present in all demosponges, and AFs are absent from calcarean, hexactinellid and homoscleromorph species included in this study. While failure to detect AF-like sequences in large datasets, particularly those generated from transcriptome libraries, is not definitive evidence of their absence from the sponge species sequenced, it is striking that candidate AFs also were not identified in non-demosponge species with full genome sequences (Oscarella carmela and Sycon ciliatum), where successful sequencing is not context- or expression-dependent. We conclude that the AFs originated in the demosponge common ancestor and evolved rapidly to produce the diversity of AF molecules that exists today.
Demosponge AFs Are Comprised of Calx-Beta, Wreath, and Other Protein Domains
In C. prolifera, MAFp3 is responsible for the formation of the AF central ring, and for self–self interactions between AF structures (Jarchow et al. 2000). A novel protein domain, which we have coined the Wreath domain, appears to be responsible for these interactions. The Wreath domain was found in all demosponge species and appears to be a demosponge-specific innovation. Wreath domains were found to co-occur with other domain types in all demosponges except Tethya wilhelma, but we also observed transcripts (including four with signal peptides) encoding single Wreath domains only. This suggests that the Wreath domain may also play an independent functional role in some species.
All demosponge species express one or more AF candidates equipped with Calx-beta domains. Calx-beta domains contain a small number of residues that are important for binding Ca2+ (Hilge et al. 2006), an ion that is critical for AF stabilization and adhesiveness (Galtsoff 1925; Cauldwell et al. 1973; Fernàndez-Busquets et al. 2009). Almost all critical residues are conserved in the Calx-beta domains in the A. queenslandica AFs (AqAFs). Outside these conserved residues, the AqAF Calx-beta domains appear to be relatively free to change without disrupting domain functionality.
von Willebrand type A (VWA) or D (VWD) domains were only found in AFs from five species distributed across the demosponges. Elsewhere, VWA domains have been proposed to mediate protein adhesion and aggregation in proteins such as integrins (Whittaker and Hynes 2002). The VWA MIDAS (metal ion-dependent adhesion site) motif has been implicated in divalent cation-dependent (usually Mg2+, but also Ca2+) ligand binding (Cantí et al. 2005). MIDAS motifs are present within each AqAF VWA domain. As AF functionality is Ca2+- and/or Mg2+-dependent (Galtsoff 1925; Humphreys et al. 1960), it is possible that the incorporation of VWA domains into some AFs aids in cation-mediated aggregation. VWD domains lack a MIDAS motif, and the role of these domains in the AFs remains unclear.
EGF-related, fn3, GPS, immunoglobulin-related, PSI, Sema and Sushi domains were present in several demosponge AF candidates, suggesting that novel protein folds may be important in some AFs, and/or that the Wreath domain may be involved in functions beyond AF-AF bridge formation. EGF-related (Campbell and Bork 1993), fn3 (Bork and Doolittle 1992), immunoglobulin-related (Williams and Barclay 1988) and Sushi (Day et al. 1989) domains are all widespread and mediate protein–protein interactions in a range of molecules, including those with cell adhesion, self–nonself recognition or immune functions. Sema and PSI domains, which are present in AFs of the freshwater sponges Ephydatia muelleri and Spongilla lacustris, are best known for their role in semaphorin-mediated axon guidance (Kolodkin et al. 1993), but have also been implicated in cell adhesion and migration processes (reviewed by Casazza et al. 2007). The function of the novel Sema-PSI-Wreath domain combination in these AFs is unknown, although it is possible that the Wreath domains allow these molecules to form circular or linear backbones, while the Sema-PSI region mediates cell–cell or cell–extracellular matrix tethering (Casazza et al. 2007).
Exon Shuffling and the Evolution of Demosponge AFs
The diversity of protein domain content and organization across the AFs indicates that these molecules evolved, in part, by domain cooption, gain and loss. This notion is further supported by the high level of structural constraint observed across the AF loci of A. queenslandica and other species, which is probably a signature of prior exon shuffling. First, despite the low sequence similarity observed between A. queenslandica AF protein domains of all types, these domains conform to precise boundaries within their encoding exon/s. These boundaries may be remnants of an ancestral domain module structure. While VWA and VWD domains are bound within single exons, most A. queenslandica AF Calx-beta and Wreath domains conform to repeated three-exon structures, with flanking and internal phase 1 introns. This suggests either that Calx-beta and Wreath domains inserted into the AFs as multi-exon modules, or that their phase 1 introns allowed the domains to be “mixed and matched” from pools of single-exon building blocks.
Another diagnostic feature of a shuffled gene is bias towards a particular intron phase (Patthy 1987, 1988). While the precise intron phase distributions differ between species (i.e., phase 1 in A. queenslandica and X. testudinaria, and phase 0 in the more distantly related C. prolifera; Fernàndez-Busquets and Burger 1999), AF exons in A. queenslandica, C. prolifera and X. testudinaria are all flanked by introns in the same phase, suggesting prior expansion and diversification of the AF gene family by exon shuffling. Despite this striking commonality, intron–exon and domain architectures of these AFs are unique in each species. Thus, although the AFs in these species show hallmarks of evolutionary relatedness, it is difficult to discern the ancestral condition of their common AF ancestor and the precise evolutionary processes leading to the extant AF repertoires in these and other species.
Non-Demosponges Lack an AF Gene Family
Candidate AFs were not identified in any analysed hexactinellid (Hyalonema populiferum, Sympagella nux, Rossella fibulata or Aphrocallistes vastus), homoscleromorph (O. carmela or Corticium candelabrum) or calcareous (Sycon coactum and S. ciliatum) sponges, suggesting that the AF gene family, at least in the form best known from C. prolifera, is an innovation restricted to demosponges. This implies either that non-demosponge poriferans do not possess the ability to discriminate self from nonself by allorecognition—implausible given functional evidence to the contrary (discussed below) and the importance of self–nonself recognition—or that reaggregation and discrimination in these poriferan classes relies on a non-AF-based allorecognition systems. This latter inference is consistent with rapid evolution of allorecognition occurring in sponges, and probably in other taxa.
