

Abstract
Streptococcus pneumoniae is a highly recombinogenic human pathogen that utilizes the competence stimulating peptide (CSP)-based quorum sensing (QS) circuitry to acquire antibiotic resistance genes from the environment and initiate its attack on the human host. Modulation of QS in this bacterium, either inhibition or activation, can therefore be used to attenuate S. pneumoniae infectivity and slow down pneumococcal resistance development. In this study we set to determine the molecular mechanism that drives CSP:receptor binding and identify CSP-based QS modulators with distinct activity profiles. To this end, we conducted systematic replacement of the amino acid residues in the two major CSP signals (CSP1 and CSP2) and assessed the ability of the mutated analogs to modulate QS against both cognate and non-cognate ComD receptors. We then evaluated the overall 3D structures of these analogs using circular dichroism (CD) to correlate between the structure and function of these peptides. Our CD analysis revealed a strong correlation between α-helicity and bioactivity for both specificity groups (CSP1 and CSP2). Furthermore, we identified the first pan-group QS activator and the most potent group-II QS inhibitor to date. These chemical probes can be used to study the role of QS in S. pneumoniae and as scaffolds for the design of QS-based anti-infective therapeutics against S. pneumoniae infections.
Graphical abstract
Introduction
Quorum sensing (QS) is a bacterial communication mechanism used to coordinate group behaviors by assessing cell density through the production, secretion, and detection of small signaling molecules.1-4 QS has been shown to be involved in the regulation of symbiotic group phenotypes such as bioluminescence and root nodulation; as well as in pathogenic behaviors including swarming, motility, competence, biofilm formation, and virulence factor production in both Gram-negative and Gram-positive bacteria.1, 4-7 Thus, QS has attracted significant attention as a potential target to control bacterial behavior.8-11 Since QS circuits are centered on signal:receptor interactions to drive QS activation, interception of this interaction using synthetic signal mimics has the potential to attenuate virulence and pathogenicity and thus mitigate infections caused by multidrug resistant pathogens. Although the same principles apply to all QS circuits, their molecular mechanisms vary significantly between species and different bacterial groups: Gram-negative bacteria generally utilize small-molecule signals such as acyl-homoserine lactones (AHLs), quinolones and autoinducer 2 (AI-2), whereas Gram-positive bacteria use peptide autoinducers (AIPs) as the chemical signals to trigger QS.1, 8, 9, 11, 12 Moreover, some bacterial species possess multiple QS circuits,1, 8 while other bacterial species produce more than one signaling molecule, leading to the classification of specificity groups within the species.6, 9, 13 For example, S. aureus, the most studied Gram-positive species, has four different specificity groups of the accessory gene regulator (agr) QS circuitry, termed agr-I through agr-IV. Each group possesses a unique AIP signal and a cognate receptor.6, 13 Most cross-group interactions between an AIP from one specificity group and a receptor from a different specificity group were found to be inhibitory, suggesting competition between the groups mediated by agr interference.12 Moreover, AIP-based inhibitors of the agr QS circuitry were found to attenuate both virulence factor production in cellular assays and