

Abstract
Pulmonary arterial hypertension (PAH) is a lung vascular disease characterized with a progressive increase of pulmonary vascular resistance and obliterative pulmonary vascular remodeling resulting in right heart failure and premature death. In this brief review, we document the recent advances in identifying genetically modified murine models of PH, with a focus on the recent discovery of the mouse model of Tie2Cre-mediated deletion of prolyl hydroxylase 2, which exhibits progressive obliterative vascular remodeling, severe PAH, and right heart failure, thus recapitulating many of the features of clinical PAH. We will also discuss the translational potential of recent findings arising from experimental studies of murine PH models.
Keywords: animal model, endothelium, hypoxia inducible factor, prolyl hydroxylases, vascular remodeling
Introduction
Pulmonary arterial hypertension (PAH) is characterized by progressive increase in pulmonary vascular resistance and arterial obliteration leading to right heart failure and premature death [1–3]. Intimal, medial and adventitial thickening, vascular fibrosis, augmented oxidative/nitrative stress, vascular occlusion, and formation of complex plexiform lesions are histopathological features of clinical PAH including idiopathic PAH (IPAH) [4–6]. Current therapies targeting abnormalities in the prostacyclin, nitric oxide, and endothelin signaling pathways result in only modest improvements in morbidity and mortality [1,3]. None of these therapeutic agents target the underlying mechanisms of obliterative vascular remodeling. Although two rat PH models induced by either monocrotaline (MCT) challenge or chronic hypoxia plus Sugen 5416 treatment (best known as an inhibitor of vascular endothelial growth factor receptors 1 and 2) exhibit severe vascular remodeling and are widely used for preclinical studies of PH, these treatments fail to induce severe PH with stable obliterative vascular remodeling in mice. Thus, the identification of mouse model(s) with severe PH and obliterative vascular remodeling (i.e. recapitulating the pathological features of clinical PAH) is critical in order to delineate the molecular mechanisms that are responsible for obliterative vascular remodeling, and thereby provide valuable druggable targets and novel therapeutic approaches for PAH patients.
Here we review recent advances in murine models of PH since 2012 (based on ref [7]) and highlight a novel mouse model established by our group with pathology resembling clinical PAH. The mouse model of Tie2Cre-mediated disruption of Egln1 [encoding prolyl-4 hydroxylase 2 (PHD2)], designated Egln1Tie2, is the first mouse model of PAH exhibiting spontaneous progressive PAH with extensive pulmonary vascular remodeling including stable vascular occlusion and complex plexiform-like lesions [8]. As seen in PAH patients, these mice also die of right heart failure. We also discuss the obligatory role of HIF-2α signaling in the pathogenesis of PH and potential novel therapeutic strategies for the treatment of PAH.
Recent Advances in Murine Models of PH
A number of genetically engineered mouse models have been generated to study the pathogenesis of PH (Table 1) since the publication of a preceding review in 2012 [7]. Hemodynamic measurement shows that 14 of the 23 mouse models have basal right ventricular systolic pressure (RVSP) of more than 30 mmHg, while 4 of these 14 have an RSVP of more than 50 mmHg. These 4 recently identified mouse models are Erg−/− mice (WT:KO, 20:50 mmHg) [9], Hif2aG536W/G536W knockin mice (28:66 mmHg) [10], Cdh5Cre-mediated Egln1 knockout mice (Egln1Cdh5) (25:54 mmHg) [11], and Tie2Cre-mediated Egln1 knockout mice (Egln1Tie2) (22:72 mmHg) [8]. The latter 3 of these models target the same pathway. Intriguingly, both Erg and its downstream target, Apelin receptor, are also downregulated in the lungs of Egln1Tie2 mice [8]. Although all of these mouse models show increased muscularization of the distal pulmonary arterioles, only a few of them show evidence of occlusive pulmonary vascular remodeling. Horita et al. reported that smooth muscle cell (SMC)-specific deletion of Pten in mice resulted in marked pulmonary vascular remodeling including occlusion of small pulmonary arteries when exposed to chronic hypoxia but not under normoxic condition [12]. The RVSP of these mice after 4-week exposure of hypoxia is less than 40 mmHg with a right ventricular hypertrophy index (RV/LV+S ratio) of 0.42. No evidence of right heart failure was observed in these mice. Lathen et al. showed that mice with deficiency of either Erg or Aplnr developed occluded pulmonary venues but not arterioles [9]. Erg−/− mice develop an RVSP as great as 50 mmHg and all die by the age of 3 months. These mice also develop RV hypertrophy (RV/LV+S ratio ~0.5), but it is unknown whether they die of right heart failure, given that no echocardiography or hemodynamic measurement of RV function have been carried out. In an experimental rat model of PAH, it has been shown that endothelial cell apoptosis is a trigger for the development of severe PAH induced by chronic hypoxia combined with Sugen5416 treatment. A recent study employing a mouse model with Fas-induced apoptosis of ECs showed evidence of occlusive vascular remodeling [13]. However, only a small portion (~20%) of these mice developed mild PH with scarce occlusive lesions. Previously, Steiner et al. have shown occlusive vascular remodeling in transgenic mice overexpressing IL-6 following 3-weeks exposure to hypoxia but not under normoxic conditions [7,14]. Although these models are helpful for us to understand the mechanisms that regulate pulmonary vascular remodeling, none of them fully resembles the pathology of clinical PAH. We recently demonstrated for the first time that Tie2Cre-mediated deletion of Egln1 in ECs and hematopoietic cells spontaneously induces severe PAH with progressive vascular remodeling including vascular occlusion and the formation of complex plexiform-like lesions, recapitulating clinical PAH.