Calcareous sponges are capable of discriminating between self and nonself at the tissue level (Amano 1990), however reaggregation of dissociated cells in this sponge class does not appear to be enabled by a soluble aggregation factor (Müller 1982). This observation is consistent with our inability to identify MAFp3/4-like AFs in calcareans, and suggests that the mechanisms underlying aggregation and self–nonself discrimination are different between demosponges and calcareous sponges. Allorecognition also exists in hexactinellids, with Rhabdocalyptus dawsoni able to discriminate between self and nonself in mixed tissue/aggregate graft experiments (Leys et al. 1999). Again, however, a different molecular self–nonself recognition system seems to be operating in this class of sponges; a C-type lectin appears to be responsible for cell reaggregation in fellow hexactinellid A. vastus, although it is unclear if it can promote species-specific differentiation (Müller et al. 1984; Gundacker et al. 2001). Finally, we did not detect AFs in homoscleromorphs. Although cellular reaggregation or allorecognition phenomena have not been investigated in this class of sponge, the presence of a circular molecule closely resembling the circular core AF has been found in Oscarella tuberculata; it is currently unknown whether this represents a true AF (Humbert-David and Garrone 1993). Overall, these observations, coupled with our inability to identify AF-like sequences from any non-demosponge species, suggest that one or more unknown allorecognition mechanisms await discovery in these other sponge lineages.
Conclusions
Although metazoan allorecognition does not appear to be under the control of conserved genes, similar molecular and genomic properties can be observed between these unrelated systems; the demosponge AFs also display these properties. First, allorecognition molecules are often encoded by genes that are clustered in the genome, a phenomenon that supports further molecular diversification (Grice and Degnan 2015a). In A. queenslandica, the five AF genes are clustered within an 80-kb genomic locus. Second, allorecognition molecules are often large extracellular or membrane-bound proteins with numerous repeated domains, facilitating the physical interactions between self and/or nonself cells (Grice and Degnan 2015a). In A. queenslandica, the five AFs are large, and most demosponge AFs are predicted to occur extracellularly—either secreted or membrane-bound—and have domains involved in cell-cell interaction or adhesion. Finally, allorecognition molecules often exhibit high degrees of sequence polymorphism, enabling the high level of specificity required of a molecule responsible for discriminating between conspecific individuals (Grice and Degnan 2015a). C. prolifera AF isoforms have been previously demonstrated to be highly polymorphic between and within individuals (Fernàndez-Busquets and Burger 1997; Fernàndez-Busquets et al. 1998), and here we have demonstrated that the same applies in A. queenslandica (fig. 3). Indeed, our identification of RDDs in AqAFB in some A. queenslandica individuals—an indicator of RNA editing—implies the contribution of an additional level of complexity to the diversification of these molecules. The existence of these commonalities between unrelated metazoan allorecognition systems suggests there are similar selective pressures driving the evolution and function of these divergent systems, regardless of the fact that allorecognition molecules vary markedly between taxa.
We found Wreath and Calx-beta domain-equipped AF candidates in all analysed demosponges. The ubiquity of these domains suggests that the ancestral AF probably encoded one Wreath domain sequence—itself a demosponge-specific novelty—or precursor, and one or more Calx-beta domains. Prior to demosponge cladogenesis, AFs appear to have acquired a self–nonself recognition role, possibly as a result of increasing specificity of a more ancient role in cell-cell interaction or adhesion activity (Grice and Degnan 2015a). The AF gene family most likely expanded via multiple duplication events, as inferred from the clustered A. queenslandica AF locus. Diversification of the demosponge AFs occurred via the inclusion of additional domain types and their subsequent gain and loss. Many of the domains present in extant AFs have been implicated elsewhere in cell adhesion and interaction processes; these domains likely play similar roles within the AFs. Finally, beyond the between-species diversification discussed above, a fundamental requirement of an allorecognition molecule is to be sufficiently variable within a species, such that every individual possesses its own “self marker”. Sponge allorecognition has been proposed to operate via self recognition, whereby binding of matching self markers is required to prevent rejection between adjacent cells (Sabella et al. 2007). Generation of this required variation could be achieved potentially in several ways, including high allelic variance as observed in the A. queenslandica and C. prolifera AFs (Fernàndez-Busquets and Burger 1997; Fernàndez-Busquets et al. 1998), alternative splicing (aided, perhaps, by frameshift-minimizing symmetrical exons as present in the A. queenslandica, C. prolifera and X. testudinaria AFs), post-transcriptional or post-translational modification, the former of which appears to occur in the A. queenslandica AFs, somatic recombination and variation in associated non-protein molecules such as glycans.
To the best of our knowledge, this study represents the first instance of a phylum wide survey of putative allorecognition genes. The AFs appear to have evolved rapidly by mechanisms including exon shuffling and nucleotide mutation, and continue to generate within-species variation required for self–nonself recognition by the means listed above. Reconstruction of the evolution of AFs in sponges exposes the evolutionary processes underpinning the disparity of allorecognition factors in the animal kingdom, and similar evolutionary mechanisms to those seen in sponges are likely operational across the Metazoa. Therefore, understanding of the sponge AF gene family sheds light on one of the central features of being an animal, self–nonself recognition.
