

Supplemental digital content is available in the text.
Key Words: binge eating disorder, extension, lisdexamfetamine dimesylate, safety and tolerability
Abstract
Background
A 12-month, open-label extension study assessed the long-term safety and tolerability of lisdexamfetamine dimesylate (LDX) in adults with binge eating disorder (BED).
Methods
Adults (aged 18–55 y) with BED who completed 1 of 3 antecedent studies were enrolled in a 52-week, open-label extension study (dose optimization, 4 weeks [initial titration dose, 30-mg LDX; target doses, 50- or 70-mg LDX]; dose maintenance, 48 weeks). Safety evaluations included the occurrence of treatment-emergent adverse events (TEAEs), vital sign and weight assessments, and Columbia-Suicide Severity Rating Scale responses.
Results
Of the 604 enrolled participants, 599 (521 women and 78 men) comprised the safety analysis set, and 369 completed the study. Mean (SD) LDX exposure was 284.3 (118.84) days; cumulative LDX exposure duration was 12 months or longer in 344 participants (57.4%). A total of 506 participants (84.5%) reported TEAEs (TEAEs leading to treatment discontinuation, 54 [9.0%]; severe TEAEs, 42 [7.0%]; serious TEAEs, 17 [2.8%]). Treatment-emergent adverse events reported in greater than or equal to 10% of participants were dry mouth (27.2%), headache (13.2%), insomnia (12.4%), and upper respiratory tract infection (11.4%). Mean (SD) changes from antecedent study baseline in systolic and diastolic blood pressure, pulse, and weight at week 52/early termination (n = 597) were 2.19 (11.043) and 1.77 (7.848) mm Hg, 6.58 (10.572) beats per minute, and −7.04 (7.534) kg, respectively. On the Columbia-Suicide Severity Rating Scale, there were 2 positive responses for any active suicidal ideations; there were no positive responses for suicidal behavior or completed suicides.
Conclusions
In this 12-month, open-label, extension study, the long-term safety and tolerability of LDX in adults with BED were generally consistent with its established profile for attention-deficit/hyperactivity disorder.
Lisdexamfetamine dimesylate (LDX) is approved by the US Food and Drug Administration (FDA) for treating adults with moderate to severe binge eating disorder (BED).1 Lisdexamfetamine dimesylate is absorbed by the gastrointestinal tract and subsequently metabolized to its active form, d-amphetamine, primarily by red blood cells after oral administration.2 The pharmacokinetics of LDX has been described in detail in a recent review.3 In brief, oral administration of LDX (30–70 mg) to adults is associated with rapid increases in plasma amphetamine levels (maximum concentrations, 32.2–88.9 ng/mL; time to maximum concentration, 3.5–5.5 hours), with the reported elimination half-life of amphetamine ranging from 9.69 to 15 hours.3
Approval of LDX was based on the results of a single phase 2 study and 2 phase 3, placebo-controlled, double-blind studies.1,4 In the phase 2 study, 50- and 70-mg LDX (but not 30-mg LDX) demonstrated efficacy versus placebo in decreasing binge eating days per week (primary efficacy end point) in adults with BED.4 In the 2 phase 3 pivotal trials,1 dose-optimized LDX (50 or 70 mg) produced statistically significant and clinically meaningful reductions compared with placebo in binge eating days per week (primary efficacy end point) in adults with BED. Furthermore, LDX demonstrated statistically greater improvement than placebo on key secondary efficacy end points in these studies, with the changes on the Clinical Global Impression-Improvement (CGI-I) scale, 4-week cessation of binge eating at end point, BED-related obsessive and compulsive psychopathology on the Yale-Brown Obsessive Compulsive Scale modified for Binge Eating, and percentage of body weight change also being considered clinically meaningful.1
In these short-term studies,1,4 the safety and tolerability profile of LDX was generally similar to its established safety profile in attention-deficit/hyperactivity disorder (ADHD).5–7 Treatment-emergent adverse events (TEAEs) reported by greater than or equal to 10% of participants with BED treated with LDX in any study were dry mouth, decreased appetite, insomnia, and headache.1,4 Across studies,1,4 increases (mean [SD]) from baseline in systolic blood pressure (SBP) and pulse at final visit/early termination (ET) with LDX ranged from 0.1 (9.85) to 1.45 (10.818) mm Hg for SBP and from 3.8 (11.57) to 6.31 (9.505) beats per minute for pulse; mean (SD) diastolic blood pressure (DBP) increases from baseline at the final visit/ET were 1.06 (7.905) and 1.83 (7.956) mm Hg in the 2 phase 3 studies.1
To understand the long-term safety and tolerability of LDX in adults with BED, a 12-month, open-label extension study was conducted. The primary objective of the study was to evaluate the long-term safety and tolerability of LDX in adults with BED as measured by the occurrence of TEAEs, responses on the Columbia-Suicide Severity Rating Scale (C-SSRS), and evaluation of vital signs, weight, and electrocardiogram (ECG). Secondary objectives included the evaluation of LDX effects on clinical outcome measures using the CGI-I and the Eating Disorder Examination Questionnaire (EDE-Q).
MATERIALS AND METHODS
Study Design and Treatment
This 52-week, open-label extension study (ClinicalTrials.gov, NCT01657019) was conducted at 86 sites in the United States, Germany, and Spain and enrolled adults with BED who completed 1 of the 3 antecedent studies (NCT01291173, NCT01718483, and NCT01718509) described previously.1,4 The study consisted of a 2-week screening phase (only for those participants with an enrollment gap that was 30 or more days from antecedent study completion), a 52-week open-label phase (4 weeks of LDX dose optimization followed by 48 weeks of LDX dose maintenance), and a 1-week follow-up period. Postbaseline on-treatment study visits were conducted during each week of the dose optimization phase (weeks 1–4) and at 4-week intervals during the dose maintenance phase (weeks 8–52).