Table 1.
Recent Mouse models of pulmonary hypertension published since 2012
| Mouse strain | Genetic modification | RVSP, mmHg | RV/LV+S | Histology | Ref | |||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Normoxia | Hypoxia | Normoxia | Hypoxia | Normoxia | Hypoxia | |||
| C57BL6/J | Prkg1−/− | 43 | N/A | N/A | N/A | ↑Musc | N/A | Zhao(2012) |
| C57BL6/J | Il13 Tg | 38 | N/A | 0.31 | N/A | ↑Musc | N/A | Cho(2013) |
| C57BL6/J | Irp1−/− | 36 | N/A | N/A | N/A | N/A | N/A | Ghosh (2013) |
| C57BL6/J | EC Smad1−/− | 28 | N/A | 0.294 | N/A | ↑Musc | N/A | Han(2013) |
| SMC Smad1−/− | 25 | N/A | 0.313 | N/A | ↑Musc | N/A | ||
| C57BL6/J | Ccr2−/− | 30 | 47 | 0.28 | 0.41 | ↑Musc | ↑Musc | Yu(2013) |
| Ccl2−/− | 26 | 42 | 0.27 | 0.42 | N/A | N/A | ||
| C57BL6/J | Sod1−/− | 34 | 34 | 0.32 | 0.41 | ↑Musc | N/A | Ramiro-Diaz(2013) |
| C57BL6/J | SMC Pten−/− | 30 | 35 | 0.27 | 0.42 | ↑Musc | Occlusive intima of small PA | Hortia (2013) |
| C57BL6/J | EC Gata6−/− | 27 | 33 | 0.31 | 0.48 | Normal | ↑Musc | Ghatneka (2013) |
| C57BL6/J | Hif2aG536w knock in | 66 | N/A | 0.61 | N/A | ↑Musc | N/A | Tan(2013) |
| C57BL6/J | Ucp2−/− | 25(mPAP) | 28(mPAP) | N/A | N/A | ↑Musc | No difference | Dromparis (2013) |
| C57BL6/J | EC Klf4−/− | 24 | 34 | 0.28 | 0.40 | Normal | ↑Musc | Shatat (2014) |
| C57BL6/J | Aplnr−/− | 35 | N/A | 0.38 | N/A | Occluded PV | N/A | Lathen (2014) |
| EC Aplnr−/− | 35 | N/A | 0.38 | N/A | Occluded PV | N/A | ||
| Erg−/− | 50 | N/A | 0.48 | N/A | Occluded PV | N/A | ||
| C57BL6/J | Sirt3−/− | 30(mPAP) | N/A | 0.33 | N/A | ↑Musc | N/A | Paulin (2014) |
| C57BL6/J | SMC Foxo1−/− | 30 | 35 | 0.24 | 0.28 | ↑Musc | ↑Musc | Savai (2014) |
| C57BL6/J | Epha2−/− | 25 | 32(Hx) 35 (SuHx) | 0.25 | 0.42(Hx) 0.47(Su Hx) | Normal | No difference | Rhodes (2015) |
| C57BL6/J | EC Fas-induced apoptosis-EFIA | 29 | N/A | 0.27 | N/A | Occlusive small PA | N/A | Goldthorp (2015) |
| C57BL6/J | Egln1Cdh5Cre | 54 | N/A | 0.5 | N/A | ↑Musc | N/A | Kapitsino (2016) |
| C57BL6/J | Egln1Tie2Cre | 72 | N/A | 0.85 | N/A | ↑Musc, occlusive small and large PA, plexiform lesions | N/A | Dai(2016) |
| C57BL6/J | EC Ampk−/− | 27 | 38 | 0.2 | 0.37 | Normal | ↑Musc | Omura (2016) |
Hx, hypoxia; Musc, muscularization of distal pulmonary vessels; mPAP, mean pulmonary arterial pressure; N/A, not analyzed; PA, pulmonary artery; PV, pulmonary vein; SuHx, Sugen5416/hypoxia.