Materials and Methods
Identification of Aggregation Factors from the Amphimedonqueenslandica Genome
Amphimedon queenslandica AF (AqAF) sequences were identified using BLASTP and TBLASTN searches with Clathria prolifera MAFp3 and MAFp4 isoforms (Genbank: AAB71890, AAB71891, AAC33162, AAC33163, CAA65098), Geodia cydonium GEOCY AF (Müller et al. 1999), and Suberites domuncula SdSLIP (Genbank: CAI68017.1) used as queries. Searches were performed against A. queenslandica genomic traces and assemblies, expressed sequence tags (Srivastava et al. 2010), and subsequently with Aqu2.1 gene model predictions (Fernandez-Valverde et al. 2015). Aqu2.1 annotations were derived from new transcriptomic data combined with the original genome assembly (Srivastava et al. 2010; Fernandez-Valverde et al. 2015) and can be accessed at http://amphimedon.qcloud.qcif.edu.au/index.html (last accessed December 16, 2015).
Protein Domain and Topology Predictions
Domain architectures of the AqAF proteins were predicted using Pfam with default parameters (Finn et al. 2014), and verified using hmmscan, available in the HMMER 3.0 software package (Eddy 1998). Domain hits were counted if they returned an expect (e)-value ≤10 − 4. For each sequence, signal peptides and transmembrane domains were also predicted using Phobius (Käll et al. 2004).
Generation of a Wreath Domain Hidden Markov Model
NCBI BLASTP searches (http://blast.ncbi.nlm.nih.gov/; last accessed November 9, 2015) between the MAFp3 region and the publicly available translated genomes of various model metazoan species (Acropora digitifera, A. queenslandica, Arabidopsis thaliana, Branchiostoma floridae, Caenorhabditis elegans, Capitella teleta, Capsaspora owczarzaki, Ciona intestinalis, Dictyostelium discoideum, Drosophila melanogaster, Helobdella robusta, Homo sapiens, Hydra magnipapillata, Lottia gigantea, Mnemiopsis leidyi, Monosiga brevicollis, Nematostella vectensis, Neurospora tetrasperma, Pleurobrachia bachei, Salpingoeca rosetta, Strongylocentrotus purpuratus, or Trichoplax adhaerens) indicated that this region is not present outside sponges. We propose that the MAFp3 region encodes a sponge-specific domain we coin the “Wreath” domain. A multiple sequence alignment of MAFp3 isoform C, SdSLIP and AqAFC (supplementary fig. S1.5 and supplementary methods, Supplementary Material online) was used to generate a profile hidden Markov model (HMM) (supplementary methods and supplementary file S5, Supplementary Material online) for the MAFp3-equivalent region (i.e., Wreath domain). The model was tested using hmmsearch as above.
Calculation of Intron Phase Distribution Frequencies
Amphimedon queenslandica intron phase values were derived by modification of a publicly available Aqu2.1 GFF3 file (http://amphimedon.qcloud.qcif.edu.au/index.html; last accessed December 4, 2015) (Fernandez-Valverde et al. 2015), as follows. The “phase” field in GFF3 files lists the number of nucleotides between the start of an exon and the first base of the next codon; this is distinct from the standard definition of intron phase, which describes where a codon is interrupted by an intron at the end of an exon (Sharp 1981). Thus a phase 1 intron would interrupt a codon after its first nucleotide, but the next exon would be given a value of “2” in a GFF file. Therefore, “phase” field entries of “2” were changed to “1” and vice versa; “0” values did not need to be changed. Second, as the first exon of any gene is not immediately preceded by an intron, the phase values erroneously associated with these first exons were removed. Amphimedonqueenslandica Calx-beta domain-containing genes were identified from a genome-wide analysis of domain content (Hatleberg WL, personal communication), performed using hmmscan (maximum e-value 10 − 3) from the HMMER 3.1b1 package (Eddy 1998). Phase values for these Calx-beta domain-containing genes, with and without AFs included, were also extracted from the aforementioned GFF3 file.
Intron phase frequencies, standard deviations of the mean, and significance values were calculated for three datasets (the AFs, all Calx-beta domain-containing genes, and all non-AF Calx-beta domain-containing genes) as per Fedorov et al. (1992, 1998). We calculated the A. queenslandica genome-wide intron phase distribution (Pgenome), and tested whether the suite of Calx-beta domain-containing genes (Pcalx) differed statistically from this distribution; values were considered statistically significantly different if |Pgenome−Pcalx| > 3σ (Fedorov et al. 1998). We repeated this analysis by comparing the suite of AF genes (PAF) to both Pgenome and Pcalx.
Genomic data from the sponge Xestospongia testudinaria is publicly available for individual genes (http://xt.reefgenomics.org/; last accessed October 7, 2015). To determine intron phase distribution for the single X. testudinaria candidate AF identified in the present work (Xt_88826), we performed a BLASTp search between this sequence and the X. testudinaria genome (http://reefgenomics.org/blast/; last accessed October 7, 2015) and accessed the appropriate gene model from Scaffold 972 of the X. testudinaria genome assembly (http://xt.reefgenomics.org/jbrowse/; last accessed October 7, 2015). The two sequences were compared using MGAlign to determine intron phase values (Lee et al. 2003).
Sanger Sequencing-Based Analysis of Polymorphism
Three AqAF polymorphism “hotspots” (within AqAFB, AqAFC and AqAFE) and one less variable region (within AqAFE) were selected for in-depth analysis of polymorphism (fig. 3), based on identification of regions of the AqAF locus exhibiting high numbers of variants (supplementary fig. S1.2 and supplementary methods, Supplementary Material online).