In the open-label, dose optimization phase, participants were titrated to 50- or 70-mg LDX regardless of their antecedent study treatment. During week 1, all participants received 30-mg LDX (for initial titration only). During week 2, all participants received 50-mg LDX. Participants were titrated to 70-mg LDX based on safety, tolerability, and clinical response based on participants' daily self-reported binge eating diaries (as assessed and confirmed by experienced and trained clinicians) during weeks 3 and 4; the overall assessment of safety and tolerability was the key factor when deciding whether a dose increase was justified. Investigators were to evaluate each participant's overall clinical condition (safety, tolerability, and clinical response) at each visit and had the option to downtitrate to 50-mg LDX if 70-mg LDX was not tolerated. During the dose maintenance phase, a single adjustment between 70- and 50-mg LDX was allowed based on investigator assessment of tolerability and clinical response. When a dose titration was required, assessments of vital signs, adverse events (AEs), the 12-lead ECG, and the C-SSRS were to be performed. Participants not tolerating 50-mg LDX were discontinued.
The study protocol, informed consent document, and relevant supporting information were approved by ethics committees and regulatory agencies before study initiation. The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice, the principles of the Declaration of Helsinki, and all applicable local ethical and legal requirements. All participants provided written informed consent before performance of any study procedures.
Participants
Inclusion and Exclusion Criteria
Study eligibility was based on the final treatment visit or the follow-up visit of the antecedent study if the enrollment gap was less than 30 days from antecedent study completion or on the extension study screening visit to reestablish baseline if the enrollment gap was 30 or more days from antecedent study completion. Eligible adults (aged 18–55 y at antecedent study consent) had completed 1 of 3 antecedent studies,1,4 with no clinically significant AEs during the antecedent study or clinically significant or relevant abnormalities that precluded LDX exposure.
Key exclusion criteria included having a history of symptomatic cardiovascular disease or serious cardiac problems; having moderate to severe hypertension; having resting sitting SBP of greater than 139 mm Hg or DBP of greater than 89 mm Hg; having a lifetime history of psychosis, mania, hypomania, dementia, or ADHD; being considered a suicide risk, previously attempting suicide, or currently having active suicidal ideation; having a lifetime stimulant abuse or dependence history or an abuse or dependence history on substances other than stimulants within the past 6 months; having known or suspected intolerance or hypersensitivity to LDX or related compounds; having a history of significant neurological or cerebrovascular disease; having used an investigational product in an observational clinical study within 30 days of screening; or having participated in a previous clinical LDX trial other than the specified antecedent studies. Female participants of childbearing potential were required to screen negative on blood and urine pregnancy tests and be willing to use acceptable contraceptive methods.
When the enrollment gap was 30 or more days from antecedent study completion, additional eligibility criteria included having a body mass index of 18 to 45 kg/m2 and having a confirmed diagnosis of BED from the antecedent study based on the eating disorder module of the Structured Clinical Interview for the Diagnostic and Statistical Manual of Mental Disorders (DSM), Fourth Edition, Text Revision and the EDE-Q. Additional exclusion criteria included having current anorexia nervosa or bulimia nervosa, receiving psychotherapy or weight loss support for BED (<3 months of screening), having a comorbid psychiatric disorder controlled with prohibited medications or uncontrolled with significant symptoms or a condition or symptom that may confound study assessments, having a Montgomery-Åsberg Depression Rating Scale score of 18 or greater at screening, initiating lipid-lowering medication less than 3 months before screening, having a positive drug test at screening, or having used stimulants or other psychoactive agents within 7 days of open-label baseline.
Study End Points
Safety and tolerability assessments included AEs, vital signs, weight changes, ECGs, laboratory evaluations, and C-SSRS scores. Adverse events were collected at all study visits from the time of informed consent through the end of follow-up and were categorized by the investigator based on their intensity (mild, moderate, or severe), seriousness (serious or not serious), and relatedness to treatment (related or not related). Vital signs (SBP, DBP, and pulse) and weight were assessed at all study visits. Vital signs were measured after the participant was seated for at least 5 minutes and were based on a mean of 3 measurements separated by approximately 2 minutes. Clinical laboratory assessments were collected at screening and weeks 12, 24, and 52/ET. A 12-lead ECG was assessed at screening (when the enrollment gap was ≥30 days from antecedent study completion); at weeks 1 through 4; at weeks 20, 36, and 52/ET; and at follow-up. Electrocardiograms were collected in triplicate (separated by approximately 2 minutes) at screening and once at all the other visits. The C-SSRS,8 which is a semistructured interview designed to assess suicidal ideation and behavior and nonsuicidal self-injurious behavior, was assessed at screening and at all study visits through follow-up.
Secondary clinical outcome–related end points included assessment of the CGI-I9 and the patient-completed EDE-Q.10 Assessments of the frequency of binge eating and of scores on the Yale-Brown Obsessive Compulsive Scale modified for Binge Eating were not included as end points in this study. The CGI-I was administered by a trained and experienced clinician at each on-treatment postbaseline visit and rated improvement on a 7-point scale (1, very much improved, to 7, very much worse). When the enrollment gap was 30 or more days from antecedent study completion, the CGI-Severity assessment was performed at screening. The EDE-Q was assessed at screening and at each on-treatment study visit from weeks 4 through 52/ET. The EDE-Q is a 28-item questionnaire derived from the Eating Disorder Examination Interview, which assesses the presence and frequency of abnormal eating behaviors (questions 13–18) and measures eating psychopathology (questions 1–12 and 19–28) for the past 28 days.10,11 The latter questions are rated on a 7-point scale ranging from 0 to 6 (higher scores indicate more severe pathology)10 and generate 4 subscales (restraint, eating concern, weight concern, and shape concern), which can be used to produce a global score.
