


Abstract
Single nucleotide polymorphisms (SNPs) are an increasingly important tool for genetic and biomedical research. However, the accumulated sequence information on allelic variation is not matched by an understanding of the effect of SNPs on the functional attributes or ‘molecular phenotype’ of a protein. Towards this aim we developed SNPeffect, an online resource of human non-synonymous coding SNPs (nsSNPs) mapping phenotypic effects of allelic variation in human genes. SNPeffect contains 31 659 nsSNPs from 12 480 human proteins. The current release of SNPeffect incorporates data on protein stability, integrity of functional sites, protein phosphorylation and glycosylation, subcellular localization, protein turnover rates, protein aggregation, amyloidosis and chaperone interaction. The SNP entries are accessible through both a search and browse interface and are linked to most major biological databases. The data can be displayed as detailed descriptions of individual SNPs or as an overview of all SNPs for a given protein. SNPeffect will be regularly updated and can be accessed at http://snpeffect.vib.be/.
INTRODUCTION
As a result of the ongoing genomic efforts worldwide an enormous amount of sequence information on human DNA variation is accumulating (1). Single nucleotide polymorphisms (SNPs) are the most common form of allelic variation observed in human populations. NCBI's dbSNP (1) currently contains 4 540 241 validated human entries out of which 45 896 are non-synonymous coding SNPs (nsSNPs). Although only 1–3% of the human genome is taken up by protein-coding regions, this small subset of coding SNPs (together with SNPs in gene regulatory regions) has the highest likelihood of being functionally relevant. Most of what is known about the genetics of diseases comes either from studies on rare monogenetic diseases or from family studies of common diseases that have identified rare high-risk variants. However, modest-risk variants probably have a higher impact on public health because they have a higher frequency in human populations than much less frequent but high-risk variants [the so-called common disease-common variant hypothesis (2)]. For instance, only 5% of all Alzheimer's cases can be attributed to more than 150 rare high-risk alleles (3) whereas the presence of the Apoɛ allele in late-onset Alzheimer's has been estimated to be 20% (4). The availability of SNPs offers the possibility to develop high-density genetic maps for whole-genome association analyses, allowing the identification of genetic polymorphisms contributing to susceptibility for common polygenetic diseases. If we aim at truly linking genetic variation with phenotypic variation and natural selection a first step will be to describe how allelic variation affects the functional attributes or molecular phenotype of a protein (5). The effect of many nsSNPs will probably be neutral as natural selection will have removed mutations on essential positions. However, a fraction of nsSNPs will display phenotypic variation at the molecular level that will, by interaction with other proteins, affect intermediate phenotypes relevant from a clinical viewpoint [for instance, low-density lipoprotein (LDL) levels for risk assessment of myocardial infarction]. Assessment of non-neutral SNPs is mainly based on phylogenetic information (i.e. correlation with residue conservation) extended to a certain degree with structural approaches [e.g. see PolyPhen (6) and the method of Chasman and Adams (7)].
Here we present SNPeffect, a web-server describing the effect of nsSNPs on the molecular phenotype of human proteins. Our goal is to assign the effect of SNPs to specific functional attributes. For each protein in which SNPs are mapped, we compare their effect on protein stability and folding, aggregation and amyloidosis, catalytic sites and binding sites, phosphorylation and glycosylation sites, cellular localization and protein turnover.
MOLECULAR PHENOTYPING OF ALLELIC VARIANTS
SNPeffect uses not only in-house but also other publicly available biocomputational tools to predict the effect of nsSNPs on functional properties defining the molecular phenotype of proteins. The functional properties evaluated in the current release of SNPeffect are classified into three categories: (i) properties affecting protein folding and stability, (ii) properties affecting functional sites and binding sites and (iii) properties affecting cellular processing of a protein. The results belonging to each of these categories can be accessed in a summarized way in the protein-centred view or can be looked up in detail for each SNP in the SNP-centred view. Easy toggling between both views is provided.