Nine larval individuals (0-h post-emergence) and small slices of tissue from eight adult A. queenslandica individuals were obtained as previously described (Leys et al. 2008) and preserved in RNA Later (Ambion). RNA and genomic DNA were simultaneously extracted from individuals using TRIzol (Thermo Fisher Scientific) following manufacturer’s guidelines. Briefly, tissue was homogenized in TRIzol, phase separated with 1-bromo-3-chloropropane (BCP), and RNA precipitated from the aqueous layers using glycogen and isopropanol. Genomic DNA was back-extracted from the interphase and organic layers using 4 M guanidine thiocyanate, 50 mM sodium citrate and 1 M Tris (pH 8) and precipitated from the resulting aqueous layer using glycogen and isopropanol. After DNase I treatment (Invitrogen), cDNA was synthesized from RNA using SuperScript III reverse transcriptase (Invitrogen) and oligo(dT)15 and random pentadecamer primers (Stangegaard et al. 2006), according to manufacturer’s guidelines. AqAF gene-specific primers were designed using Primer3 (Untergasser et al. 2012) to cover < 2 kb of genomic and cDNA sequences (supplementary table S1.8, Supplementary Material online). Primer specificity was checked via BLAT search on an in-house UCSC genome browser and via BLAST search on the NCBI nucleotide database. AqAF fragments were amplified from genomic and cDNA from each individual using Phusion high-fidelity DNA polymerase (New England Biolabs) and touch-down PCR cycling. Size-correct amplicons were gel purified using the QIAquick gel extraction kit (Qiagen) and sequenced using the Big Dye Terminator 3.1 Cycle Sequencing kit (Applied Biosystems) at the Australian Genome Research Facility (AGRF). Internal and overlapping primers were used to obtain double sequence coverage of the entire fragment to ensure precision of base calling and eliminate ambiguity.
Raw sequencing chromatograms were trimmed using Geneious Pro v6.1.8 (Kearse et al. 2012). Genomic and cDNA sequences from each individual were assembled to the AqAFB gene model, and sites exhibiting read mismatches or low-quality base calls were flagged. Sites displaying putative polymorphism, heterozygosity or an RDD were identified by manual inspection. RDD calls were only made for sites where a single allele was identified in both the genomic and cDNA.
Statistical comparisons of nucleotide or amino acid change frequencies are described in supplementary methods, Supplementary Material online.
Analysis of RDDs and Gene Duplication in AqAFB
We sought to determine the number of AqAFB alleles in Larvae 3.1, 3.5, 6, 7 and 9 and Adults 5 and 9. We amplified the AqAFB hotspot region from the genomic and cDNA of each individual as described above and cloned the amplicons into pGEM-T Easy Vectors (Promega). Clones were screened for inserts and plasmid inserts were Sanger sequenced. Raw sequencing chromatograms were trimmed using Geneious Pro. Matched pairs of forward and reverse sequences from individual clones were aligned to one another using the De Novo Assemble tool. Consensus sequences were extracted for each alignment, and were manually annotated to flag read mismatches or low-quality base calls. Genomic and cDNA sequences from each individual were assembled to the AqAFB gene model, and were manually inspected to identify polymorphic sites.
Sequence Data Used for AF Identification
The sponge referred to here as “Niphatidae indet.” has tentatively been identified as belonging to the demosponge family Niphatidae based on spicule morphology, and its transcriptome has been sequenced and assembled (Gaiti F, Kocot KM, Degnan BM, unpublished data). Transcripts were assembled as per Whelan et al. (2015) and best open reading frame (ORF) predictions were generated using TransDecoder (http://transdecoder.github.io/; last accessed September 28, 2015).
Ephydatia muelleri translated messenger RNA sequences (“T-PEP”) were downloaded from Compagen (http://www.compagen.org; last accessed September 5, 2013) (Hemmrich and Bosch 2008). Gene models for Oscarella carmela were prepared as described by Grice and Degnan (2015b). Translated peptide sequences from the Sycon ciliatum genome (Fortunato et al. 2015) were provided by M. Adamska and M. Adamski.
For A. queenslandica (developmental transcriptomes) (Fernandez-Valverde et al. 2015), Aphrocallistes vastus, Chondrilla nucula, C. prolifera (Fernandez-Valverde SL, Degnan BM, unpublished data), Corticium candelabrum, Crella elegans, Ircinia fasciculata, Petrosia ficiformis, Spongilla lacustris, Pseudospongosorites suberitoides and Sycon coactum (Riesgo et al. 2012), we determined the longest ORF between stop codons for each assembled transcript using the program getorf from the EMBOSS 6.5.7 software package (Rice et al. 2000). For A. queenslandica and Cr. elegans, sequences from all available developmental stages per species were pooled prior to further analysis.
Best ORF predictions for Hyalonema populiferum, Kirkpatrickia variolosa, Latrunculia apicalis, Rossella fibulata, Sympagella nux, Tethya wilhelma (Whelan et al. 2015) were computed using TransDecoder (Kocot KM, personal communication). Cliona varians (Riesgo et al. 2014), Haliclona amboinensis (SRR1619429) and X. testudinaria (SRR1738101) transcriptomes were assembled as per Whelan et al. (2015) and ORF predictions were determined using TransDecoder (Kocot KM, personal communication).
Identification of AF-Like Sponge Sequences
Sequences of at least 100 amino acids in length from the translated transcriptomes or genomes listed in the previous section were filtered to generate a list of AF-like sequences using criteria described in figure 5. An A. queenslandica developmental transcriptome dataset was included as a point of comparison between genomic and transcriptomic sequences. Sequences encoding Calx-beta (Pfam PF03160), von Willebrand type A (VWA; Pfam PF00092), von Willebrand type D (VWD; Pfam PF00094) or Wreath domains (supplementary file S5, Supplementary Material online) were identified using hmmsearch. Sushi domains, as seen in the putative G. cydonium AF, GEOCY AF, were not included as search criteria, as this domain combination has only been observed in the AFs of one species to date and has not been well characterized. To remove redundancies, sequences within each species were clustered into groups sharing at least 90% amino acid sequence identity, using the default parameters of the cd-hit tool (Li and Godzik 2006), run using the CD-HIT Suite server (Huang et al. 2010) or, in the case of the A. queenslandica transcriptomes, the cd-hit v4.6.1 command line application. Only the representative sequence from each cluster (as determined by cd-hit; equivalent to the longest sequence) was passed through for further analysis.