Data Presentation
Safety and tolerability are presented descriptively in the safety analysis set (participants taking ≥1 study drug dose and having ≥1 postdose safety assessment). For vital sign and ECG changes, baseline refers to antecedent study baseline (if the enrollment gap was <30 days from antecedent study completion) or the last vital sign value collected at or before the extension study baseline visit and the average of the first 3 readable ECGs at or before the extension study baseline visit (if the enrollment gap was ≥30 days from antecedent study completion). For weight changes, baseline refers to the antecedent study baseline (if the enrollment gap was <30 days from antecedent study completion) or the last value collected at or before the extension study baseline visit (if the enrollment gap was ≥30 days from antecedent study completion).
Data from the CGI-I and EDE-Q are presented descriptively in the full analysis set (FAS; participants from the safety analysis set having ≥1 postdose secondary end point assessment [CGI-I or EDE-Q]). Scores on the CGI-I were dichotomized as improved (scores of 1 [very much improved] or 2 [much improved]) or not improved (scores of 3 [minimally improved] to 7 [very much worse]). The percentage of participants who improved at each assessment on the CGI-I was reported; participants who discontinued for any reason before week 52 were categorized as not improved on the CGI-I. For the EDE-Q, mean changes from baseline in EDE-Q global and subscale scores were summarized at weeks 52 and 52/ET. Baseline for the EDE-Q refers to antecedent study baseline (if the enrollment gap was <30 days from antecedent study completion) or to extension study baseline or the last value collected at or before the extension study baseline (if the enrollment gap was ≥30 days from antecedent study completion). A post hoc summary was also conducted to descriptively summarize responses to question 15 of the EDE-Q, which asks “Over the past 28 days, on how many days have such episodes of overeating occurred (ie, you have eaten an unusually large amount of food and have had a sense of loss of control at the time)?”10
RESULTS
Participant Disposition, Demographics, and LDX Exposure
Of the 604 enrolled participants, 599 were included in the safety analysis set, and 597 were included in the FAS; 369 participants (61.1%) completed the study, and 235 participants (38.9%) did not complete the study (Supplemental Digital Content 1, http://links.lww.com/JCP/A437). Among the participants in the safety analysis set, the most frequently reported reasons for discontinuation were participant withdrawal (10.8% [65/599]), other (9.6% [58/599]), and AEs (9.1% [55/599]); 3 participants (0.5%) discontinued because of lack of efficacy. For reasons for discontinuation categorized as “other,” most resulted from a site closure, and none were related to AEs. Most participants were women, white, and overweight or obese (Supplemental Digital Content 2, http://links.lww.com/JCP/A438).
The mean (SD) duration of LDX exposure was 284.3 (118.84) days. Of the 599 participants with exposure data, the cumulative duration of LDX exposure was 3 or more months in 524 participants (87.5%), 6 or more months in 455 participants (76.0%), and 12 or more months in 344 participants (57.4%). Total LDX exposure (person-time in days) for the 599 participants in the safety analysis set was 170,544 days. At the end of dose optimization, 179 participants (29.9%) had an optimized dose of 50-mg LDX, and 389 (64.9%) had an optimized dose of 70-mg LDX.
Safety and Tolerability
Treatment-Emergent AEs
Most participants reported TEAEs (Table 1), and most of the reported TEAEs were of mild or moderate intensity. There were no deaths during the study. Listings of serious or severe TEAEs and of TEAEs leading to treatment discontinuation are provided in the footnote of Table 1. Cholecystitis was the only serious AE (SAE) reported in more than 1 participant (n = 3). A detailed review of these events did not suggest a direct association with LDX, and none was considered to be related to LDX by the investigator. The only SAEs considered to be related to LDX by the investigator were coincident events of supraventricular tachycardia (mild intensity) and acute coronary syndrome (moderate intensity) reported in 1 participant who indicated that a double dose of 50-mg LDX may have been taken on the day of the events. Both events resolved 1 day after study discontinuation after metoprolol (for the supraventricular tachycardia) and acetylsalicylic acid, atorvastatin calcium, and enoxaparin sodium (for the acute coronary syndrome).
TABLE 1.