Discovery of the First Murine Model of Progressive PAH Recapitulating Clinical PAH
As pointed out by Gomez-Arroyo, et al., an ideal animal model of clinical PAH should display some or all of the key features found in humans, including markedly increased RVSP (>60 mmHg), pulmonary artery lumen obliteration, formation of plexiform-like lesions, severe RV hypertrophy (RV/LV+S ratio > 0.45), RV chamber dilation, and RV failure [7]. The Egln1Tie2 mouse model is the only mouse model that closely resembles many clinical features of severe PAH in patients [8]. Under basal conditions, Egln1Tie2 mice develop progressive PAH with RVSP levels ranging from 60 to 90 mmHg at the age of 3.5 months. As seen in patients, these mice die progressively (i.e. starting at the age of 2 months and with 80% mortality by the age of 6 months). Remarkably, these mice also develop unprecedented RV hypertrophy (RV/LV+S ratio ranging from 0.75 to 1.1). Echocardiography reveals a 3-fold increase in RV wall thickness, a marked dilatation of the RV chambers, and a decrease in RV fraction area change indicating RV dysfunction. Pulmonary artery dysfunction is evidenced by a marked decrease in the ratio of pulmonary artery acceleration time:ejection time (Figure 1 and ref [8]). Molecular analysis also shows induced expression of atrial natriuretic factor and skeletal α-actin in the RV, indicating heart failure [15]. These data demonstrate that Egln1Tie2 mice develop spontaneous progressive PAH that results in RV failure and premature death as seen in patients with severe PAH.
Figure 1. Spontaneous severe PAH with obliterative pulmonary vascular remodeling in Egln1Tie2 mice.
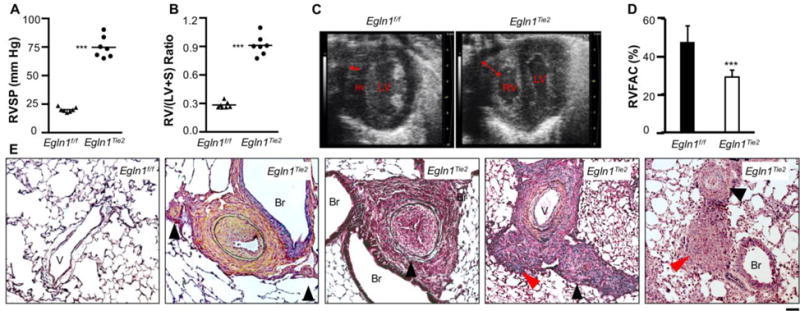
(A) Marked increase of RVSP. (B) Unprecedented RV hypertrophy. (C) Echocardiography demonstrating marked thickness of RV wall (double arrow) and enlarged RV chamber. LV, left ventricle. (D) Decreased RV fraction area indicating RV dysfunction. (E) Representative micrographs of Russel-Movat pentachrome staining demonstrating thickening of the intima, media, and adventitia; occlusion of the large and small vessels (black arrowheads); and plexiform-like lesions (red arrowheads) in 3.5-month-old Egln1Tie2 mice. Data are expressed as mean ± SD. ***, P < 0.001. (Adapted from ref. 8).
Histological examination of lung sections from Egln1Tie2 mice demonstrates various forms of vascular remodeling, including thickening of intima, media and adventitia, and neointima occlusion as well as the formation of plexiform-like lesions (Figure 1). Pulmonary vascular obliterative remodeling is prominent in both large and small vessels, and this remodeling is progressive and irreversible. Proliferation of vascular cells including both ECs and SMCs is evident in the lesions. Besides CD11b+ monocyte infiltration in the lesions, we also observed increased expression of IL-6 (indicating inflammation), vascular fibrosis, and increased oxidative/nitrative stress in Egln1Tie2 lungs (unpublished observations). These are also the characteristic features of the pathology of clinical PAH [4,6,16–18]. In human, it was shown that female gender is a risk factor for PAH, but that men with PAH have high mortality [19,20]. In Egln1Tie2 mice, however, we did not observe significant differences in RVSP or RV hypertrophy between genders.