Overall domain architecture for each sequence was determined using HMMER 3.0 (Eddy 1998) within the DoMosaics environment (Moore et al. 2014). Batch BLASTp searches were performed using BLAST+ v2.2.28+ and a local nr database, to identify the top hit with a maximum e-value of 10 − 4. Sequences were assigned to one of three groups based on these results (fig. 5), as follows. Group 1 sequences are those possessing one or more Wreath domains (which, for sequences not predicted to encode additional domain types or sequence features such as transmembrane domains or signal peptides, had to span 60% or more of the Wreath HMM model). Sequences possessing either three or more Calx-beta domains or at least one each of a Calx-beta and VWA or VWD domain were sorted into Group 2 if they also showed a top BLAST hit to an AqAF, GEOCY AF, MAFp3, MAFp4 or SdSLIP sequence, or into Group 3 if they did not. Only sequences in Groups 1 or 2 were considered candidate AFs and passed onto further analysis.
Presence and absence of AF candidates were mapped to a phylogenetic tree showing relationships between analysed sponge species, with tree topology based on analyses by Thacker et al. (2013), Whelan et al. (2015) and Kocot KM (personal communication).
Supplementary Material
Supplementary data and methods are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Australian Research Council (grant numbers DP0985995 to S.D. and FL110100044 to B.D.). We thank Federico Gaiti, Kevin Kocot and Selene Fernandez-Valverde for generation and assembly of some of the transcriptomes included in this analysis, and William Hatleberg for providing genome-wide Pfam annotation results for Amphimedon queenslandica. We also thank Maja Adamska and Marcin Adamski for access to genomic data for Sycon ciliatum prior to publication.
References
- Amano S. 1990. Self and non-self recognition in a calcareous sponge, Leucandra abratsbo. Biol Bull. (Woods Hole) 179:272–278. [DOI] [PubMed] [Google Scholar]
- Bork P, Doolittle RF.. 1992. Proposed acquisition of an animal protein domain by bacteria. Proc Natl Acad Sci U S A. 89:8990–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadavid LF, Powell AE, Nicotra ML, Moreno M, Buss LW.. 2004. An invertebrate histocompatibility complex. Genetics 167:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell ID, Bork P.. 1993. Epidermal growth factor-like modules. Curr Opin Struct Biol. 3:385–392. [Google Scholar]
- Cantí C, Nieto-Rostro M, Foucault I, Heblich F, Wratten J, Richards MW, Hendrich J, Douglas L, Page KM, Davies A, et al. 2005. The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A. 102:11230–11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A, Fazzari P, Tamagnone L.. 2007. Semaphorin signals in cell adhesion and cell migration: functional role and molecular mechanisms. Adv Exp Med Biol. 600:90–108. [DOI] [PubMed] [Google Scholar]
- Cauldwell CB, Henkart P, Humphreys T.. 1973. Physical properties of sponge aggregation factor. A unique proteoglycan complex. Biochemistry 12:3051–3055. [DOI] [PubMed] [Google Scholar]
- Day AJ, Campbell RD, Reid K.. 1989. The mosaic nature of the complement proteins In: Melchers F, Albert ED, Boehmer von H, Dierich MP, Pasquier Du L, Eichmann K, Gemsa D, Götze O, Kalden JR, Kaufmann S, et al. editors. Progress in Immunology. Springer-Verlag Berlin Heidelberg; p. 209–212. [Google Scholar]
- De Tomaso AW, Nyholm SV, Palmeri KJ, Ishizuka KJ, Ludington WB, Mitchel K, Weissman IL.. 2005. Isolation and characterization of a protochordate histocompatibility locus. Nature 438:454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham P, Anderson C, Rich AM, Weissmann G.. 1983. Stimulus-response coupling in sponge cell aggregation: evidence for calcium as an intracellular messenger. Proc Natl Acad Sci U S A. 80:4756–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14:755–763. [DOI] [PubMed] [Google Scholar]
- Erwin DH, Laflamme M, Tweedt SM, Sperling EA, Pisani D, Peterson KJ.. 2011. The Cambrian conundrum: early divergence and later ecological success in the early history of animals. Science 334:1091–1097. [DOI] [PubMed] [Google Scholar]
- Fedorov A, Fedorova L, Starshenko V, Filatov V, Grigor’ev E.. 1998. Influence of exon duplication on intron and exon phase distribution. J Mol Evol. 46:263–271. [DOI] [PubMed] [Google Scholar]
- Fedorov A, Suboch G, Bujakov M, Fedorova L.. 1992. Analysis of nonuniformity in intron phase distribution. Nucleic Acids Res. 20:2553–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Valverde SL, Calcino AD, Degnan BM.. 2015. Deep developmental transcriptome sequencing uncovers numerous new genes and enhances gene annotation in the sponge Amphimedon queenslandica. BMC Genomics 16:387.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernàndez-Busquets X, Burger MM.. 1997. The main protein of the aggregation factor responsible for species-specific cell adhesion in the marine sponge Microciona prolifera is highly polymorphic. J Biol Chem. 272:27839–27847. [DOI] [PubMed] [Google Scholar]
- Fernàndez-Busquets X, Burger MM.. 1999. Cell adhesion and histocompatibility in sponges. Microsc Res Tech. 44:204–218. [DOI] [PubMed] [Google Scholar]