TEAEs, Vital Sign Outliers, and ECG Changes From Baseline: Safety Analysis Set
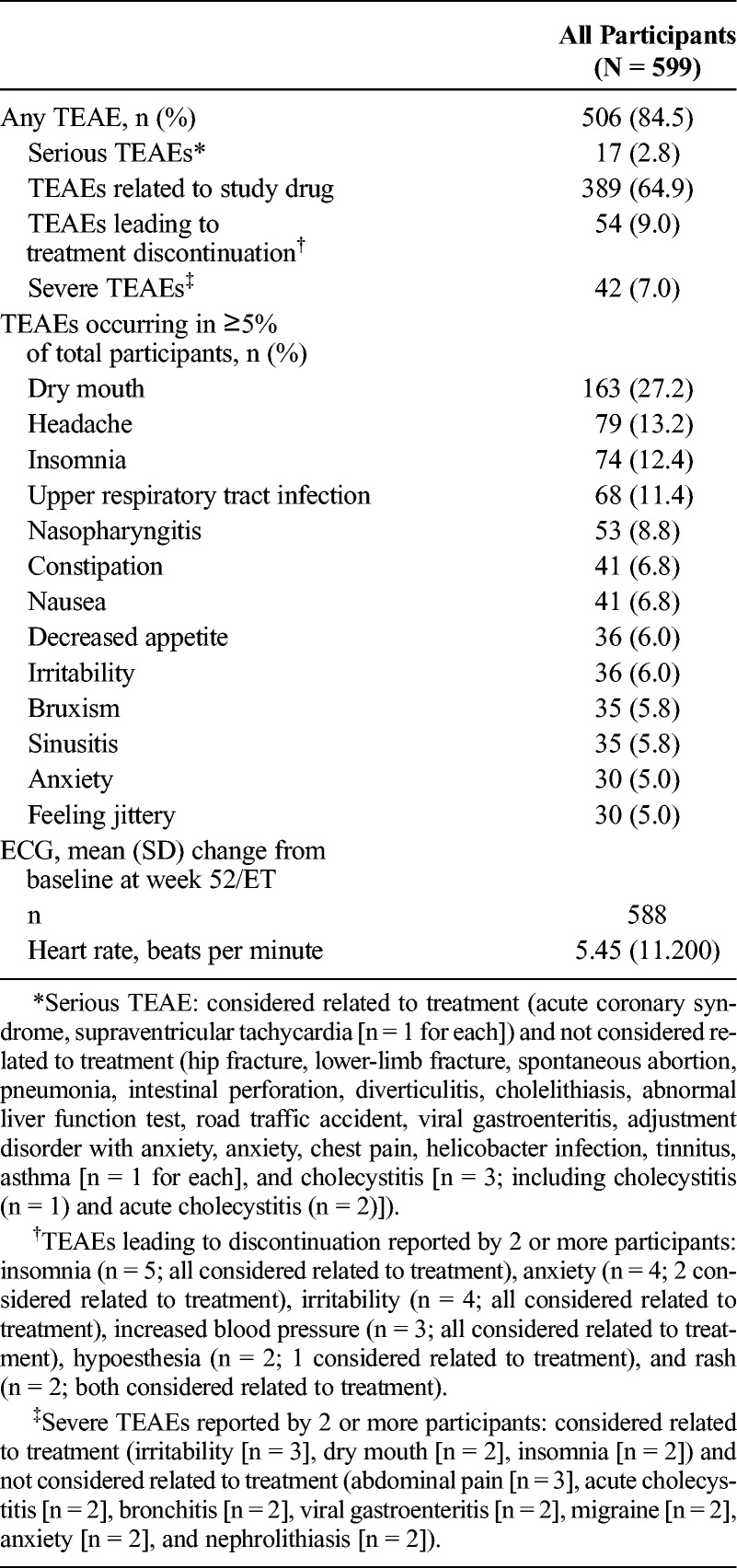
Most serious TEAEs, severe TEAEs, and TEAEs leading to treatment discontinuation resolved. Treatment-emergent AEs that were serious, or severe, or that led to treatment discontinuation that had not resolved or from which the participant had not fully recovered by the follow-up visit and were considered not related to treatment (n = 1 participant for each) were hip fracture/lower limb fracture (serious, severe intensity, led to treatment discontinuation), a case of adjustment disorder with anxiety (serious, severe intensity, did not lead to treatment discontinuation), an intervertebral disk protrusion (not serious, severe intensity, did not lead to treatment discontinuation), and hypoesthesia (not serious, mild intensity, led to treatment discontinuation). Treatment-emergent AEs that were serious, or severe, or led to treatment discontinuation that had not resolved or from which the participant had not fully recovered by the follow-up visit and were considered related to treatment (n = 1 participant for each) were a case of increased severe headaches (not serious, severe intensity, led to treatment discontinuation), increased blood pressure (not serious, moderate intensity, led to treatment discontinuation), fatigue (not serious, moderate intensity, led to treatment discontinuation), alopecia/hair loss (not serious, moderate intensity, led to treatment discontinuation), and intermittent drug craving (not serious, moderate intensity, led to treatment discontinuation). The participant reporting intermittent drug craving was a man with no history of drug dependence (lifetime or current), as assessed by a structured diagnostic interview, who had been titrated to 70-mg LDX. During the maintenance phase, the participant reported intermittent drug craving and was subsequently discontinued; the event was ongoing at the time of the last assessment.
The most frequently reported TEAEs (occurring in ≥10% of participants) were dry mouth, headache, insomnia, and upper respiratory tract infection (Table 1). Treatment-emergent AEs reported with preferred terms that were part of the psychiatric disorder system organ class (SOC) were observed in 31.2% (187/599) of the participants. During the study, TEAEs related to psychosis, hallucination, or mania were reported in 9 participants (abnormal behavior, 1 [0.2%]; affect lability, 2 [0.3%]; apathy, 1 [0.2%]; emotional poverty, 1 [0.2%]; hypomania, 1 [0.2%]; and logorrhea, 3 [0.5%]). Treatment-emergent AEs reported with preferred terms that were part of the cardiac disorder SOC were observed in 5.3% (32/599) of the participants. The most frequently occurring TEAEs with preferred terms from the psychiatric disorder SOC (occurring in ≥2% of participants) were insomnia (74/599 [12.4%]), bruxism (35/599 [5.8%]), anxiety (30/599 [5.0%]), and initial insomnia (25/599 [4.2%]). The most frequently reported TEAE with preferred terms from the cardiac disorder SOC (occurring in ≥2% of participants) was tachycardia (14/599 [2.3%]).
Vital Signs
During the study, mean (SD) SBP and DBP (Fig. 1A) and pulse (Fig. 1B) increased compared with baseline. The time points at which the greatest mean (SD) changes from baseline were observed were week 28 for SBP (2.78 [10.560] mm Hg, n = 438), week 16 for DBP (2.09 [8.215] mm Hg, n = 517), and week 28 for pulse (8.85 [10.246] beats per minute, n = 438), after which values tended to decrease slightly but remain elevated relative to baseline. In an outlier analysis of potentially clinically significant vital sign changes (Supplemental Digital Content 3, http://links.lww.com/JCP/A439), fewer than 5% of the participants exhibited changes in SBP, DBP, or pulse that were deemed to be of potential clinical significance.