pathogenicity in different staphylococcal infection models.14-16 The extensive work that has been conducted on the agr circuitry in S. aureus highlights the therapeutic potential associated with QS interception and has sparked numerous studies of QS circuits in other Gram-positive bacterial pathogens, including Staphylococcus epidermidis,17, 18 Enterococcus faecalis,19-21 Clostridium perfringens,22, 23 and Streptococcus pneumoniae.24-27 S. pneumoniae is a commensal bacterium that predominately colonizes in the nasopharynx of many humans, but is also an opportunistic pathogen that causes diverse acute and chronic infections such as pneumonia, meningitis, and bacteremia.28, 29 Pathogenic S. pneumoniae strains are a significant threat to the health of individuals, leading to over 400,000 hospitalizations, 22,000 deaths, and a financial toll of $3.5 billion a year in the United States.30 Furthermore, S. pneumoniae is of particular danger to children because it colonizes 40-60% of this vulnerable population worldwide resulting in the death of over 800,000 children under the age of 5 each year.29, 31 S. pneumoniae utilizes a QS circuit that is centered on a 17-amino acid signaling peptide termed the competence stimulating peptide (CSP) to govern, among other phenotypes, the acquisition of genetic information through competence.24, 32 This QS circuit is comprised of 5 components termed ComA through E: The CSP pro-peptide (ComC) is processed and secreted extracellularly by the ComAB transporter as the mature CSP signal. The mature signal accumulates and, once a threshold concentration is achieved, the CSP signal can effectively bind and activate its cognate transmembrane histidine-kinase receptor (ComD). This binding triggers the phosphorylation of the response regulator (ComE) and results in the autoinduction of the QS circuitry (ComABCDE) and CSP synthesis. The expression of QS-regulated phenotypes, such as competence, is initiated by the transcription of the effector molecule of the QS circuit, comX (Figure 1).32-34 Thus far, two major chemical signals utilized by pneumococci have been identified, CSP1 and CSP2, along with their cognate receptors, ComD1 and ComD2 respectively, leading to the classification of two specificity groups within this species.35 Interestingly, in contrast to the agr-mediated QS circuitry of S. aureus,12cross-group interactions between the two specificity groups in S. pneumoniae are not inhibitory, rather the signal molecules have a significantly higher affinity to their cognate receptors leading to substantial changes in their ability to activate the different receptors (>40-fold difference in EC50 between cognate and non-cognate receptor for both signal molecules).25
Figure 1. S. pneumoniae CSP-mediated QS circuit.
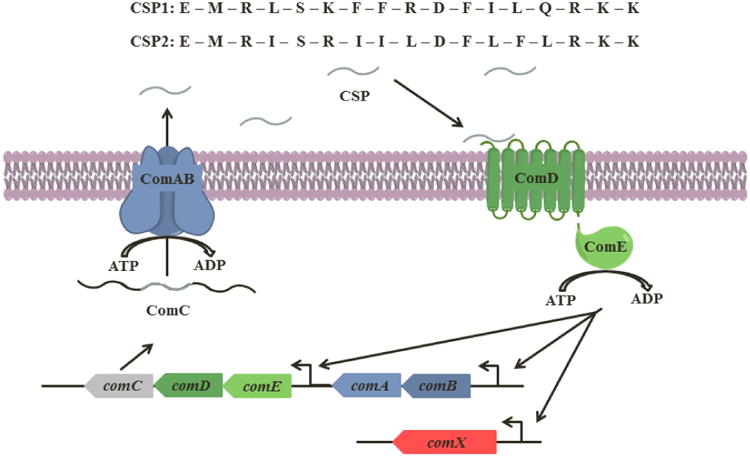
The CSP pro-peptide, ComC, is processed intracellularly and secreted by the ABC transporter (ComAB) as the mature CSP. When the extracellular CSP concentration reaches a threshold concentration, it can effectively bind and activate its cognate receptor, ComD. Activation of ComD leads to phosphorylation of ComE, a response regulator, and results in the autoinduction of the QS circuitry (ComABCDE) and expression of comX, which codes for QS-regulated phenotypes. Thus far, two major specificity groups of S. pneumoniae have been identified, each having a unique signaling peptide (CSP1 and CSP2) and corresponding cognate receptor (ComD1 and ComD2). Both CSPs are 17-amino acid peptide signals (written in one-letter code) and vary mainly in their central region.