- Fernàndez-Busquets X, Burger MM.. 2003. Circular proteoglycans from sponges: first members of the spongican family. Cell Mol Life Sci. 60:88–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernàndez-Busquets X, Gerosa D, Hess D, Burger MM.. 1998. Accumulation in marine sponge grafts of the mRNA encoding the main proteins of the cell adhesion system. J Biol Chem. 273:29545–29553. [DOI] [PubMed] [Google Scholar]
- Fernàndez-Busquets X, Kammerer RA, Burger MM.. 1996. A 35-kDa protein is the basic unit of the core from the 2 x 104-kDa aggregation factor responsible for species-specific cell adhesion in the marine sponge Microciona prolifera. J Biol Chem. 271:23558–23565. [DOI] [PubMed] [Google Scholar]
- Fernàndez-Busquets X, Körnig A, Bucior I, Burger MM, Anselmetti D.. 2009. Self-recognition and Ca2+-dependent carbohydrate-carbohydrate cell adhesion provide clues to the Cambrian explosion. Mol Biol Evol. 26:2551–2561. [DOI] [PubMed] [Google Scholar]
- Fernàndez-Busquets X, Kuhns WJ, Simpson TL, Ho M, Gerosa D, Grob M, Burger MM.. 2002. Cell adhesion-related proteins as specific markers of sponge cell types involved in allogeneic recognition. Dev Comp Immunol. 26:313–323. [DOI] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. 2014. Pfam: the protein families database. Nucleic Acids Res. 42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunato SAV, Adamski M, Ramos OM, Leininger S, Liu J, Ferrier DEK, Adamska M.. 2015. Calcisponges have a ParaHox gene and dynamic expression of dispersed NK homeobox genes. Nature 514:620–623. [DOI] [PubMed] [Google Scholar]
- Galtsoff PS. 1925. Regeneration after dissociation (An experimental study on sponges) I. Behavior of dissociated cells of Microciona prolifera under normal and altered conditions. J Exp Zool B. 42:183–221. [Google Scholar]
- Gauthier M, Degnan BM.. 2008. Partitioning of genetically distinct cell populations in chimeric juveniles of the sponge Amphimedon queenslandica. Dev Comp Immunol. 32:1270–1280. [DOI] [PubMed] [Google Scholar]
- Gloria-Soria A, Moreno MA, Yund PO, Lakkis FG, Dellaporta SL, Buss LW.. 2012. Evolutionary genetics of the hydroid allodeterminant alr2. Mol Biol Evol. 29:3921–3932. [DOI] [PubMed] [Google Scholar]
- Grice LF, Degnan BM.. 2015a. How to build an allorecognition system: a guide for prospective multicellular organisms In: Ruiz-Trillo I, Nedelcu AM, editors. Evolutionary transitions to multicellular life. Vol. 2 Springer Dordrecht Heidelberg; New York London: p. 395–424. [Google Scholar]
- Grice LF, Degnan BM.. 2015b. The origin of the ADAR gene family and animal RNA editing. BMC Evol Biol. 15:4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundacker D, Leys SP, Schröder HC, Müller IM.. 2001. Isolation and cloning of a C-type lectin from the hexactinellid sponge Aphrocallistes vastus: a putative aggregation factor. Glycobiology 11:21–29. [DOI] [PubMed] [Google Scholar]
- Hemmrich G, Bosch TCG.. 2008. Compagen, a comparative genomics platform for early branching metazoan animals, reveals early origins of genes regulating stem-cell differentiation. BioEssays 30:1010–1018. [DOI] [PubMed] [Google Scholar]
- Henkart P, Humphreys S, Humphreys T.. 1973. Characterization of sponge aggregation factor. A unique proteoglycan complex. Biochemistry 12:3045–3050. [DOI] [PubMed] [Google Scholar]
- Hildemann WH, Johnson IS, Jokiel PL.. 1979. Immunocompetence in the lowest metazoan phylum: transplantation immunity in sponges. Science 204:420–422. [DOI] [PubMed] [Google Scholar]
- Hilge M, Aelen J, Vuister GW.. 2006. Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol Cell 22:15–25. [DOI] [PubMed] [Google Scholar]
- Huang Y, Niu B, Gao Y, Fu L, Li W.. 2010. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert-David N, Garrone R.. 1993. A six-armed, tenascin-like protein extracted from the Porifera Oscarella tuberculata (Homosclerophorida). Eur J Biochem. 216:255–260. [DOI] [PubMed] [Google Scholar]
- Humphreys T, Humphreys S, Moscona AA.. 1960. A procedure for obtaining completely dissociated sponge cells. Biol Bull. (Woods Hole) 119:294–294. [Google Scholar]
- Jarchow J, Fritz J, Anselmetti D, Calabro A, Hascall VC, Gerosa D, Burger MM, Fernàndez-Busquets X.. 2000. Supramolecular structure of a new family of circular proteoglycans mediating cell adhesion in sponges. J Struct Biol. 132:95–105. [DOI] [PubMed] [Google Scholar]
- Jumblatt JE, Schlup V, Burger MM.. 1980. Cell-cell recognition: specific binding of Microciona sponge aggregation factor to homotypic cells and the role of calcium ions. Biochemistry 19:1038–1042. [DOI] [PubMed] [Google Scholar]
- Karadge UB, Gosto M, Nicotra ML.. 2015. Allorecognition proteins in an invertebrate exhibit homophilic interactions. Curr Biol. 25:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Krogh A, Sonnhammer ELL.. 2004. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 338:1027–1036. [DOI] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Matthes DJ, Goodman CS.. 1993. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell 75:1389–1399. [DOI] [PubMed] [Google Scholar]
- Lee BT, Tan TW, Ranganathan S.. 2003. MGAlignIt: a web service for the alignment of mRNA/EST and genomic sequences. Nucleic Acids Res. 31:3533–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leys SP, Larroux C, Gauthier M, Adamska M, Fahey B, Richards GS, Degnan SM, Degnan BM.. 2008. Isolation of Amphimedon developmental material. Cold Spring Harb Protoc. 2008:pdb.prot5095. [DOI] [PubMed] [Google Scholar]