FIGURE 1.
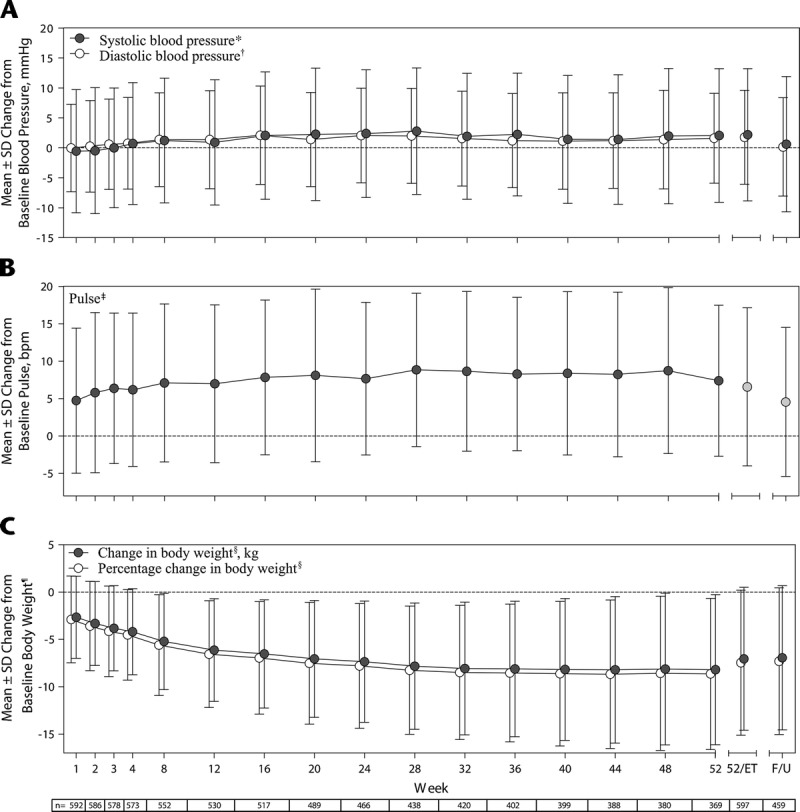
Change from baseline (Note: For vital sign changes, baseline refers to the antecedent study baseline [if enrollment gap was <30 days from antecedent study completion] or the last value collected at or before the extension study baseline visit [if enrollment gap was ≥30 days from antecedent study completion]). For weight changes, baseline refers to the antecedent study baseline [if the enrollment gap was <30 days from antecedent study completion] or the last value collected at or before the extension study baseline [if the enrollment gap was ≥30 days from antecedent study completion] in (A) SBP and DBP, (B) pulse, and (C) body weight by treatment week, safety analysis set. F/U indicates follow-up. *SBP baseline, 116.14 (10.309) mm Hg (n = 599). †DBP baseline, 76.32 (7.725) mm Hg (n = 599). ‡Pulse baseline, 72.18 (9.637) beats per minute (n = 599). §For change in body weight and percentage change in body weight, n = 529 at week 12, n = 516 at week 16, n = 387 at week 44, and n = 458 at follow-up.
The ECG-based heart rate changes are summarized in Table 1. The time point at which the greatest mean (SD) change from baseline for ECG-based heart rate was observed was week 36 (7.26 [10.21] beats per minute, n = 393). The mean (SD) change from baseline for the Fridericia-corrected QT interval (QTcF) at week 52/ET was −2.23 (12.974) (n = 588). No participant had a QTcF of 500 milliseconds or greater or an increase from baseline QTcF of 60 milliseconds or greater.
Other Safety and Tolerability Assessments
Mean decreases in weight from baseline (mean [SD] baseline weight, 94.06 [20.052] kg) were observed during the study (Fig. 1C). The time point at which the greatest mean (SD) weight decrease from baseline was observed was week 44 (−8.21 [7.733] kg, n = 387), which corresponded to an 8.67% (7.848%) decrease from baseline, after which weight remained stable. Mean changes from baseline in clinical laboratory assessments were small in magnitude and generally not of clinical significance.
There were no suicidal behaviors reported on the C-SSRS. A positive response for “wish to be dead” (passive ideation) on the C-SSRS was reported by 7 participants. Two of these 7 individuals also reported a positive response for any active suicidal ideation on the C-SSRS, of which one also reported a positive response to “nonspecific active suicidal thoughts” that was not considered a TEAE by the investigators. The other individual also reported positive responses to “nonspecific suicidal thoughts” and “active suicidal ideation with any method” on the C-SSRS and was the only study participant to report a suicide-related TEAE (suicidal ideation of moderate intensity that led to treatment discontinuation). This TEAE was not considered by the investigator to be related to the investigational product or to be an SAE, and it resolved after treatment discontinuation. Four participants had a positive response for “nonsuicidal self-injurious behavior”; none of these individuals reported suicide-related TEAEs during the study.
Secondary End Points
Changes in the percentage of participants from the FAS categorized as improved on the CGI-I are summarized in Figure 2. During the study, more than half of the participants in the FAS were categorized as improved on the CGI-I. At week 52/ET, 89.8% (536/597) of the participants were categorized as improved on the CGI-I, with most participants having scores of 1 (“very much improved,” 67.0% [400/597]). At week 52/ET, 4 participants exhibited worsening on the CGI-I (“minimally worse,” n = 3; “much worse,” n = 1).
FIGURE 2.