In support of the concept that Egln1Tie2 mice may be the long-sought-after murine model of clinical PAH, expression of many of the PH-causing genes is altered in Egln1Tie2 lungs. Expression of Arg1, Lcn2, Slc39α12, Il13, Retnla, Ngf, Serpine 1, Il6, cxcl12, Csf2, Ptger3, Arg2, Eln, Edn1, Trpv4, and Sphk1 is upregulated while Aplnr, Ccr7, Ccr2, Bmpr2, Cav1, EphA1, Erg, Apln, Prkg1, Acvr2b, Acvr11, and Eng expression is downregulated. It remains unclear whether altered expression of these genes collectively causes severe PAH in Egln1Tie2 mice. Nevertheless, we have shown that upregulated Cxcl12 (also named SDF-1) expression in lung ECs (secondary to PHD2 deficiency) is partially responsible for increases in SMC proliferation and the development of severe PAH in Egln1Tie2 mice, given that genetic deletion of Cxcl12 attenuates PAH in Egln1Tie2 mice. Consistently, altered expression of some of these genes has been reported in lung tissues of IPAH patients, and some of them such as BMPR2 [21], ACVRL1 [22], ENG [23], and CAV1 [24], have been shown mutations in IPAH patients. Importantly, PHD2 expression is diminished in ECs of occlusive pulmonary vessels of IPAH patients, but not in ECs of vessels without occlusive remodeling [8]. These findings provide clear evidence about the important pathogenic role of PHD2 deficiency in ECs in obliterative vascular remodeling in Egln1Tie2 mice and PAH patients.
In summary, different from other mouse models, Egln1Tie2 mice exhibit markedly elevated RVSP (> 60 mmHg), progressive pulmonary vascular remodeling including vascular occlusion and formation of plexiform-like lesions, severe RV hypertrophy (RV/LV+S >0.75) and RV failure, irreversible PAH and progressive mortality. As seen in IPAH lungs, expression of many of the PH-causing genes is also altered in Egln1Tie2 lungs. Thus, the Egln1Tie2 mice appear to be the long-sought-after mouse model of clinical PAH. Discovery of this unique mouse model of clinical PAH is potentially useful for delineating the molecular mechanisms underlying the complex vascular remodeling of clinical PAH, and thereby enabling identification of valuable druggable targets and development of novel therapeutic approaches for PAH patients.
Synergistic Role of PHD2 Deficiency in ECs and Hematopoietic Cells in Mediating Obliterative Vascular Remodeling
Several lines of evidence suggest that multiple cell types derived from bone marrow (including macrophages, T cells and progenitor cells) are recruited to the plexiform lesions in PH, and contribute to the progression of pulmonary vascular remodeling [25–28]. Bone marrow abnormality is also shown in clinical PAH. Asosingh et al. demonstrate that CD133+ cells from PAH patients exhibit more multipotency and greater myeloid commitment and engraftment in the pulmonary vascular bed compared with healthy controls [29]. They also show that hypoxia inducible factor (HIF) and its transcription targets such as CXCL12, HGF and SCF may cause the proliferation of the multipotent hemangioblasts in the bone marrow, and mobilization of progenitor cells from bone marrow [30].
Kapitsinou, et. al. have studied the Egln1Cdh5 mice with Cdh5Cre-mediated disruption of Egln1[11]. Surprisingly, Egln1Cdh5 mice exhibit much weaker PH compared to Egln1Tie2 mice. Egln1Cdh5 mice don’t exhibit obliterative vascular remodeling except increased muscularization of distal pulmonary arterioles. RV hypertrophy is much weaker in the Egln1Cdh5 mice (i.e. RV/LV+S ratio is less than 0.5) compared with the Egln1Tie2 mice, and there is no evidence of RV failure in the Egln1Cdh5 mice. The more complex vascular remodeling and severe PAH phenotype observed in Egln1Tie2 mice versus Egln1Cdh5 mice indicates the importance of the bone marrow abnormality in the severity of PH given that both of them have PHD2 deficiency in ECs. In fact, transplantation of bone marrow cells from wild type mice to lethally irradiated Egln1Tie2 mice results in marked decreases of RVSP, pulmonary vascular remodeling, and RV hypertrophy. Furthermore, no occlusive vascular lesions are observed in these chimeric mice. These data provide unequivocal evidence that PHD2 deficiency in bone marrow cells induced by Tie2Cre markedly contributes to the severity of PAH in Egln1Tie2 mice. The difference between these two models might be due to the distinct expression patterns of Tie2 versus Cdh5 promoters. Although both promoters drive Cre expression in ECs, Tie2Cre also drives gene expression in the majority of hematopoietic cells in adult mice [31], whereas Cdh5Cre used in Kapitsinou et al. studies affects only 50% of adult hematopoietic lineages [11,32]. Thus, future studies are warranted to identify the distinct cell subpopulation(s) in the bone marrow responsible for the obliterative vascular remodeling.
