

Abstract
Background
Androgen receptor (AR) splice variants have been clinically associated with progressive cancer, castration-resistance, and resistance to AR antagonists and androgen synthesis inhibitors. AR variants can be generated by genomic alterations and alternative splicing, and their expression is androgen-regulated. There has been a suggestion that AR variants bearing premature termination codons and coding for truncated proteins should be regulated by the nonsense-mediated decay (NMD) mRNA surveillance pathway, suggesting that either the NMD pathway is dysfunctional in variant-expressing cell lines or that variants are somehow able to evade degradation by NMD.
Methods
We first used siRNA knockdown of the NMD regulator, UPF1, in an NMD reporter assay to determine if this surveillance pathway is functioning normally in AR variant-expressing cell lines. We then used UPF1 knockdown to determine if expression of the AR variants ARV3 and ARV7 is affected by inhibition of NMD. Next, we analyzed androgen regulation of UPF1 and used transcript expression analysis to determine if there is any association between UPF1 expression, resistance, and ARV3 or ARV7 expression.
Results
We found that the NMD pathway functions normally in the AR variant-expressing cell line 22Rv1 and that inhibition of NMD does not increase expression of ARV3 or ARV7. Furthermore, we found that expression of UPF1 is not androgen-regulated. We also found that UFP1 expression levels do not differentiate castration-sensitive from resistant cell line and that UPF1 expression does not correlate with expression of ARV3 or ARV7 in cells in which these variants are highly expressed.
Conclusion
This study eliminates a possible mechanism of regulation of certain AR variants. Future research into the regulation of AR variants should focus on other mechanisms to better understand the origin of these variants and to possibly inhibit their expression for the resensitization of resistant cancers.
Keywords: prostate cancer, androgen receptor, splice variants, nonsense-mediated decay, UPF1
Introduction
The androgen receptor (AR) is a modular ligand-inducible transcription factor. The AR protein, translated from a mature 8-exon mRNA, is comprised of an amino-terminal transactivation domain followed by a DNA-binding domain, hinge region, and ligand-binding domain. Upon androgen-binding, AR is released from cytoplasmic interaction with heat shock protein-90 and translocates to the nucleus. Conformational changes reveal protein-protein interaction surfaces that facilitate homodimerization and DNA-binding. AR gene regulation has myriad consequences for prostate homeostasis, function, and cancer progression [1,2]. AR, which regulates cell proliferation, survival, and apoptosis [3], is the primary therapeutic target in advanced prostate cancer. Inhibition of the AR pathway through surgical or pharmacologic castration, competitive AR antagonists, and biosynthesis inhibitors results in decreased serum prostate specific antigen (PSA), increased tumor regression, and increased overall survival [4-6].
Recently, the expression of AR splice variants has been proposed as a possible mechanism of resistance to AR-targeted therapies. Some of these variants seem to be capable of constitutive biological activity, are able to partially recapitulate the AR transcriptional program [7-9], and have been associated with clinical resistance to AR pathway-targeted therapies [10-12]. AR variants are produced by the use of cryptic splice sites and are part of the growing interest in the role of alternative and/or aberrant splicing in cancer [13,14].
It has been noted that alternative splicing of AR results in the generation of premature termination codons (PTCs) in some AR variant transcripts. The expression of these variants would suggest that deficiencies in or evasion of the mRNA surveillance nonsense-mediated decay (NMD) pathway could be involved in the expression of some variant transcripts. NMD is an mRNA surveillance pathway that degrades PTC-bearing or nonsense transcripts before they are translated into potentially nonfunctional or toxic truncated proteins. Nonsense transcripts are detected during the pioneering round of translation. After mRNA splicing, the resulting exon-exon junctions are marked by exon junction complexes (EJCs). If ribosome-associated NMD proteins encounter a stop codon upstream of an EJC, translation is terminated and nucleases are recruited to degrade the transcript [15].
In this study, we focused on two variants, ARV3 and ARV7. Although ARV3 is not a major AR splice variant, it does have the structure of nonsense target and serves as a good marker for NMD. ARV7 is the most studied AR variant and one of the most clinically relevant. To determine the role of NMD in the expression of AR variants ARV3 and ARV7, we examined the expression and androgen regulation of up-frame shift protein 1 (UPF1), which is an RNA helicase required for NMD [16], as well as consequence of UPF1 knockdown on an NMD reporter and ARV3 and ARV7 expression. We also assessed the whether UPF1 is androgen-regulated and whether there is an association between UPF1 expression and resistance or betweenUPF1 expression and ARV3 or ARV7 expression. We found that while NMD is functional and capable of degrading nonsense transcripts in the AR variant-expressing cell line 22Rv1, the mRNA expression of ARV3 and ARV7 is not regulated by NMD. Our results also show that UPF1 is androgen-independent and that UPF1 is not a marker of resistance or correlated with ARV3 or ARV7 expression in cells that express high levels of these variants.