Protein folding and stability
Whenever a high-quality structure (better than 3 Å) is available, the mutation is modelled and its energetic effect is evaluated with the FoldX force field (8). The FoldX force field was developed for the fast and accurate estimation of the free change upon mutation on the stability of a protein or a protein complex. FoldX has been validated on a test database of more than 1000 mutants from more than 20 different proteins. FoldX currently yields a correlation of 0.78 with a SD of 0.41 kcal/mol. Changes in protein aggregation and amyloidosis are evaluated with the algorithms TANGO (9) and AmyScan (10,11), respectively. TANGO is a statistical mechanics algorithm that identifies the regions of a protein sequence involved in the process of β-sheet aggregation. TANGO was validated on a set of 250 peptides. AmyScan scans a protein sequence for six-residue amyloidogenic stretches. Amyloid fragment identification relies on a sequence pattern extracted from a saturation mutagenesis analysis on a de novo designed amyloid peptide and has been validated experimentally. In addition, PROF (12,13) was used to predict secondary structure, solvent accessibility and transmembrane regions.
Functional sites
Integrity of active sites is checked for all enzymes having an entry in the CSA database (14). The Catalytic Site Atlas (CSA) is a database documenting enzyme active sites and catalytic residues in enzymes for which three-dimensional structures are available. The authors defined a classification of catalytic residues, which includes only those residues thought to be directly involved in some aspect of the reaction catalysed by an enzyme. The CSA contains two types of entries: (i) original hand-annotated entries, derived from the primary literature and (ii) homologous entries, found by PSI-BLAST alignment (using an E-value cut-off of 0.00005) to one of the original entries. The equivalent residues, which align in sequence to the catalytic residues found in the original entry are documented. To further analyse functional sites, we are currently in the process of mapping disruption in protein–protein interaction sites using the FoldX force field described above.
Cellular processing
Two tools are used to predict subcellular localization: PA Subcellular and Psort II. PA Subcellular uses established machine learning techniques to predict the localization of the protein, namely, mitochondrion, nucleus, endoplasmic reticulum, extracellular, cytoplasm, plasma membrane, Golgi, lysosome or peroxisome. Rather than using sequence information alone, this method uses database text annotations from homologues and machine learning to substantially improve the prediction of subcellular location. The authors report an accuracy of 92–94% (15). Psort II uses a k-nearest neighbour classifier that is trained on yeast sequences from Swiss-Prot (16). The subcellular localizations that can be predicted are cytoskeletal, cytoplasmic, nuclear, mitochondrial, vesicles of secretory system, endoplasmic reticulum, Golgi, vacuolar, plasma membrane, peroxisomal and extracellular localizations including the cell wall. Effects on post-translational modification sites are also screened: phosphorylation is checked with PhosphoBase (17) and glycosylation with O-Glycobase (18). PhosphoBase contains information about phosphorylated residues in proteins and data about peptide phosphorylation by a variety of protein kinases. The data are collected from literature and compiled into a common format. PhosphoBase covers phosphorylatable serine, threonine and tyrosine residues. O-GLYCBASE is a database of glycoproteins with O-linked glycosylation sites. Entries with at least one experimentally verified O-glycosylation site have been compiled from protein sequence databases and literature. Finally, protein turnover rates depend strongly on the identity of the N-terminal residue. A highly destabilizing N-terminal residue such as arginine can lead to a half-life as short as 2 min, whereas other amino acids produce half-lives of around a day. We predict protein turnover rates using the N-terminal rules as described by Varshavsky and coworkers (19,20).
RESULTS AND DISCUSSION
The current version of SNPeffect contains 31 659 entries. As shown in Table 1 most of these SNPs are neutral, producing no change of molecular phenotype (e.g. only 0.01% of nsSNPs cause a change in subcellular localization; Figure 1 and Table 1). This is to be expected since large changes in molecular phenotype such as cellular localization, turnover-rate or disruption of active sites are very likely to be deleterious and will be eliminated by natural selection. Whenever they are present, however, they form interesting targets of investigation since they could be associated with disease. The only molecular phenotype that is significantly affected in more than 50% of the cases analysed is the stability of the protein. Even if, due to a lack of structural information, the set of SNPs analysed for stability is very small, it is to be expected that this trend will be maintained. Change in stability of a protein is not necessarily deleterious for protein function and is much more likely to create a more subtle range of functional effects by modulating protein–protein interactions. To supplement the lack of structural information, we are currently generating high-quality homology models using the FoldX force field.