- Leys SP, Mackie GO, Meech RW.. 1999. Impulse conduction in a sponge. J Exp Biol. 202:1139–1150. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A.. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- Lin S, Zhang H, Spencer DF, Norman JE, Gray MW.. 2002. Widespread and extensive editing of mitochondrial mRNAS in dinoflagellates. J Mol Biol. 320:727–739. [DOI] [PubMed] [Google Scholar]
- McKitrick TR, De Tomaso AW.. 2010. Molecular mechanisms of allorecognition in a basal chordate. Sem Immunol. 22:34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misevic GN, Burger MM.. 1990. Involvement of a highly polyvalent glycan in the cell-binding of the aggregation factor from the marine sponge Microciona prolifera. J Cell Biochem. 43:307–314. [DOI] [PubMed] [Google Scholar]
- Misevic GN, Burger MM.. 1993. Carbohydrate-carbohydrate interactions of a novel acidic glycan can mediate sponge cell adhesion. J Biol Chem. 268:4922–4929. [PubMed] [Google Scholar]
- Mokady O, Buss LW.. 1996. Transmission genetics of allorecognition in Hydractinia symbiolongicarpus (Cnidaria: Hydrozoa). Genetics 143:823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AD, Held A, Terrapon N, Weiner J, Bornberg-Bauer E.. 2014. DoMosaics: software for domain arrangement visualization and domain-centric analysis of proteins. Bioinformatics 30:282–283. [DOI] [PubMed] [Google Scholar]
- Moscona AA. 1968. Cell aggregation: properties of specific cell-ligands and their role in the formation of multicellular systems. Dev Biol. 18:250–277. [DOI] [PubMed] [Google Scholar]
- Müller WEG, Conrad J, Zahn RK, Steffen R, Uhlenbruck G, Müller I.. 1984. Cell adhesion molecule in the hexactinellid Aphrocallistes vastus: species-unspecific aggregation factor. Differentiation 26:30–35. [Google Scholar]
- Müller WEG, Gamulin V, Rinkevich B, Spreitzer I, Weinblum D, Schröder HC.. 1994. Ubiquitin and ubiquitination in cells from the marine sponge Geodia cydonium. Biol Chem Hoppe-Seyler 375:53–60. [DOI] [PubMed] [Google Scholar]
- Müller WEG, Koziol C, Müller IM, Wiens M.. 1999. Towards an understanding of the molecular basis of immune responses in sponges: the marine demosponge Geodia cydonium as a model. Microsc Res Tech. 44:219–236. [DOI] [PubMed] [Google Scholar]
- Müller WEG, Müller I, Zahn R.. 1976. Species-specific aggregation factor in sponges V. Influence on programmed syntheses. Biochim Biophys Acta 418:217–225. [DOI] [PubMed] [Google Scholar]
- Müller WEG, Rottmann M, Diehl-Seifert B, Kurelec B, Uhlenbruck G, Schröder HC.. 1987. Role of the aggregation factor in the regulation of phosphoinositide metabolism in sponges: possible consequences on calcium efflux and on mitogenesis. J Biol Chem. 262:9850–9858. [PubMed] [Google Scholar]
- Müller WEG, Zahn RK.. 1973. Purification and characterization of a species-specific aggregation factor in sponges. Exp Cell Res. 80:95–104. [DOI] [PubMed] [Google Scholar]
- Müller WEG. 1982. Cell membranes in sponges In: Bourne GH, Danielli JF, editors. International review of cytology. Vol. 77 New York: International Review of Cytology; p. 129–181. [Google Scholar]
- Mungpakdee S, Shinzato C, Takeuchi T, Kawashima T, Koyanagi R, Hisata K, Tanaka M, Goto H, Fujie M, Lin S, et al. 2014. Massive gene transfer and extensive RNA editing of a symbiotic dinoflagellate plastid genome. Genome Biol Evol. 6:1408–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicotra ML, Powell AE, Rosengarten RD, Moreno M, Grimwood J, Lakkis FG, Dellaporta SL, Buss LW.. 2009. A hypervariable invertebrate allodeterminant. Curr Biol. 19:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nydam ML, Hoang TA, Shanley KM, De Tomaso AW.. 2013a. Molecular evolution of a polymorphic HSP40-like protein encoded in the histocompatibility locus of an invertebrate chordate. Dev Comp Immunol. 41:128–136. [DOI] [PubMed] [Google Scholar]
- Nydam ML, Netuschil N, Sanders E, Langenbacher A, Lewis DD, Taketa DA, Marimuthu A, Gracey AY, De Tomaso AW.. 2013b. The candidate histocompatibility locus of a basal chordate encodes two highly polymorphic proteins. PLoS One 8:e65980.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholm SV, Passegue E, Ludington WB, Voskoboynik A, Mitchel K, Weissman IL, De Tomaso AW.. 2006. fester, a candidate allorecognition receptor from a primitive chordate. Immunity 25:163–173. [DOI] [PubMed] [Google Scholar]
- Patthy L. 1987. Intron-dependent evolution: preferred types of exons and introns. FEBS Lett. 214:1–7. [DOI] [PubMed] [Google Scholar]
- Patthy L. 1988. Detecting distant homologies of mosaic proteins: analysis of the sequences of thrombomodulin, thrombospondin complement components C9, C8 alpha and C8 beta, vitronectin and plasma cell membrane glycoprotein PC-1. J Mol Biol. 202:689–696. [DOI] [PubMed] [Google Scholar]
- Pfeifer K, Frank W, Schröder HC, Gamulin V, Rinkevich B, Batel R, Müller IM, Müller WEG.. 1993. Cloning of the polyubiquitin cDNA from the marine sponge Geodia cydonium and its preferential expression during reaggregation of cells. J Cell Sci. 106:545–554. [DOI] [PubMed] [Google Scholar]
- Powell AE, Moreno M, Gloria-Soria A, Lakkis FG, Dellaporta SL, Buss LW.. 2011. Genetic background and allorecognition phenotype in Hydractinia symbiolongicarpus. G3 (Bethesda) 1:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AE, Nicotra ML, Moreno MA, Lakkis FG, Dellaporta SL, Buss LW.. 2007. Differential effect of allorecognition loci on phenotype in Hydractinia symbiolongicarpus (Cnidaria: Hydrozoa). Genetics 177:2101–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Richardson JS. 1981. The anatomy and taxonomy of protein structure. Adv Protein Chem. 34:167–339. [DOI] [PubMed] [Google Scholar]