As both Tie2Cre and Cdh5Cre induce gene deletion in ECs, these studies demonstrate that PHD2 deficiency in ECs is essential for inducing PH. Indeed, transplantation of bone marrow cells from Egln1Tie2 mice to lethally irradiated WT mice fail to induce PH. Mechanistically, we have shown that PHD2-deficient human lung vascular ECs release CXCL12, which induces pulmonary vascular SMC proliferation (likely via activation of the CXCL12 receptor, CXCR4, which is also expressed in SMCs). Tie2Cre-mediated genetic deletion of Cxcl12 in Egln1Tie2 mice attenuates PH and pulmonary vascular remodeling. One possible mechanism for this observation is that deletion of Cxcl12 results in inhibition of SMC proliferation and pulmonary vascular remodeling induced by PHD2-deficient ECs. However, it is also possible that Cxcl12 released from PHD2-deficiecnt ECs plays an important role in recruiting bone marrow cells to the lesions, and promoting their proliferation within the lesions. Other factors such as Endothelin-1 (upregulated) or Apelin (downregulated), which are released by PHD2-deficient ECs are also likely to be involved in the pathogenesis of severe PAH seen in Egln1Tie2 mice. Both preclinical and clinical studies have demonstrated the pathogenic role of increased Endothelin-1 in PAH [33]. Defective Apelin/Apelin receptor signaling is also shown to induce PH in mice [9,34]. These data together suggest endothelial PHD2 deficiency plays a prerequisite role in initiating PH, and that bone marrow PHD2 deficiency enhances pulmonary vascular remodeling thereby promoting the severity of PH (Figure 2).
Figure 2. Synergistic role of PHD2 deficiency in ECs and hematopoietic cells in mediating obliterative vascular remodeling and severe PAH.
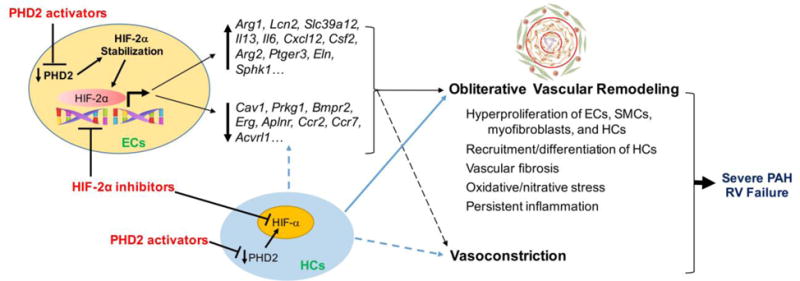
Through HIF-2α activation, PHD2 deficiency induces dysregulation of multiple signaling pathways (directly or indirectly) in mouse lungs. PHD2 deficiency in hematopoietic cells also contribute to the molecular changes. Collectively, these molecular changes coupled with the effects of PHD2 deficiency on hematopoietic cell (inflammatory cells, progenitor cells, etc.) recruitment, engraftment, differentiation, proliferation, and cross-talk with pulmonary vascular cells and adventitial fibroblasts induce obliterative pulmonary vascular remodeling as well as vasoconstriction and thereby severe PAH, which leads to RV failure and premature death. Thus, targeting the dysregulated PHD2/HIF-2α signaling may represent a novel therapeutic strategy to reverse obliterative vascular remodeling and thereby effectively treat PAH and promote survival.
Obligatory Role of HIF-2α Not HIF-1α in the Pathogenesis of Severe PH
As oxygen sensors, PHDs (PHD1-3) use molecular O2 as a substrate to hydroxylate specific proline residues of the α subunit of HIFs (including HIF-1α, HIF-2α, and HIF-3α). Hydroxylation promotes HIF-α binding to the von Hippel-Lindau ubiquitin E3 ligase, which leads to HIF-α ubiquitination and subsequent degradation by the proteasome [35,36]. Inhibition of PHD activity, such as under hypoxic conditions, results in stabilization and accumulation of the α-subunit of HIF in the nucleus, and therefore formation of a HIF heterodimer with the constitutive HIF-1β subunit. The HIF heterodimer consequently activates expression of multiple HIF target genes that regulate angiogenesis, erythropoiesis, metabolism, inflammation, and vascular responses [37,38]. As expected, genetic deletion of Egln1 results in stabilization of both HIF-1α and HIF-2α, the 2 major HIF-α isoforms, in lungs of Egln1Tie2 mice. Although previous studies have shown the important role of HIF-1α in mediating hypoxia-induced PH, genetic deletion of Hif2a but not Hif1a normalizes the PH phenotype in Egln1Tie2 mice [8] as well as in Egln1Cdh5 mice [11]. Expression of PH-causing genes including Bmpr2, Cav1, Edn1, Il6, Cxcl12, Apln/Aplnr, and Erg are normalized in the Egln1Tie2/Hif2aTie2 double knockout mice. These studies provide unequivocal evidence for the critical role of HIF-2α in the pathogenesis of PH. Consistently, mice with genetic knockin of an activation mutation (G536W) of Hif2a also develop severe PH with RVSP of 66 mmHg [10]. The residue G536 is located close to the primary prolyl hydroxylation site for PHDs. G537W mutation of human HIF2A (equivalent to G536W in mice) is also shown to be associated with severe PAH in patients [39]. A recent study also shows the superior role of endothelial HIF-2α versus endothelial HIF-1α in mediating chronic hypoxia-induced pulmonary vascular remodeling and PH. Mice lacking HIF-2α but not HIF-1α in lung endothelium (L1Cre) are completely resistant to chronic hypoxia-induced PH [40]. Chuvash polycythemia patients who have an R200W mutation in the von Hippel-Lindau (VHL) tumor suppressor protein are susceptible to development of PAH [41]. Consistently, mice with Vhl R200W mutation (VhlR200W) develop PH, which is rescued by a heterozygous deletion of Hif2a but not Hif1a [42]. Intriguingly, genome-wide studies of Tibetan high attitude adaptation have identified mutations at both EGLN1 and HIF2A loci [43,44]. Tibetans have blunted pulmonary response to hypoxia probably because of the coding region variants in EGLN1, which result in increased hydroxylase activity under hypoxic conditions [45,46]. Taken together, these studies suggest that HIF hydroxylase signaling, specifically the PHD2/HIF-2α axis, is critical in regulating pulmonary vascular homeostasis. Although endothelial HIF-2α plays a critical role in the pathogenesis of PH, it remains unclear which HIF-α isoform(s) (HIF-2α or HIF-1α or both) in the bone marrow cells whose activation secondary to PHD2 deficiency contributes to obliterative vascular remodeling and severe PH.