Materials and Methods
Cell Lines
CWR22 is an androgen-dependent cell line derived from a primary prostate tumor that is serially passaged in mice [17]. 22Rv1 was derived from a CWR22 tumor that regressed after castration then recurred in this castrate setting. The resulting cell line expresses AR and the AR variant 7 (ARV7), demonstrates androgen-independent growth, enzalutamide-resistance, and castration-resistance in vivo [7,18]. LN95 and LAPC4-cr are resistant cell lines derived from LNCaP and LAPC4, respectively, by in vitro passaging in androgen-depleted media (LN95) or after xenograft outgrowth in castrated nude mice followed by subsequent adaptation to cell culture (LAPC4-cr).
22Rv1, LNCaP, and VCaP cell lines were purchased from ATCC (Manassas, VA). LAPC4 and frozen CWR22 mouse tumors were generous gifts from John Isaacs (Johns Hopkins School of Medicine, Baltimore, MD). LN95 and LAPC4-cr cell lines were generous gifts from Alan Meeker and Michael Haffner, respectively, (Johns Hopkins School of Medicine). All cell lines were authenticated by short tandem repeat (STR) profiling using the GenePrint 10 System (Promega, Madison, WI). STR analysis was conducted by the Genetic Research Core Facility (Johns Hopkins School of Medicine, Baltimore, MD) in May 2016.
All media was supplemented with 1% Pen/Strep and 1% L-glutamine (Life Technologies, Grand Island, NY) as well as the additional indicated reagents. RPMI 1640 and IMDM were purchased from Life Technologies. Fetal bovine serum (FBS) was obtained from Sigma-Aldrich (St. Louis, MO) and charcoal-stripped FBS (CSS) from Gemini Bio-Products (West Sacramento, CA). LAPC4 cells were cultured in IMDM supplemented with 10% FBS and 1 nM R1881 (Sigma-Aldrich). 22Rv1 and LNCaP cells were cultured in RPMI 1640 supplemented with 10% FBS. VCaP cells were cultured in DMEM (P/N: 30-2002, ATCC) supplemented with 10% FBS. LN95 cells were cultured in phenol-red free RPMI 1640 supplemented with CSS and B27 supplement (Life Technologies, Grand Island, NY). LAPC4-cr cells were cultured in phenol-red free IMDM supplemented with 10% CSS and B27 supplement.
Nonsense-mediated decay reporter assay
NMD reporter, pb510-HA-TCRβ-ZsG PTC+ (hereon referred to as ZSG PTC+), was a generous gift from Oliver Mühlemann (University of Bern, Bern, Switzerland) and was generated as previously described with the ZsGreen1 (ZsG) open reading frame (ORF) replacing the green fluorescent protein ORF [19]. Cells were transfected in 10 mm tissue culture plates with ZsG PTC+ using FuGene HD Transfection Reagent (Promega) according to the manufacturer's instructions. After 48h, cells were trypsinized and evenly divided into two 60 mm plates for transfection with negative control (Silencer® Negative Control 1, Life Technologies, Grand Island, NY) or UPF1 siRNA. Knockdown of UPF1 was achieved using the target sequence 5′-AAGAGAAUCGCCUACUUCACU-3′ [19] (Silencer® Select siRNA, Life Technologies) using RNAiMax (Life Technologies), according to the manufacturer's instructions. Knockdown was verified 48h post-transfection by semiquantitative qRT-PCR. ZsG mRNA expression was determined by semiquantitative qRT-PCR of the ZsG ORF as described below.
Androgen regulation assay
Cells were deprived of androgen and other hormones as previously described [20]. Cells were treated with vehicle or 1 nM R1881 for 24h in phenol-red free RPMI 1640 or DMEM containing 10% CSS, after which cells were harvested by trypsinization. Total RNA was isolated and subsequently used for qRT-PCR as described below.
Reverse Transcription Quantitative Polymerase Chain Reaction
Total RNA was harvested by RNeasy Plus Mini Kit (Qiagen, Venlo, Netherlands). On column DNA digestion was performed using RNase-Free DNase Set (Qiagen) for lysates from cells transfected with ZsG PTC+, according to the instructions found in the RNeasy Mini Kit handbook (Qiagen). Equivalent amounts of total RNA were used between conditions for cDNA synthesis by the First Strand cDNA Synthesis Kit (Life Technologies). For the NMD reporter assay, a 1:1000 dilution of the resulting cDNA was used for quantification of ZsG, whereas expression of UPF1 and TBP was quantified from the neat cDNA reaction. 2 μl of the diluted or neat cDNA reaction was used for semi-quantitative qRT-PCR using iQ SYBR® Green Supermix and iCycler iQ Real-Time PCR Detection System (Bio-Rad, Hercules, CA), using TATA-binding protein (TBP) as the reference gene. The following primers were used for qPCR:
| ZsG | Fwd 5′-GACCGCTCCTTCCTGTTC-3′Rev 5′-GAACTTGGACTCGTGGTACAT-3′ |
| TBP | Fwd 5′-CACGAACCACGGCACTGATT-3′Rev 5′-TTTTCTTGCTGCCAGTCTGGAC-3′ |
| UPF1 | Fwd 5′-GACCTGGGCCTTAACAAGAA-3′Rev 5′-TGAGCCGCATGTCTCTTAAC-3′ |
| AR[9] | Fwd 5′-CCATCTTGTCGTCTTCGGAAATGTTATGAAGC-3′Rev 5′-AGCTTCTGGGTTGTCTCCTCAGTGG-3′ |
| ARV3 | Fwd 5′-AAGAGCCGCTGAAGGATTT-3′Rev 5′-TTCTGTCAGTCCCATTGGTG-3′ |
| ARV7[9] | Fwd 5′-CCATCTTGTCGTCTTCGGAAATGTTATGAAGC-3′Rev 5′-TTTGAATGAGGCAAGTCAGCCTTTCT-3′ |
The cycling protocol was as follows: 1 cycle at 95°C for 3 min, 35 cycles at 95°C for 30s, 60°C for 30s, and 72°C for 45s, followed by melt curve analysis from 65 to 95°C in 0.5°C increments for 5s each.