- Riesgo A, Andrade SC, Sharma PP, Novo M, Pérez-Porro AR, Vahtera V, González VL, Kawauchi GY, Giribet G.. 2012. Comparative description of ten transcriptomes of newly sequenced invertebrates and efficiency estimation of genomic sampling in non-model taxa. Front Zool. 9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesgo A, Peterson K, Richardson C, Heist T, Strehlow B, McCauley M, Cotman C, Hill M, Hill A.. 2014. Transcriptomic analysis of differential host gene expression upon uptake of symbionts: a case study with Symbiodinium and the major bioeroding sponge Cliona varians. BMC Genomics 15:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich B, Porat R, Goren M.. 1995. Allorecognition elements on a urochordate histocompatibility locus indicate unprecedented extensive polymorphism. Proc R Soc Lond B. 259:319–324. [Google Scholar]
- Rosa SFP, Powell AE, Rosengarten RD, Nicotra ML, Moreno MA, Grimwood J, Lakkis FG, Dellaporta SL, Buss LW.. 2010. Hydractinia allodeterminant alr1 resides in an immunoglobulin superfamily-like gene complex. Curr Biol. 20:1122–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann M, Schröder HC, Gramzow M.. 1987. Specific phosphorylation of proteins in pore complex-laminae from the sponge Geodia cydonium by the homologous aggregation factor and phorbol ester. Role of protein kinase C in the phosphorylation of DNA topoisomerase II. Embo J. 6:3939–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabella C, Faszewski E, Himic L, Colpitts KM, Kaltenbach J, Burger MM, Fernàndez-Busquets X.. 2007. Cyclosporin a suspends transplantation reactions in the marine sponge Microciona prolifera. J Immunol. 179:5927–5935. [DOI] [PubMed] [Google Scholar]
- Schröder HC, Kuchino Y, Gramzow M, Kurelec B, Friese U, Uhlenbruck G, Müller WEG.. 1988. Induction of ras gene expression by homologous aggregation factor in cells from the sponge Geodia cydonium. J Biol Chem. 263:16334–16340. [PubMed] [Google Scholar]
- Scofield VL, Schlumpberger JM, West LA, Weissman IL.. 1982. Protochordate allorecognition is controlled by a MHC-like gene system. Nature 295:499–502. [DOI] [PubMed] [Google Scholar]
- Sharp P. 1981. Speculations on RNA splicing. Cell 23:643–646. [DOI] [PubMed] [Google Scholar]
- Smith L, Hildemann WH.. 1986. Allograft rejection, autograft fusion and inflammatory responses to injury in Callyspongia diffusa (Porifera; Demospongia). Proc R Soc Lond B. 226:445–464. [DOI] [PubMed] [Google Scholar]
- Smith LC, Ghosh J, Buckley KM, Clow LA, Dheilly NM, Haug T, Henson JH, Li C, Lun CM, Majeske AJ, et al. 2010. Echinoderm immunity In: Söderhäll K, editor. Invertebrate innate immunity. Vol. 708 Austin, TX: Landes Bioscience and Springer Science+Business Media; p. 260–301. [DOI] [PubMed] [Google Scholar]
- Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier MEA, Mitros T, Richards GS, Conaco C, Dacre M, Hellsten U, et al. 2010. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466:720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangegaard M, Høgh Dufva I, Dufva M.. 2006. Reverse transcription using random pentadecamer primers increases yield and quality of resulting cDNA. Biotechniques 40:649–657. [DOI] [PubMed] [Google Scholar]
- Thacker RW, Hill AL, Hill MS, Redmond NE, Collins AG, Morrow CC, Spicer L, Carmack CA, Zappe ME, Pohlmann D, et al. 2013. Nearly complete 28S rRNA gene sequences confirm new hypotheses of sponge evolution. Integr Comp Biol. 53:373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG.. 2012. Primer3 – new capabilities and interfaces. Nucleic Acids Res. 40:e115–e115.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskoboynik A, Newman AM, Corey DM, Sahoo D, Pushkarev D, Neff NF, Passarelli B, Koh W, Ishizuka KJ, Palmeri KJ, et al. 2013. Identification of a colonial chordate histocompatibility gene. Science 341:384–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan NV, Kocot KM, Moroz LL, Halanych KM.. 2015. Error, signal, and the placement of Ctenophora sister to all other animals. Proc Natl Acad Sci U S A. 112:5773–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker C, Hynes R.. 2002. Distribution and evolution of von Willebrand/Integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell 13:3369–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens M, Korzhev M, Krasko A, Thakur NL, Perovic-Ottstadt S, Breter HJ, Ushijima H, Diehl-Seifert B, Müller IM, Müller WEG.. 2005. Innate immune defense of the sponge Suberites domuncula against bacteria involves a MyD88-dependent signaling pathway. J Biol Chem. 280:27949–27959. [DOI] [PubMed] [Google Scholar]
- Williams AF, Barclay AN.. 1988. The immunoglobulin superfamily – domains for cell surface recognition. Annu Rev Immunol. 6:381–405. [DOI] [PubMed] [Google Scholar]
- Wilson HV. 1907. On some phenomena of coalescence and regeneration in sponges. J Exp Zool. 5:245–258. [Google Scholar]
- Wimmer W, Blumbach B, Diehl-Seifert B, Koziol C, Batel R, Steffen R, Müller IM, Müller WEG.. 1999. Increased expression of integrin and receptor tyrosine kinase genes during autograft fusion in the sponge Geodia cydonium. Cell Adhes Commun. 7:111–124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.