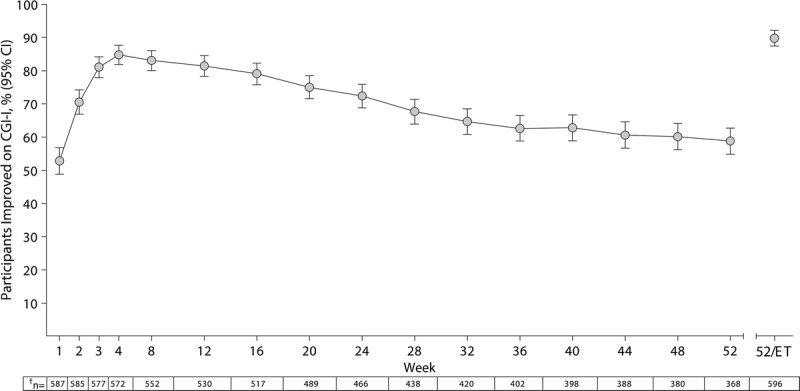
Percentage (95% confidence interval [CI]) of improved* participants by treatment week on the CGI-I, FAS. *Participants categorized as improved on the CGI-I had scores of 1 (very much improved) or 2 (much improved); those who discontinued from the study for any reason before week 52 were categorized as not improved, which accounted for study attrition in the most conservative fashion. †Number of participants with a CGI-I assessment at the given treatment week (FAS, n = 597).
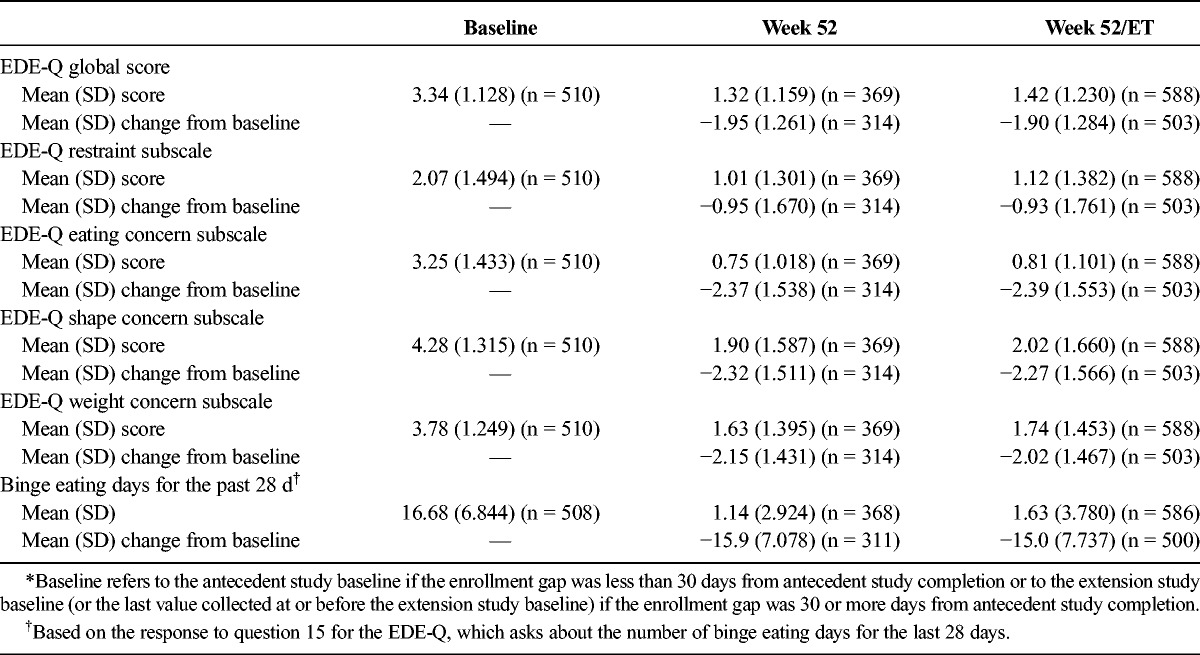
The EDE-Q scores and the number of binge eating days for the past 28 days are summarized in Table 2. Mean (SD) EDE-Q global and subscale scores and the number of binge eating days for the past 28 days at weeks 52 and 52/ET were numerically lower than those at baseline.
TABLE 2.
EDE-Q Baseline Scores and Changes From Baseline*: FAS
DISCUSSION
In this 52-week, open-label extension study, the safety and tolerability profile associated with 12 months of exposure to LDX (50 or 70 mg) was consistent with previous observations from short-term studies of LDX in adults with moderate to severe BED1,4 and with the established profile of LDX for the treatment of ADHD.5–7
Most participants in the study reported TEAEs. Most TEAEs were of mild to moderate intensity and did not preclude continuation of treatment, and with few exceptions, most serious or severe TEAEs resolved during the study or after discontinuation of treatment. Treatment with LDX was associated with mean increases from baseline in SBP, DBP, and pulse. The assessment of ECG did not reveal patterns of changes that were of clinical concern, and there was no evidence of clinically significant QT interval prolongation. One suicide-related TEAE (suicidal ideation of moderate intensity) led to study treatment discontinuation. No suicidal behaviors were reported during the study, and C-SSRS responses did not indicate that there were associations between LDX treatment and active or passive suicidal ideation or suicidal behavior. No deaths were reported in the study.