Statistical Analyses
Paired parametric t-tests and linear regression analyses were performed using GraphPad (La Jolla, CA). Statistical significance is indicated according to the following key: *p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, ****p ≤ 0.0001. Error bars represent standard error of the mean.
Results
NMD pathway is functioning in an AR variant-expressing cell line
To determine if the NMD pathway is functional in an AR variant-expressing cell line, we used a reporter assay in which the ZsG ORF was inserted in-frame into a T-cell receptor β (TCRβ) exon upstream of an intron and the TCRβ stop codon (Figure 1A). After excision of the intron, the ZsG stop codon is located >50 nucleotides upstream of an exon-exon junction, is recognized as a PTC, and is therefore a substrate for NMD. Inhibition of NMD by knockdown of UPF1 should increase expression of this reporter if the NMD pathway is functional [19].
Figure 1. Nonsense-mediated decay assay in 22Rv1 cells.
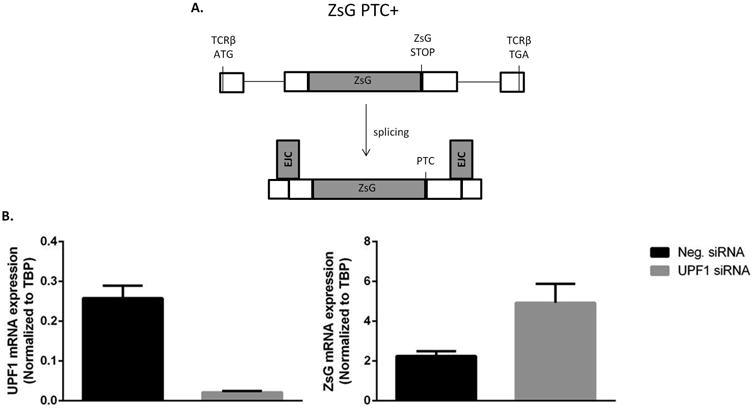
A) ZsG green fluorescent protein is inserted in-frame into a TCRβ exon such that when the minigene is transcribed and spliced, the ZsG stop codon is located upstream of an EJC, is recognized as a PTC, and is subject to degradation by NMD. (Cartoon adapted from Paillusson et al. [19].) B) siRNA inhibition of UPF1 in 22Rv1 cells causes a significant (p = 0.03) increase in reporter expression. Shown are the mean +/- SEM of three replicate experiments.
22Rv1 cells, which express several AR variants, were serially transfected with the PTC-bearing reporter, ZsG PTC+, followed by negative control or UPF1-targeting siRNA. qRT-PCR of ZsG was used to determine reporter mRNA expression. Expression of ZsG PTC+ increased significantly following knockdown of UPF1 (Figure 1B). These data indicate that the NMD pathway is intact in 22Rv1 cells.
AR variants are not NMD pathway substrates
It was previously noted that alternative splicing of AR results in the creation of PTCs in some variants, which should render such transcripts vulnerable to degradation by NMD [9]. NMD is activated when a PTC is located >50 nucleotides upstream of an exon-exon junction, as marked by EJCs, and has at least one intron downstream [21,22]. The ARV7 stop codon is located in the terminal exon, so no intron or EJC would be located downstream of this codon, and it is not expected that this stop codon would activate the NMD pathway (Figure 2A). However, ARV3, which has a PTC 124 nucleotides upstream of an exon-exon junction (Figure 2B) as well as a proper stop codon in the terminal exon, has the structure of a canonical nonsense transcript, and we predicted that ARV3 would be a more potent activator of NMD than ARV7.
Figure 2. Nonsense-mediated decay regulation of ARV3 and ARV7.