Translational Potential
Current therapies for PAH mainly focus on the abnormalities in the prostacyclin, nitric oxide, and endothelin signaling pathways, including five classes of drugs, endothelin receptor antagonists, phosphodiesterase-5 inhibitors, soluble guanylate cyclase activators, prostacyclin analogues, and prostacyclin receptors agonists [1,3]. Although substantial therapeutic advances have been achieved over the past 25 years in PAH patients, currently-approved drugs fail to markedly improve morbidity and mortality. The identification of novel druggable targets therefore remains critical for the development of novel effective therapeutic agents. The essential role of PHD2/HIF-2α signaling in the development of obliterative pulmonary vascular remodeling and severe PAH suggest that targeting the dysregulated PHD2/HIF-2α signaling represents a novel effective therapeutic strategy.
Given the critical role of HIF-2α in the pathogenesis of PAH, it will be crucial to identify HIF-2α-selective inhibitors for the treatment of PAH. Although transcription factors are difficult to target, there are several studies reporting the development of small molecules targeting HIF-2α. Zimmer, et al. have identified small molecules (e.g., compound 76) that selectively decrease HIF-2α translation by enhancing the binding of iron-regulatory protein 1 to the 5′-untranslated region of HIF2A but not HIF1A [47]. It has been shown that inhibition of HIF-2α by compound 76 suppresses VHL-associated diseases including erythrocytosis, and pathologic angiogenesis in zebrafish [48]. Our recent studies also show that compound 76 attenuates RVSP and RV hypertrophy as well as pulmonary vascular remodeling in Egln1Tie2 mice (unpublished observation). It would be interesting to determine whether compound 76 is also effective in inhibiting PH in other animal models of PAH such as the Sugen/Hypoxia rat model. Other groups have also developed different classes of HIF-2α inhibitors [49–51]. It would be pertinent to determine whether these HIF-2α inhibitors also inhibit PAH in Egln1Tie2 mice.
Pharmacological activation of PHD2 by small molecules may be another potential therapeutic approach to suppress HIF in the pathogenesis of PH. Choi et al. screened a small molecular library based on an in vitro hydroxylation assay and identified KRH102053 as a potent PHD2 activator. KRH102053 and its more effective analogue, KRH102140, downregulate HIF-1α protein and its downstream target genes [52]. It is unclear whether KRH102140 also downregulates HIF-2α. In another study, Temes et al. identified a diacylglycerol kinase inhibitor, R59949, that stimulates the activity of PHDs without affecting PHD expression levels [53]. One possible caveat with PHD2 activators is their limited ability to activate PHD2 in PAH patients, given that PHD2 expression is diminished in occlusive pulmonary vascular ECs of IPAH lungs. Delineation of the signaling mechanisms underlying downregulation of PHD2 in the occlusive pulmonary vascular ECs may provide an important approach to restore PHD2 expression and activity in occlusive pulmonary vascular ECs, and thereby potentially reverse obliterative vascular remodeling.
Conclusions and Perspectives
Recent studies have provided fundamentally important information regarding the molecular mechanisms of obliterative pulmonary vascular remodeling. Most excitingly, the Egln1Tie2 mice may be the long-sought-after mouse model of clinical PAH. These mice exhibit many of the pathological features of clinical PAH including severely elevated progressive RVSP, extensive pulmonary vascular occlusion, formation of complex plexiform-like lesions associated with vascular fibrosis, augmented oxidative/nitrative stress and inflammation, increased proliferation of pulmonary vascular cells, and severe right heart hypertrophy and failure. Egln1Tie2 mice also exhibit marked alteration of expression of many PH-causing genes as seen in lung tissues of clinical PAH, and ultimately, progressive mortality. Discovery of the first mouse model of clinical PAH will help us to understand the molecular basis of obliterative vascular remodeling and develop novel therapeutic approaches to effectively treat PAH in patients and promote survival.