The tolerability and safety profile of LDX observed in this long-term extension trial was similar to the profiles observed in short-term trials of LDX in individuals with BED1,4; the observed occurrence of intercurrent illnesses (eg, sinusitis, nasopharyngitis, and upper respiratory infection) was also generally consistent with previous reports of long-term exposure to LDX in adults with ADHD.5–7 The most commonly reported TEAEs in this extension study were dry mouth, headache, insomnia, and upper respiratory tract infection. Dry mouth, headache, and insomnia were also among the most frequently reported TEAEs across the short-term LDX studies1,4; the overall frequency of these TEAEs was comparable with short-term BED studies1,4 and long-term ADHD studies.5–7 Insomnia, bruxism, anxiety, and initial insomnia were the most frequently occurring TEAEs with preferred terms from the psychiatric disorder SOC, and tachycardia was the most frequently reported TEAE with a preferred term from the cardiac disorder SOC. Anxiety and palpitations were also reported by more than 2% of study participants in the short-term studies.1,4 The increases from baseline in SBP and pulse observed in this study are also consistent with observations from the short-term studies of LDX1,4; DBP was increased in this study and in the pivotal phase 3 studies of LDX.1 Across all studies of LDX for BED, weight loss has been reported during LDX treatment.1,4
Long-term exposure to LDX in this population of adults with BED was not associated with new safety signals compared with the established profile of LDX in ADHD, the other indication for which it is approved.5–7 In long-term, open-label studies in children, adolescents, and adults with ADHD,5–7 the most commonly reported TEAEs associated with any dose of LDX included decreased appetite (14.3%–33%), insomnia (12.1%–19.5%), headache (17.2%–20.8%), and upper respiratory tract infection (11%–21.9%). Increases in SBP, DBP, and pulse have also been observed in long-term studies of LDX in individuals with ADHD.5–7 The increases in blood pressure and pulse in the current study were similar in magnitude to those reported in adolescents and adults with ADHD5,7 but higher than those reported in children with ADHD6 (perhaps due to differences in the age of the study populations). Although LDX is not an FDA-approved weight loss treatment and is not recommended for weight loss,12 weight loss was observed in the current study. Decreases in weight have also been reported in adults with ADHD after long-term exposure to LDX.5 The larger magnitude decreases in weight with LDX in the current study may be partially attributable to the higher mean baseline weight (94.06 kg) and higher proportion of participants meeting the criteria for being overweight or obese (92.2%) in this study compared with the study of LDX in adults with ADHD (approximately 79 kg and 70%, respectively).5
Lisdexamfetamine dimesylate is the only pharmacotherapy approved by the FDA for the treatment of BED, and data on the long-term safety and tolerability of other pharmacologic agents that have been investigated for potential use in BED (but are not FDA approved) are limited.13 In a 42-week, open-label topiramate extension study conducted in individuals with BED,14 greater than or equal to 30% of participants reported multiple AEs, including paresthesia, dry mouth, headache, taste perversion, cognitive problems, and dizziness; there were no substantive changes in vital signs.14 In individuals with BED treated with orlistat or placebo for 24 weeks,15 orlistat was associated with a significantly greater decrease in DBP than placebo (from 81.1 [12.3] mm Hg at baseline to 78.0 [11.8] mm Hg at week 24 with orlistat; from 80.5 [12.0] mm Hg at baseline to 81.7 [12.4] mm Hg with placebo) but no differential change in SBP compared with placebo (from 123.2 [18.6] mm Hg at baseline to 122.0 [18.2] mm Hg at week 24 with orlistat; from 122.1 [18.2] mm Hg at baseline to 121.5 [18.0] with placebo). Overall, there are limited data on the long-term effects of pharmacotherapy for BED. The current study addresses this data gap and suggests that the long-term safety and tolerability profile of LDX for the treatment of BED is similar to the profiles observed in short-term LDX trials in BED1,4 and with previous long-term studies of LDX for the treatment for adults with ADHD.5–7
Examination of the CGI-I and EDE-Q provided supportive evidence for decreased global BED severity and eating pathology with LDX treatment. During the course of the 52-week extension study, more than half of the study participants were categorized as improved on the CGI-I. Reductions in EDE-Q global scores, EDE-Q subscale scores, and the number of binge eating days for the past 28 days were also observed at the end of the study. However, it is important to note that the study attrition rate and the lack of a placebo control group should be considered when interpreting both the CGI-I and EDE-Q data. To address the issue of study attrition for the CGI-I data, the conservative approach of categorizing all participants who discontinued from the study (regardless of the reason) as not improved was taken. Because this was an open-label safety study with no comparator arm, the CGI-I and EDE-Q data should not be interpreted as being indicative of the long-term effectiveness or efficacy of LDX treatment.
Several other study limitations should also be considered when interpreting the data presented in this report. Specifically, participants were 87% women, 76.8% white individuals, and more than 90% overweight or obese individuals; had a confirmed BED diagnosis based on DSM, Fourth Edition, Text Revision criteria (rather than DSM, Fifth Edition criteria); and did not have current psychiatric (including psychosis and mania) or medical comorbidities that were significant and required treatment. Thus, it is not known (as has been commented upon16) how these findings would generalize to a more heterogeneous population of adults with BED. Furthermore, there were design differences (including varying exposure times to different LDX doses across the placebo and LDX treatment arms) between the phase 2 and 3 studies. The presented results, which pooled all participants, therefore may have created the possibly erroneous impression of consistency of findings across all the design features of these studies.16
In conclusion, in this open-label extension study, the 12-month safety and tolerability of LDX in adults with BED were generally consistent with the safety profile observed in 3 short-term antecedent studies in adults with protocol-defined moderate to severe BED1,4 and with the established safety profile of LDX for the treatment of ADHD.5–7 The most frequently reported TEAEs were those known to be associated with LDX or with commonly occurring intercurrent illnesses. Lisdexamfetamine dimesylate treatment was associated with increased blood pressure and pulse. Clinical laboratory and ECG assessments did not indicate new safety concerns or clinically meaningful trends. For those adults remaining in the study, the CGI-I and EDE-Q findings were supportive of decreased global BED severity and eating pathology.