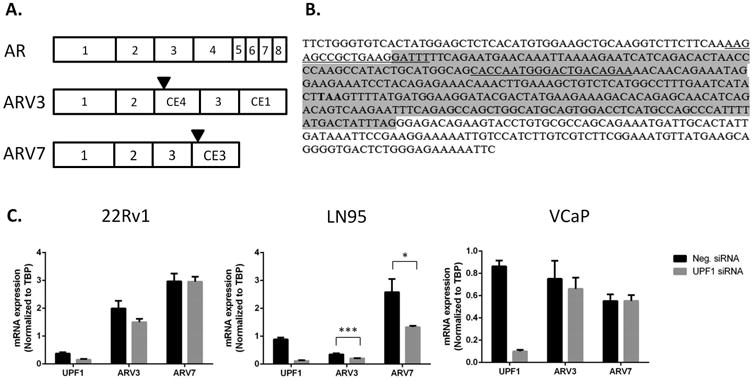
A) Structures of AR, ARV3, and ARV7. Stop codons are indicated by black triangles. B) Sequence structure of ARV3. Cryptic exon 4 is highlighted in gray. The sequences before and after cryptic exon 4 are of exons 2 and 3, respectively. The ARV3 stop codon is indicated in bold text. qPCR primers, spanning the exon 2-cryptic exon 4 junction and within cryptic exon 3, are underlined. There are 124 nucleotides between the terminal nucleotide of the stop codon and the putative EJC at the junction of cryptic exon 4 and exon 3. Because the stop codon occurs >50 nucleotides upstream of an EJC, ARV3 would be expected to be a substrate of NMD. C) siRNA knockdown of UPF1 did not alter expression of ARV3 or ARV7 in 22Rv1 (p = 0.1 and 1, respectively) or VCaP (p = 0.6 and 1.0, respectively) but did decrease expression of ARV3 and ARV7 in LN95 (p = 0.005 and 0.02, respectively). Shown are the mean +/- SEM of three replicate experiments.
Because knockdown of UPF1 was sufficient to inhibit NMD in the above reporter assay, we again used UPF1 knockdown to investigate the effect of NMD inhibition on the expression of ARV3 and ARV7 in 22Rv1, LN95, and VCaP. ARV3 and ARV7 expression in 22Rv1 and VCaP was unaltered by knockdown of UPF1, but was statistically decreased in LN95 (Figure 2C).
UPF1 not regulated by androgen
Expression of AR and AR are variants androgen-regulated, decreasing with high levels of androgen and increasing in low androgen[23]. We wondered if in low androgen, NMD activity could be reduced to allow for the expression of nonsense transcripts. To that end, we investigated how UPF1 expression changes with androgen. Our data show that, as expected, the expression of AR and ARV7 significantly decreased in 22Rv1, LN95, and VCaP with androgen. However, UPF1 expression did not change significantly in LN95 or VCaP, but 22Rv1 cells showed a statistical decrease in UPF1 expression (Figure 3).
Figure 3. Androgen regulation of AR, ARV7, and UPF1 expression.
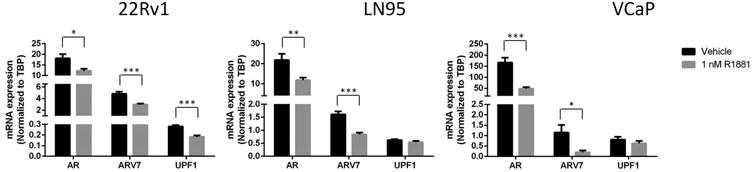
AR and ARV7 expression was significantly decreased in all cell lines by R1881 (AR: p = 0.02, 0.01, and 0.004; ARV7: p = 0.001, 0.0002, and 0.03 for 22Rv1, LN95, and VCaP, respectively). UPF1 expression in LN95 and VCaP was unaffected by androgen (p = 0.2 and 0.3, respectively), but expression of UPF1 was significantly decreased by androgen in 22Rv1 (p = 0.0003) cells. Shown are the mean +/- SEM of three replicate experiments.
UPF1 expression did not differ between sensitive and resistant cells and did not correlate with AR variant expression
Conversion of cells to a castration-resistant phenotype is frequently accompanied by increased expression of AR and/or de novo expression of ARV7 [9,24]. Although, NMD is functional in AR variant-expressing cell lines, we examined UPF1 expression in sensitive, resistant, variant-negative or -positive cell lines for a correlation between resistance or variant status and UPF1 expression. Decreased UPF1 expression between sensitive parent cell lines and the resistant derivatives or between variant-negative and variant-positive cell lines would suggest that a dysfunctional NMD pathway could be characteristic of resistance, such that significant levels of PTC-bearing transcripts could be expressed.
As expected, AR mRNA was significantly increased from LAPC4 to LAPC4-cr. There was a trend toward increased expression from CWR22 and 22Rv1, but this difference did not reach statistical significance. AR expression between LNCaP and LN95 was stable. ARV3 expression was statistically elevated between CWR22 and 22Rv1 and between LNCaP and LN95 but was similar between LAPC4 and LAPC4-cr. ARV7 expression in CWR22 and LAPC4 was barely detectable and low in LNCaP. 22Rv1 and LN95 expressed many times more ARV7 than the parental cell lines. While expression of ARV7 in LAPC4-cr was statistically higher than that of LAPC4, expression was still barely detectable (Figure 4).
Figure 4. Expression of UPF1 in prostate cancer cell lines.