Table 1. Number of differences between wild type (WT) and SNP per property analysed in SNPeffect.
| Phenotypic property | Number of SNPs analysed | Number of SNPs with significant change | Percentage of SNPs with significant change |
|---|---|---|---|
| Aggregation—TANGO | 30 738a | 907 | 2.95 |
| Amylogenic regions—AmyScan | 31 659 (of which 28 693 had amylogenic regions) | 897 | 2.83 |
| Stability—FoldX | 93b | 52 | 55.91 |
| Subcellular localization—PA Subcellular | 31 659 | 290 | 0.92 |
| Turnover-rate | 31 659 | 28 | 0.09 |
| Phosphorylation sites—PhosphoBase | 18 214c | 2 | 0.01 |
| Glycosylation sites—O-GlycBase | 18 214c | 0 | 0 |
| Active sites—CSA | 2101d | 0 | 0 |
| Hsp70 binding | 31 659 (of which 20 376 had Hsp70 binding regions) | 399 | 1.3 |
aThe remaining 921 SNP entries caused a runtime error in the TANGO execution.
bOnly high-resolution PDB structures were used to predict stability changes for the SNPs. This number will increase in the future as we will continue by using models for the FoldX prediction.
cQuerying the PhospoBase and O-GLYCBASE was limited by a non-complete mapping between RefSeq and Swiss-Prot identifiers.
dOnly the SNPs with PDB identifiers could be used to query the CSA.
Figure 1.

Examples of disruptive effects caused by allelic variation. From the 31 659 SNPs analysed by PA Subcellular, 290 show a clear change in subcellular localization. Arrows signify differences in localization between wild type (WT) and SNP. The label of each arrow shows how many times the transition from one classification to another occurs in the SNPeffect dataset.
DATABASE ACCESS
SNPeffect can be accessed at http://snpeffect.vib.be/. Both a search and a browse interface are offered, which give the option to look for SNPs or for proteins; this leads either to a SNP-centred view or a protein-centred view, respectively (Figure 2). The SNP view reports all the information of one specific SNP in tabs according to the classification of functional properties described earlier. These tabs include a graphical as well as detailed numerical overview of the different phenotypic effects, as well as a link to the properties of the wild type. The data can be displayed as detailed descriptions of individual SNPs or as an overview of all SNPs for a given protein. Summaries of phenotypic properties are given at the first tab of the SNP-centred view and the ‘SNPs’ tab of the protein centred view. The meaning of output values and scores for the various properties and how they are translated to ‘significant change’ flags is described in the manual section of the website and in the Supplementary Materials of this paper.
Figure 2.
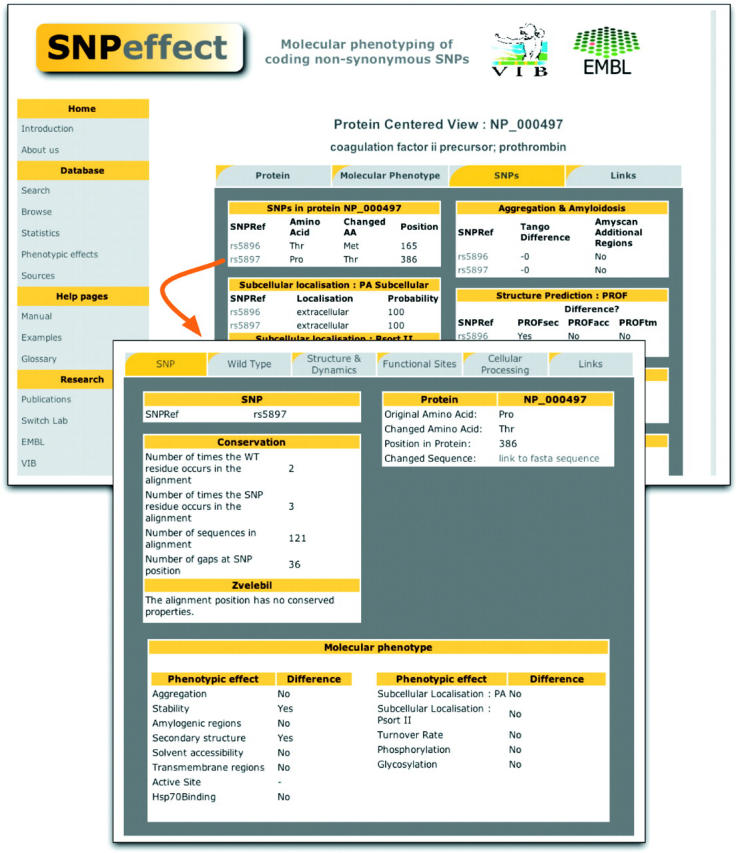
From protein centred to SNP centred view. SNPeffect can be searched for proteins or for SNPs. In the protein centred view (background) an overview is given of all known nsSNPs for a given protein as well as of all phenotypic effects of those SNPs on the function of the wild type. By clicking on a particular SNP a detailed description of the phenotypic effects and the general information of that variant is displayed in six tabs (foreground).