Supplementary Material
ACKNOWLEDGMENTS
Under author direction, Stefan Kolata, PhD (a former employee of Complete Healthcare Communications, LLC [CHC]), and Craig Slawecki, PhD (a current employee of CHC), provided writing and formatting assistance for this manuscript. Editorial assistance in the form of proofreading, copyediting, and fact checking was also provided by CHC. The content of this manuscript, the ultimate interpretation, and the decision to submit it for publication in the Journal of Clinical Psychopharmacology were made by the authors.
AUTHOR DISCLOSURE INFORMATION
M. Gasior was a full-time employee of Shire as of December 2015 and is currently an employee of BTG International; she holds stock and/or stock options in Shire and BTG International. J. Hudson has received consulting fees and grant support from Shire; has received consulting fees from Genentech, Pronutria, Roche, and Sunovion; and has received grant support from Genentech and Sunovion. J. Quintero has received consulting fees from Shire, Janssen, Grünenthal, and Lilly and has received grant support from Shire, Otsuka, and Mutua Madrileña. M.C. Ferreira-Cornwell is a former employee of Shire and a current employee of GlaxoSmithKline; she holds stock and/or stock options in Shire and GlaxoSmithKline. J. Radewonuk is a former employee of Shire and a current employee of GlaxoSmithKline; she holds stock and/or stock options in Shire and GlaxoSmithKline. S.L. McElroy is a consultant to and has received grant support from Shire and is also a consultant to or a member of the scientific advisory boards of Alkermes, Bracket, Corcept, F. Hoffmann-LaRoche Ltd, Ironshore, MedAvante, Myriad, Naurex, Novo Nordisk, Sunovion, and Teva and has received grant support from the Agency for Healthcare Research & Quality, Allergan, Alkermes, AstraZeneca, Azevan, Cephalon (now Teva), Forest, Lilly, Marriott Foundation, National Institute of Mental Health, Orexigen, Pfizer, Takeda, and Transcept. She is also an inventor on US patent no. 6,323,236 B2, “Use of Sulfamate Derivatives for Treating Impulse Control Disorders,” and, along with the patent's assignee, University of Cincinnati (Cincinnati, OH), has received payments from Johnson & Johnson, which has exclusive rights under the patent.
Footnotes
This study was funded by the study sponsor, Shire Development LLC (Lexington, MA).
Supplemental digital contents are available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal’s Web site (www.psychopharmacology.com).
REFERENCES
- 1.McElroy SL, Hudson J, Ferreira-Cornwell MC, et al. Lisdexamfetamine dimesylate for adults with moderate to severe binge eating disorder: results of two pivotal phase 3 randomized controlled trials. Neuropsychopharmacology. 2015;41:1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pennick M. Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr Dis Treat. 2010;6:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comiran E, Kessler FH, Froehlich PE, et al. Lisdexamfetamine: a pharmacokinetic review. Eur J Pharm Sci. 2016;89:172–179. [DOI] [PubMed] [Google Scholar]
- 4.McElroy SL, Hudson JI, Mitchell JE, et al. Efficacy and safety of lisdexamfetamine for treatment of adults with moderate to severe binge-eating disorder: a randomized clinical trial. JAMA Psychiatry. 2015;72:235–246. [DOI] [PubMed] [Google Scholar]
- 5.Weisler R, Young J, Mattingly G, et al. Long-term safety and effectiveness of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. CNS Spectr. 2009;14:573–585. [DOI] [PubMed] [Google Scholar]
- 6.Findling RL, Childress AC, Krishnan S, et al. Long-term effectiveness and safety of lisdexamfetamine dimesylate in school-aged children with attention-deficit/hyperactivity disorder. CNS Spectr. 2008;13:614–620. [DOI] [PubMed] [Google Scholar]
- 7.Findling RL, Cutler AJ, Saylor K, et al. A long-term open-label safety and effectiveness trial of lisdexamfetamine dimesylate in adolescents with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2013;23:11–21. [DOI] [PubMed] [Google Scholar]
- 8.Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168:1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guy W. Clinical Global Impressions. Rockville, MD: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse and Mental Health Administration; NIMH Psychopharmacology Research Branch; 1976. [Google Scholar]
- 10.Fairburn CG. Cognitive Behavior Therapy and Eating Disorders. New York, NY: Guilford Press; 2008. [Google Scholar]
- 11.Fairburn CG, Beglin SJ. Assessment of eating disorders: interview or self-report questionnaire? Int J Eat Disord. 1994;16:363–370. [PubMed] [Google Scholar]
- 12.McElroy SL, Guerdjikova AI, Mori N, et al. Overview of the treatment of binge eating disorder. CNS Spectr. 2015;20:546–556. [DOI] [PubMed] [Google Scholar]
- 13.Berkman ND, Brownley KA, Peat CM, et al. Management and Outcomes of Binge-Eating Disorder. Rockville, MD: Agency for Healthcare Research and Quality; 2015. [PubMed] [Google Scholar]
- 14.McElroy SL, Shapira NA, Arnold LM, et al. Topiramate in the long-term treatment of binge-eating disorder associated with obesity. J Clin Psychiatry. 2004;65:1463–1469. [DOI] [PubMed] [Google Scholar]
- 15.Golay A, Laurent-Jaccard A, Habicht F, et al. Effect of orlistat in obese patients with binge eating disorder. Obes Res. 2005;13:1701–1708. [DOI] [PubMed] [Google Scholar]
- 16.Fornaro M, Solmi M, Perna G, et al. Lisdexamfetamine in the treatment of moderate-to-severe binge eating disorder in adults: systematic review and exploratory meta-analysis of publicly available placebo-controlled, randomized clinical trials. Neuropsychiatr Dis Treat. 2016;12:1827–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.