Future studies should be directed to: 1) delineating the molecular mechanisms of obliterative vascular remodeling, including identification of the subpopulation(s) of bone marrow cells responsible for obliterative vascular remodeling; 2) elucidating the signaling pathways that mediate the transcriptional regulation of PHD2 expression; 3) determining whether the obliterative vascular remodeling seen in Egln1Tie2 mice is reversible by pharmacotherapies such as HIF-2α inhibitors; and 4) developing novel HIF-2α inhibitors and/or PHD2 activators/inducers to test their efficacies in inhibiting PAH and promoting survival in Egln1Tie2 mice as well as in rat models of PAH such as the Sugen/hypoxia model. Collectively, these studies will lead to a comprehensive understanding of the mechanisms that control obliterative vascular remodeling, and subsequently, development of novel therapeutic agents for effective treatment of PAH and promotion of survival of PAH patients.
Acknowledgments
Funding Sources
This work was supported in part by NIH grants R01HL123957, R01HL125350, R01HL133951, and P01HL077806 to Y.Y. Z and American Heart Association postdoctoral fellowship grant 15POST25700124 to Z.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Subject codes: Pulmonary hypertension, animal models of human disease, vascular disease, pathophysiology
There is no conflict of interest.
References
- 1.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: A report of the american college of cardiology foundation task force on expert consensus documents and the american heart association: Developed in collaboration with the american college of chest physicians, american thoracic society, inc., and the pulmonary hypertension association. Circulation. 2009;119:2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 2.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65:1976–1997. doi: 10.1016/j.jacc.2015.03.540. [DOI] [PubMed] [Google Scholar]
- 4.Tuder RM, Stacher E, Robinson J, Kumar R, Graham BB. Pathology of pulmonary hypertension. Clin Chest Med. 2013;34:639–650. doi: 10.1016/j.ccm.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–272. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, et al. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, et al. A brief overview of mouse models of pulmonary arterial hypertension: Problems and prospects. Am J Physiol Lung Cell Mol Physiol. 2012;302:L977–91. doi: 10.1152/ajplung.00362.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai Z, Li M, Wharton J, Zhu MM, Zhao YY. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2alpha. Circulation. 2016;133:2447–2458. doi: 10.1161/CIRCULATIONAHA.116.021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lathen C, Zhang Y, Chow J, Singh M, Lin G, Nigam V, et al. ERG-APLNR axis controls pulmonary venule endothelial proliferation in pulmonary veno-occlusive disease. Circulation. 2014;130:1179–1191. doi: 10.1161/CIRCULATIONAHA.113.007822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan Q, Kerestes H, Percy MJ, Pietrofesa R, Chen L, Khurana TS, et al. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem. 2013;288:17134–17144. doi: 10.1074/jbc.M112.444059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapitsinou PP, Rajendran G, Astleford L, Michael M, Schonfeld MP, Fields T, et al. The endothelial prolyl-4-hydroxylase domain 2/Hypoxia-inducible factor 2 axis regulates pulmonary artery pressure in mice. Mol Cell Biol. 2016;36:1584–1594. doi: 10.1128/MCB.01055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horita H, Furgeson SB, Ostriker A, Olszewski KA, Sullivan T, Villegas LR, et al. Selective inactivation of PTEN in smooth muscle cells synergizes with hypoxia to induce severe pulmonary hypertension. J Am Heart Assoc. 2013;2:e000188. doi: 10.1161/JAHA.113.000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldthorpe H, Jiang J-Y, Taha M, Deng Y, Sinclair T, Ge CX, et al. Occlusive lung arterial lesions in endothelial-targeted, fas-induced apoptosis transgenic mice. Am J Respir Cell Mol Biol. 2015;53:712–718. doi: 10.1165/rcmb.2014-0311OC. [DOI] [PubMed] [Google Scholar]
- 14.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–44. doi: 10.1161/CIRCRESAHA.108.182014. 28p following 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, et al. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 17.Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, et al. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–2018. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao YY, Malik AB. A novel insight into the mechanism of pulmonary hypertension involving caveolin-1 deficiency and endothelial nitric oxide synthase activation. Trends Cardiovasc Med. 2009;19:238–242. doi: 10.1016/j.tcm.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mair KM, Johansen AK, Wright AF, Wallace E, MacLean MR. Pulmonary arterial hypertension: basis of sex differences in incidence and treatment response. Br J Pharmacol. 2014;171:567–579. doi: 10.1111/bph.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ventetuolo CE, Praestgaard A, Palevsky HI, Klinger JR, Halpern SD, Kawut SM. Sex and haemodynamics in pulmonary arterial hypertension. Eur Respir J. 2014;43:523–530. doi: 10.1183/09031936.00027613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.International PPH Consortium. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 22.Girerd B, Montani D, Coulet F, Sztrymf B, Yaici A, Jaïs X, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181:851–861. doi: 10.1164/rccm.200908-1284OC. [DOI] [PubMed] [Google Scholar]