Search interface
The search interface allows searching for various identifiers (DB cross-references), full-text search in the protein description. Filters for proteins with structure (PDB file) or disease-related proteins can be applied to the search result.
Browse interface
Through this interface the possibility to browse enzyme codes, Protein Data Bank (PDB) entries, OMIM entries and Swiss-Prot accession codes is provided. After choosing a browse category a specific entry can be clicked on and a list of SNPs or proteins for that entry are shown. The same filters as in the search interface can be applied.
DATA SOURCES AND LINKS
The raw data source for SNPeffect is NCBI's dbSNP (1). From dbSNP nsSNPs were extracted, whereas related protein entries where extracted from the NCBI Protein resources. These NCBI accessions (rs for SNP entries and RefSeq for protein entries) are the key identifiers in the SNPeffect database. Where available protein structures were retrieved from the PDB (21). For accurate prediction only crystal structures with a resolution better than 3 Å were selected; NMR structures and low-resolution crystal structures were rejected. The protein entries were also checked with two protein indexes, EBI's International Protein Index (22) and PDBSprotEC (23), to maximize cross-references between the different biological databases. Proteins for which no cross-reference was available in these indexes, were blasted against the Swiss-Prot database (24) to obtain the Swiss-Prot accession number where possible. SNPeffect entries are linked to several important biological databases including OMIM (25), Gene Ontology (26), the Brenda Enzyme database (27), Swiss-Prot/TREMBL (24), Pfam (28) and structural databases such as SCOP (29), CATH (30), CSA (31) and PDBSUM (32).
DATABASE STATUS AND FUTURE WORK
Currently SNPeffect contains data on 31 659 human nsSNPs. Future work will aim at including high-quality homology models generated using FoldX to increase the structural information available in SNPeffect. The list of properties analysed is continually being extended and will soon include protein folding and dynamics, metal ion binding, protein–protein interactions and protein–DNA interactions. Subsequently, we will also create a murine SNPeffect database. The database will be regularly updated as new SNP data and information on phenotypic effects become available.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Manuela Lopez de la Paz for pre-publication use of AmyScan. We would also like to thank Luc Van Wiemeersch and Els Herregods for help with setting up the computing facilities.
REFERENCES
- 1.Thorisson G.A. and Stein,L.D. (2003) The SNP Consortium website: past, present and future. Nucleic Acids Res., 31, 124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reich D.E. and Lander,E.S. (2001) On the allelic spectrum of human disease. Trends Genet., 17, 502–510. [DOI] [PubMed] [Google Scholar]
- 3.Rocchi A., Pellegrini,S., Siciliano,G. and Murri,L. (2003) Causative and susceptibility genes for Alzheimer's disease: a review. Brain Res. Bull., 61, 1–24. [DOI] [PubMed] [Google Scholar]
- 4.Slooter A.J., van Duijn,C.M., Bots,M.L., Ott,A., Breteler,M.B., De Voecht,J., Wehnert,A., de Knijff,P., Havekes,L.M., Grobbee,D.E. et al. (1998) Apolipoprotein E genotype, atherosclerosis, and cognitive decline: the Rotterdam study. J. Neural Transm. Suppl., 53, 17–29. [DOI] [PubMed] [Google Scholar]
- 5.Bork P., Jensen,L.J., von Mering,C., Ramani,A.K., Lee,I. and Marcotte,E.M. (2004) Protein interaction networks from yeast to human. Curr. Opin. Struct. Biol., 14, 292–299. [DOI] [PubMed] [Google Scholar]
- 6.Ramensky V., Bork,P. and Sunyaev,S. (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res., 30, 3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chasman D. and Adams,R.M. (2001) Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J. Mol. Biol., 307, 683–706. [DOI] [PubMed] [Google Scholar]
- 8.Guerois R., Nielsen,J.E. and Serrano,L. (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol., 320, 369–387. [DOI] [PubMed] [Google Scholar]
- 9.Escamilla-Fernandez A.M., Rousseau,F., Schymkowitz,J.W. and Serrano,L. (2004) Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol., 22, 1302–1306. [DOI] [PubMed] [Google Scholar]