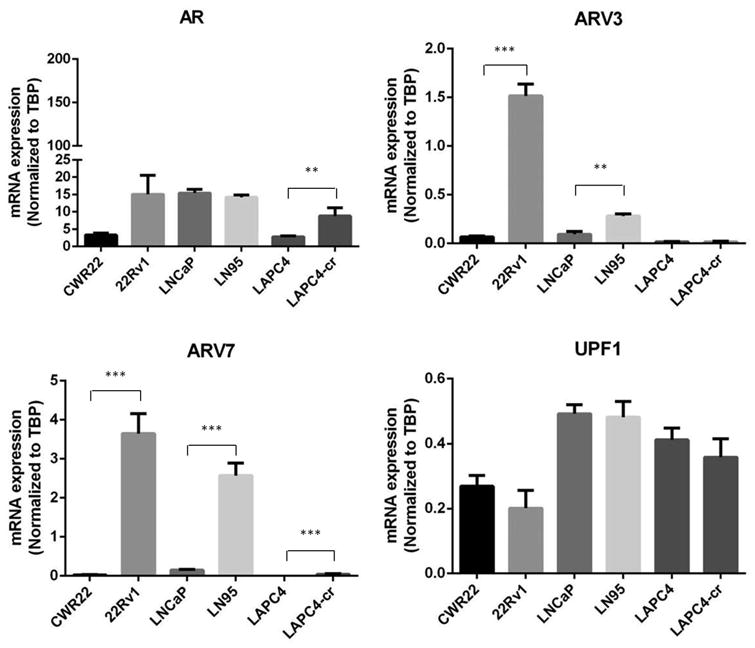
AR expression is statistically increased in resistant cell line LAPC4-cr relative to the sensitive parent cell line, LAPC4 (p = 0.01). There was no statistical difference in AR expression between CWR22 and LNCaP and its resistant derivatives, 22Rv1 and LN95, respectively (p = 0.08 and 0.4, respectively). Both ARV3 and ARV7 expression are increased between CWR22 and 22Rv1 (p < 0.0001 and p = 0.0004, respectively) and between LNCaP and LN95 (p = 0.002 and 0.0003, respectively). UPF1 expression is unchanged between sensitive and resistance cell lines (p = 0.3 CWR22/22Rv1, p = 0.9 LNCaP/LN95, and p = 0.5 LAPC4/LAPC4-cr).
Whereas AR, ARV3, and/or ARV7 expression was significantly different between some pairs of sensitive and resistant lines, UPF1 expression was stable between all sensitive and resistant lines (Figure 4).
Finally, we asked if UPF1 was in any way correlated to expression of ARV3 or ARV7 using linear regression analysis of ARV3 or ARV7 v. UPF1. If diminished expression of UPF1 was correlated to increased expression of ARV3 or ARV7, we would expect a negative slope. Linear regression analysis revealed a significant deviation from a slope of zero for ARV3 v. UPF1 in LAPC4-cr and LAPC4 as well as for ARV7 v. UPF1 in CWR22 and LAPC4-cr (Figure 5). However, all of these cell lines express very low levels of both ARV3 and ARV7. Furthermore, these analyses reveal positive relationships, i.e. the more UPF1, the more AR variant, which is not what one would predict if UPF1 was a negative regulator of these variants. The remaining cell lines showed no significant deviation from a slope of zero, indicating no correlation between ARV3 or ARV7 and UPF1 expression.
Figure 5. Linear Regression Analysis of ARV3 and ARV7 v. UPF1.
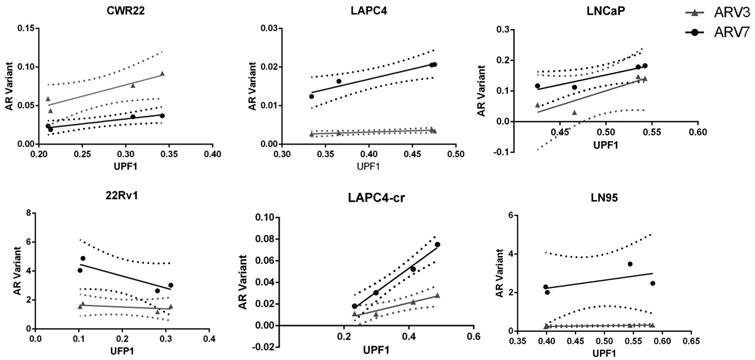
Correlation between expression levels of ARV3 or ARV7 vs. UPF1 was determined by linear regression analysis. Solid lines indicate linear regression, and dashed lines indicate confidence interval bands. Significant deviations from slopes of zero (indicated by p-values < 0.0.5) were found in CWR22 (ARV7 v. UPF1, p = 0.04), LAPC4 (ARV3 v. UPF1, p = 0.03), and LAPC4-cr (ARV3 v. UPF1, p =0.03 and ARV7 v. UPF1, p = 0.006).
Discussion
We investigated whether a dysfunctional mRNA surveillance pathway could be involved in the progression from a sensitive to resistant phenotype. Resistant prostate cancer is often marked by increased expression of AR and/or AR variants. It had previously been noted that alternative splicing of AR results in the generation of PTCs in some variants, which should activate the NMD pathway and would suggest that the existence of certain AR variants is due to either dysfunction or evasion of this pathway [9]. We investigated whether NMD dysfunction is an additional marker of resistance.