- 23.Harrison RE, Flanagan JA, Sankelo M, Abdalla SA, Rowell J, Machado RD, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40:865–871. doi: 10.1136/jmg.40.12.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA, 3rd, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5:336–343. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davie NJ, Crossno JT, Frid MG, Hofmeister SE, Reeves JT, Hyde DM, et al. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L668–78. doi: 10.1152/ajplung.00108.2003. [DOI] [PubMed] [Google Scholar]
- 26.Hayashida K, Fujita J, Miyake Y, Kawada H, Ando K, Ogawa S, et al. Bone marrow-derived cells contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension. Chest. 2005;127:1793–1798. doi: 10.1378/chest.127.5.1793. [DOI] [PubMed] [Google Scholar]
- 27.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asosingh K, Farha S, Lichtin A, Graham B, George D, Aldred M, et al. Pulmonary vascular disease in mice xenografted with human BM progenitors from patients with pulmonary arterial hypertension. Blood. 2012;120:1218–1227. doi: 10.1182/blood-2012-03-419275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood. 2011;117:3485–3493. doi: 10.1182/blood-2010-09-306357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Constien R, Forde A, Liliensiek B, Gröne HJ, Nawroth P, Hämmerling G, et al. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. doi: 10.1002/gene.1030. [DOI] [PubMed] [Google Scholar]
- 32.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, et al. VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759–767. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 33.Channick RN, Sitbon O, Barst RJ, Manes A, Rubin LJ. Endothelin receptor antagonists in pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:62S–67S. doi: 10.1016/j.jacc.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 34.Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus Perez V, et al. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2011;31:814–820. doi: 10.1161/ATVBAHA.110.219980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 36.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 37.Bishop T, Ratcliffe PJ. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ Res. 2015;117:65–79. doi: 10.1161/CIRCRESAHA.117.305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gale DP, Harten SK, Reid CDL, Tuddenham EGD, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood. 2008;112:919–921. doi: 10.1182/blood-2008-04-153718. [DOI] [PubMed] [Google Scholar]
- 40.Cowburn AS, Crosby A, Macias D, Branco C, Colaço RDDR, Southwood M, et al. HIF2α–arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci USA. 2016;113:8801–8806. doi: 10.1073/pnas.1602978113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, et al. Mutation of von Hippel–Lindau Tumour Suppressor and Human Cardiopulmonary Physiology. Plos Med. 2006;3:e290. doi: 10.1371/journal.pmed.0030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, et al. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest. 2010;120:827–839. doi: 10.1172/JCI36362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329:72–75. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- 44.Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci USA. 2010;107:11459–11464. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petousi N, Croft QPP, Cavalleri GL, Cheng H-Y, Formenti F, Ishida K, et al. Tibetans living at sea level have a hyporesponsive hypoxia-inducible factor system and blunted physiological responses to hypoxia. J Appl Physiol. 2014;116:893–904. doi: 10.1152/japplphysiol.00535.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorenzo FR, Huff C, Myllymäki M, Olenchock B, Swierczek S, Tashi T, et al. A genetic mechanism for Tibetan high-altitude adaptation. Nat Genet. 2014;46:951–956. doi: 10.1038/ng.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, et al. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol Cell. 2008;32:838–848. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Metelo AM, Noonan HR, Li X, Jin Y, Baker R, Kamentsky L, et al. Pharmacological HIF2alpha inhibition improves VHL disease-associated phenotypes in zebrafish model. J Clin Invest. 2015;125:1987–1997. doi: 10.1172/JCI73665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol. 2013;9:271–276. doi: 10.1038/nchembio.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016 doi: 10.1038/nature19796. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, et al. On-Target Efficacy of a HIF2alpha Antagonist in Preclinical Kidney Cancer Models. Nature. 2016 doi: 10.1038/nature19795. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nepal M, Gong Y-D, Park YR, Soh Y. An activator of PHD2, KRH102140, decreases angiogenesis via inhibition of HIF-1α. Cell Biochem Funct. 2011;29:126–134. doi: 10.1002/cbf.1732. [DOI] [PubMed] [Google Scholar]
- 53.Temes E, Martin-Puig S, Acosta-Iborra B, Castellanos MC, Feijoo-Cuaresma M, Olmos G, et al. Activation of HIF-prolyl hydroxylases by R59949, an inhibitor of the diacylglycerol kinase. J Biol Chem. 2005;280:24238–24244. doi: 10.1074/jbc.M414694200. [DOI] [PubMed] [Google Scholar]