- 10.de la Paz M.L. and Serrano,L. (2004) Sequence determinants of amyloid fibril formation. Proc. Natl Acad. Sci. USA, 101, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez De La Paz M., Goldie,K., Zurdo,J., Lacroix,E., Dobson,C.M., Hoenger,A. and Serrano,L. (2002) De novo designed peptide-based amyloid fibrils. Proc. Natl Acad. Sci. USA, 99, 16052–16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rost B., Fariselli,P. and Casadio,R. (1996) Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci., 5, 1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rost B. and Sander,C. (1993) Prediction of protein secondary structure at better than 70-percent accuracy. J. Mol. Biol., 232, 584–599. [DOI] [PubMed] [Google Scholar]
- 14.Porter C.T., Bartlett,G.J. and Thornton,J.M. (2004) The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res., 32, D129–D133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Z., Szafron,D., Greiner,R., Lu,P., Wishart,D.S., Poulin,B., Anvik,J., Macdonell,C. and Eisner,R. (2004) Predicting subcellular localization of proteins using machine-learned classifiers. Bioinformatics, 20, 547–556. [DOI] [PubMed] [Google Scholar]
- 16.Nakai K. and Horton,P. (1999) PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci., 24, 34–35. [DOI] [PubMed] [Google Scholar]
- 17.Kreegipuu A., Blom,N. and Brunak,S. (1999) PhosphoBase, a database of phosphorylation sites: release 2.0. Nucleic Acids Res., 27, 237–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta R., Birch,H., Rapacki,K., Brunak,S. and Hansen,J. (1999) O-GLYCBASE version 4.0: a revised database of O-glycosylated proteins. Nucleic Acids Res., 27, 370–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonda D.K., Bachmair,A., Wunning,I., Tobias,J.W., Lane,W.S. and Varshavsky,A. (1989) Universality and structure of the N-end rule. J. Biol. Chem., 264, 16700–16712. [PubMed] [Google Scholar]
- 20.Bachmair A., Finley,D. and Varshavsky,A. (1986) In vivo half-life of a protein is a function of its amino-terminal residue. Science, 234, 179–186. [DOI] [PubMed] [Google Scholar]
- 21.Bourne P.E., Westbrook,J.D., Berman,H.M., Gilliland,G.L., Flippen-Anderson,J.L. and Team,P. (2003) The Protein Data Bank (PDB) as a research tool. Abstr. Pap. Am. Chem. Soc., 226, U302–U302. [Google Scholar]
- 22.Kersey P., Duarte,J., Williams,A., Karavidopoulou,Y., Birney,E. and Apweiler,R. (2004) The International Protein Index: an integrated database for proteomics experiments. Proteomics, 4, 1985–1988. [DOI] [PubMed] [Google Scholar]
- 23.Martin A.C.R. (2004) PDBSprotEC: a web-accessible database linking PDB chains to EC numbers via SwissProt. Bioinformatics, 20, 986–988. [DOI] [PubMed] [Google Scholar]
- 24.Boeckmann B., Bairoch,A., Apweiler,R., Blatter,M.C., Estreicher,A., Gasteiger,E., Martin,M.J., Michoud,K., O'Donovan,C., Phan,I. et al. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res., 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamosh A., Scott,A.F., Amberger,J., Bocchini,C., Valle,D. and McKusick,V.A. (2002) Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res., 30, 52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris M.A., Clark,J., Ireland,A., Lomax,J., Ashburner,M., Foulger,R., Eilbeck,K., Lewis,S., Marshall,B., Mungall,C. et al. (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res., 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schomburg I., Chang,A., Ebeling,C., Gremse,M., Heldt,C., Huhn,G. and Schomburg,D. (2004) BRENDA, the enzyme database: updates and major new developments. Nucleic Acids Res., 32, D431–D433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bateman A., Coin,L., Durbin,R., Finn,R.D., Hollich,V., Griffiths-Jones,S., Khanna,A., Marshall,M., Moxon,S., Sonnhammer,E.L. et al. (2004) The Pfam protein families database. Nucleic Acids Res., 32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreeva A., Howorth,D., Brenner,S.E., Hubbard,T.J., Chothia,C. and Murzin,A.G. (2004) SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res., 32, D226–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orengo C.A., Pearl,F.M. and Thornton,J.M. (2003) The CATH domain structure database. Methods Biochem. Anal., 44, 249–271. [DOI] [PubMed] [Google Scholar]
- 31.Bartlett G.J., Porter,C.T., Borkakoti,N. and Thornton,J.M. (2002) Analysis of catalytic residues in enzyme active sites. J. Mol. Biol., 324, 105–121. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski R. (2001) PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res., 29, 221–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.