First, we used an NMD reporter assay to show that NMD is functional in 22Rv1 cells, recognizing nonsense transcripts and degrading them before translation into protein. Next, we investigated the regulation of ARV3 and ARV7 by siRNA knockdown of UPF1 as in the NMD reporter assay. We hypothesized that if ARV3 and ARV7 were NMD substrates then expression of ARV3 and ARV7 would increase. No change in expression of ARV3 or ARV7 was observed in 22Rv1 or VCaP. Curiously, expression of ARV3 and ARV7 decreased in LN95. Decrease of non-NMD substrates in response to UPF1 knockdown is not unusual (S. Brenner, personal communication, July 4, 2015), but at this time it is unclear why ARV3 and ARV7 expression decreased in LN95 and not in 22Rv1 and VCaP. Finally, we showed, through linear regression analysis, that UPF1 expression is not correlated with ARV3 or ARV7. From these data, we conclude that the expression of ARV3 and ARV7 is likely not dependent upon UPF1 or NMD.
However, inclusion of cryptic exon 4 into the ARV3 transcript does create a canonical NMD target. The resulting ARV3 PTC is 124 nucleotides upstream of an exon-exon junction site and has two introns downstream, which should trigger degradation of the transcript during translation. Our data shows that ARV3 is not regulated by NMD. There are several mechanisms by which putative nonsense transcripts evade NMD. In-frame translation reinitiation at a start codon downstream from the PTC can cause nonsense transcript evasion of NMD [25]. In addition to another PTC in cryptic exon 4 two codons downstream from the 5′-most PTC, there is an in-frame start codon 9 codons downstream from the 5′-most PTC in cryptic exon 4 followed by a proper stop codon in the terminal exon (Figure 6). Therefore, it is possible that termination reinitation is the mechanism of NMD evasion for ARV3.
Figure 6. Translation reinitiation of ARV3.

Putative nonsense transcripts and NMD substrates can evade NMD by a variety of mechanisms, one of which is reinitiation of translation downstream of the PTC. ARV3 harbors an in-frame translation start codon (outlined in black) downstream of the PTC (in bold text) located in cryptic exon 4, which could explain why inhibition of NMD does not effect expression.
Other mechanisms of evasion are not predicted to apply to ARV3. These mechanisms involve proximity of the PTC to other mRNA features. Transcripts with nonsense codons in close proximity to the AUG translational start site are not regulated by NMD due to temporal interference of ribosomal initiation, elongation, and termination and loading of associated factors, as has been noted with β-globin transcript variants. Fewer than 18-20 codons between the AUG and nonsense codons is thought to render nonsense transcripts resistant to NMD [26]. There are over 600 codons between the AUG and nonsense codons in ARV3, so evasion of NMD is not expected to be an issue of proximity. Positioning of poly(A)-binding protein (PABPC1) in close proximity to the PTC also suppresses degradation of nonsense transcripts. Positioning of PABPC1 15 codons downstream from the PTC increased a NMD-resistant transcript by 3-fold, whereas when the distance between the PTC and the PABPC1 binding site was tripled, the transcript was only increased 1.5-fold [27]. The 3′UTR of ARV3 is >500 codons downstream of the PTC, so it is unlikely that a short 3′UTR is the mechanism of NMD resistance. Finally, as noted above, NMD substrates must have PTCs >50 nucleotides from the exon-exon junction, and the ARV3 PTC is 124 nucleotides upstream of the putative exon-exon junction.
While the ARV3 transcript is able to evade mRNA surveillance, ARV3 is not currently known to produce a protein[28]. If there was a corresponding protein, the putative ARV3 protein would be truncated in the DBD, would probably be severely, functionally impaired either as a constituitive transcription factor or dominant negative protein, and was determined most likely to be clinically irrelevant and was not further pursued in studies about AR variants and their role in prostate cancer [9].
NMD evasion and dysfunction can be important contributing factors to the etiology of disease. Many of the rules for NMD targets are based on studies on β-globin. Nonsense mutations in β-globin can cause a blood disorder called β-thalassemia. Mutations in exons 1 and 2 of β-globin trigger NMD and result in a mild form of β-thalassemia in people heterozygous for these mutations. However, nonsense mutations in exon 3, the terminal exon, do not trigger NMD, produce a dominant negative form of β-globin, and a form of β-thalassemia that can lead to severe anemia [22,29]. Mutations in the components of NMD can also result in upregulation NMD targets. Recently, mutations in intronic and exonic splicing enhancer regions of UPF1 were identified in the malignant regions of pancreatic adenosquamous carcinoma tumors. The resulting alternatively spliced UPF1 mature mRNA produced deletions of the helicase domain or C-terminal domain, which is phosphorylated during NMD. Impaired NMD correlated with increased expression of a long form of the tumor suppressor p53 [30].
There are 9 different proteins that are involved in NMD. We focused on UPF1 in this study because UPF1 is required for NMD to go forward and inactivating mutations in UPF1 are sufficient to inhibit NMD [16,31] and cause a decreased rate of decay of nonsense transcripts. Furthermore, inhibition of NMD by translation inhibitors, such as cycloheximide or emetine,[32] or knockdown of UPF1 [33] is a proven method of identifying nonsense targets. NMD proteins play many roles [34], and we cannot rule out the possibility that some are affected and may mediate the rise of resistant cancer. However, at this time, we do not expect that deficiencies in the NMD pathway are involved in expression of AR splice variants.
Conclusion
AR variants have arisen as possible markers and mediators of resistance to castration, AR antagonists, and androgen synthesis inhibitors. As such, many have undertaken to understand the biological origin and their roles in mediating resistance to androgen therapy with the hope that inhibition of the synthesis or activity of these variants will resensitize caners to AR-directed therapies. In this study, we showed that NMD does not regulate the ARV3 or ARV7 variants, that UPF1 expression does not differentiate sensitive from resistant cell lines, and that there is no correlation between UPF1 and ARV3 or ARV7 expression in cell lines in which these variants are expressed at a significant level. Future research should focus on other mechanisms of regulation and mRNA surveillance to understand pathways that can be targeted for modulation of expression or activity of these variants.
Acknowledgments
The authors would like to acknowledge Stephan Engel and Oliver Mühlemann, PhD (University of Bern) for their provision of reagents and technical assistance in the nonsense-mediated decay assay, and Michael Haffner and Nicolas Wyhs for the generation of the castration resistant LAPC4-cr cell line.
Financial Support: This work was supported by NIH grant R01 CA184012-02 to SRD.
Footnotes
Disclosure Statement: The authors have no conflict of interest to report.
References
- 1.Centenera MM, Harris JM, Tilley WD, Butler LM. Minireview: The Contribution of Different Androgen Receptor Domains to Receptor Dimerization and Signaling. Molecular Endocrinology. 2008;22(11):2373–2382. doi: 10.1210/me.2008-0017. [DOI] [PubMed] [Google Scholar]
- 2.Clegg N, Nelson PS. Androgen-Regulated Genes in the Prostate. In: Mohler J, Tindall D, editors. Androgen Action in Prostate Cancer. New York, NY: Springer US; 2009. pp. 631–661. [Google Scholar]
- 3.Yang Q, Fung KM, Day WV, Kropp BP, Lin HK. Androgen receptor signaling is required for androgen-sensitive human prostate cancer cell proliferation and survival. Cancer Cell International. 2005;5:8–8. doi: 10.1186/1475-2867-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, Davis ID, de Bono JS, Evans CP, Fizazi K, Joshua AM, Kim CS, Kimura G, Mainwaring P, Mansbach H, Miller K, Noonberg SB, Perabo F, Phung D, Saad F, Scher HI, Taplin ME, Venner PM, Tombal B. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. New England Journal of Medicine. 2014;371(5):424–433. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PFA, Basch E, Small EJ, Saad F, Schrijvers D, Van Poppel H, Mukherjee SD, Suttmann H, Gerritsen WR, Flaig TW, George DJ, Yu EY, Efstathiou E, Pantuck A, Winquist E, Higano CS, Park Y, Kheoh T, Griffin T, Scher HI, Rathkopf DE on behalf of the COUAAI. Randomized Phase 3 Trial of Abiraterone Acetate in Men with Metastatic Castration-Resistant Prostate Cancer and No Prior Chemotherapy. The New England journal of medicine. 2013;368(2):138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tolis G, Ackman D, Stellos A, Mehta A, Labrie F, Fazekas AT, Comaru-Schally AM, Schally AV. Tumor growth inhibition in patients with prostatic carcinoma treated with luteinizing hormone-releasing hormone agonists. Proceedings of the National Academy of Sciences. 1982;79(5):1658–1662. doi: 10.1073/pnas.79.5.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KAT, Dehm SM. Androgen Receptor Splice Variants Mediate Enzalutamide Resistance in Castration-Resistant Prostate Cancer Cell Lines. Cancer Research. 2013;73(2):483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, Coarfa C, Frigo DE, Putluri N, Sreekumar A, Weigel NL. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. 2015 doi: 10.18632/oncotarget.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-Independent Androgen Receptor Variants Derived from Splicing of Cryptic Exons Signify Hormone-Refractory Prostate Cancer. Cancer Research. 2009;69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. New England Journal of Medicine. 2014;371(11):1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AMH, Edwards J, Qiu Y. A Novel Androgen Receptor Splice Variant Is Up-regulated during Prostate Cancer Progression and Promotes Androgen Depletion–Resistant Growth. Cancer Research. 2009;69(6):2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, Wikström P. Expression of Androgen Receptor Splice Variants in Prostate Cancer Bone Metastases is Associated with Castration-Resistance and Short Survival. PLoS ONE. 2011;6(4):e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33(46):5311–5318. doi: 10.1038/onc.2013.533. [DOI] [PubMed] [Google Scholar]
- 14.Sebestyén E, Zawisza M, Eyras E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Research. 2015 doi: 10.1093/nar/gku1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardner LB. Nonsense-Mediated RNA Decay Regulation by Cellular Stress: Implications for Tumorigenesis. Molecular Cancer Research. 2010;8(3):295–308. doi: 10.1158/1541-7786.MCR-09-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun X, Perlick HA, Dietz HC, Maquat LE. A mutated human homologue to yeast Upf1 protein has a dominant-negative effect on the decay of nonsensecontaining mRNAs in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(17):10009–10014. doi: 10.1073/pnas.95.17.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wainstein MA, He F, Robinson D, Kung HJ, Schwartz S, Giaconia JM, Edgehouse NL, Pretlow TP, Bodner DR, Kursh ED, Resnick MI, Seftel A, Pretlow TG. CWR22: Androgen-dependent Xenograft Model Derived from a Primary Human Prostatic Carcinoma. Cancer Research. 1994;54(23):6049–6052. [PubMed] [Google Scholar]
- 18.Sramkoski RM, Pretlow TG, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, Marengo SR, Rhim JS, Zhang D, Jacobberger JW. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cellular & Developmental Biology - Animal. 35(7):403–409. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 19.Paillusson A, Hirschi N, Vallan C, Azzalin CM, Mühlemann O. A GFP-based reporter system to monitor nonsense-mediated mRNA decay. Nucleic Acids Research. 2005;33(6):e54–e54. doi: 10.1093/nar/gni052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, Meeker AK, Netto G, De Marzo AM, Nelson WG, Yegnasubramanian S. Androgen-induced TOP2B mediated double strand breaks and prostate cancer gene rearrangements. Nature genetics. 2010;42(8):668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Sun X, Qian Y, LaDuca JP, Maquat LE. At Least One Intron Is Required for the Nonsense-Mediated Decay of Triosephosphate Isomerase mRNA: a Possible Link between Nuclear Splicing and Cytoplasmic Translation. Molecular and Cellular Biology. 1998;18(9):5272–5283. doi: 10.1128/mcb.18.9.5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thermann R, Neu-Yilik G, Deters A, Frede U, Wehr K, Hagemeier C, Hentze MW, Kulozik AE. Binary specification of nonsense codons by splicing and cytoplasmic translation. The EMBO Journal. 1998;17(12):3484–3494. doi: 10.1093/emboj/17.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the National Academy of Sciences. 2010;107(39):16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science. 2009;324(5928):787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neu-Yilik G, Amthor B, Gehring NH, Bahri S, Paidassi H, Hentze MW, Kulozik AE. Mechanism of escape from nonsense-mediated mRNA decay of human β-globin transcripts with nonsense mutations in the first exon. RNA. 2011;17(5):843–854. doi: 10.1261/rna.2401811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silva AL, Pereira FJC, Morgado A, Kong J, Martins R, Faustino P, Liebhaber SA, RomÃo L. The canonical UPF1-dependent nonsense-mediated mRNA decay is inhibited in transcripts carrying a short open reading frame independent of sequence context. RNA. 2006;12(12):2160–2170. doi: 10.1261/rna.201406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva AL, Ribeiro P, Inácio Â, Liebhaber SA, Romão L. Proximity of the poly(A)-binding protein to a premature termination codon inhibits mammalian nonsense-mediated mRNA decay. RNA. 2008;14(3):563–576. doi: 10.1261/rna.815108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan SC, Li Y, Dehm SM. Androgen Receptor Splice Variants Activate Androgen Receptor Target Genes and Support Aberrant Prostate Cancer Cell Growth Independent of Canonical Androgen Receptor Nuclear Localization Signal. Journal of Biological Chemistry. 2012;287(23):19736–19749. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conditions. Vol. 2015. Bethesda: National Library of Medicine; 2015. Beta thalassemia. [Google Scholar]
- 30.Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, Xu G, Lu L, Wang C, Song M, Zhu J, Wang Y, Zhao Y, Foo WC, Zuo M, Valasek MA, Javle M, Wilkinson MF, Lu Y. The UPF1 RNA Surveillance Gene is Commonly Mutated in Pancreatic Adenosquamous Carcinoma. Nature medicine. 2014;20(6):596–598. doi: 10.1038/nm.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leeds P, Wood JM, Lee BS, Culbertson MR. Gene products that promote mRNA turnover in Saccharomyces cerevisiae. Molecular and Cellular Biology. 1992;12(5):2165–2177. doi: 10.1128/mcb.12.5.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noensie EN, Dietz HC. A strategy for disease gene identification through nonsense-mediated mRNA decay inhibition. Nat Biotech. 2001;19(5):434–439. doi: 10.1038/88099. [DOI] [PubMed] [Google Scholar]
- 33.Hansen KD, Lareau LF, Blanchette M, Green RE, Meng Q, Rehwinkel J, Gallusser FL, Izaurralde E, Rio DC, Dudoit S, Brenner SE. Genome-Wide Identification of Alternative Splice Forms Down-Regulated by Nonsense-Mediated mRNA Decay in Drosophila. PLoS Genet. 2009;5(6):e1000525. doi: 10.1371/journal.pgen.1000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isken O, Maquat LE. The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nature reviews Genetics. 2008;9(9):699–712. doi: 10.1038/nrg2402. [DOI] [PMC free article] [PubMed] [Google Scholar]