Recent studies have shown that in addition to competence, the CSP-mediated QS circuit also regulates virulence and biofilm formation.32, 36-39 The involvement of the CSP-mediated QS circuit in pneumococcal pathogenesis has led to several studies investigating the therapeutic potential of modulating this circuitry. Pozzi and co-workers used a comD-null strain as well as an exogenous addition of the native CSP to determine the role of the CSP-based circuitry in different infection models.38, 40 In more recent studies, Lau and co-workers performed a systematic alanine scanning of CSP1 and found that a single mutation, Glu1 to Ala, converts CSP1 into a competitive inhibitor, CSP1-E1A.26, 27 Lau and co-workers further tested this CSP1 analog and found that it inhibited the expression of virulence factors in vitro, as well as attenuated virulence in a mouse lung infection model. Combined, these studies highlight the therapeutic potential of targeting the CSP-mediated QS circuit in S. pneumoniae. To better understand the molecular mechanism that drives this QS circuitry, provide an in-depth and quantitative analysis of the structural motifs required for ComD binding and activation, and develop new CSP-based QS modulators with desired activity profiles, we set to study the structure-activity relationships (SARs) of both CSP1 and CSP2. To this end, we performed full alanine and D-amino acid scans of both native signals and determined the activity profiles of all the analogs against the two ComD receptors, ComD1 and ComD2. We then analyzed the overall structural features of all the CSP analogs using circular dichroism (CD) spectroscopy. Our analysis revealed a strong correlation between α-helicity and biological activity for the CSP1 analogs and suggested that the α-helix is also the bioactive conformation of CSP2. Furthermore, we used our initial SAR analysis to design a second-generation library of analogs with desired activity profiles. These analogs are the most potent QS modulators identified to date in S. pneumoniae and could be utilized as scaffolds for the design of CSP-based therapeutics.
Materials and Methods
Chemical Reagents and Instrumentation
All chemical reagents and solvents were purchased from Sigma-Aldrich and used without further purification. Water (18 MΩ) was purified using a Millipore Analyzer Feed System. Solid-phase resins were purchased from Advanced ChemTech and Chem-Impex International.
Reversed-phase high-performance liquid chromatography (RP-HPLC) was performed using a Shimadzu system equipped with a CBM-20A communications bus module, two LC-20AT pumps, an SIL-20A auto sampler, an SPD-20A UV/VIS detector, a CTO-20A column oven, and an FRC-10A fraction collector. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) data were obtained on a Bruker Microflex spectrometer equipped with a 60 Hz nitrogen laser and a reflectron. In positive ion mode, the acceleration voltage on Ion Source 1 was 19.01 kV. Exact mass (EM) data were obtained on an Agilent Technologies 6230 TOF LC/MS spectrometer. The samples were sprayed with a capillary voltage of 3500 V and the electrospray ionization (ESI) source parameters were as follows: gas temperature of 325 °C at a drying gas flow rate of 8 L/min at a pressure of 35 psi.
Solid Phase Peptide Synthesis
All the CSP analogs were synthesized using standard Fmoc-based solid-phase peptide synthesis (SPPS) procedures on 4-Benzyloxybenzyl alcohol (Wang) resin. Pre-loaded Fmoc-L-Lys(Boc) Wang resin (0.59 mmol/g) was used for peptides that required a lysine at the C-terminus and preloaded Fmoc-L-Ala Wang resin (0.8 mmol/g) was used for peptides that required an alanine at the C-terminus. For peptides that have amino acids other than L-lysine or L-alanine at the C-terminus, loading of the first amino acid to the Wang resin linker was done by using the symmetrical anhydride procedure as previously described (for full procedure, see Supporting Information).41 For the D-amino acid library, L-Ile was replaced by D-allo-Ile.
Peptide Purification
Crude peptides were purified with RP-HPLC. A semi-preparative Phenomenex Kinetex C18 column (5 μm, 10 mm × 250 mm, 110 Å) was used for preparative RP-HPLC work, while an analytical Phenomenex Kinetex C18 column (5 μm, 4.6 mm × 250 mm, 110 Å) was used for analytical RP-HPLC work. Standard RP-HPLC conditions were as follows: flow rates = 5 mL min-1 for semi-preparative separations and 1 mL min-1 for analytical separations; mobile phase A = 18 MΩ water + 0.1% TFA; mobile phase B = ACN + 0.1% TFA. Purities were determined by integration of peaks with UV detection at 220 nm. Preparative HPLC methods were used to separate the crude peptide mixture to different chemical components using a linear gradient (first prep 5% B → 45% B over 40 min and second prep 20% B → 30% B over 30 min). Then, an analytical HPLC method was used to quantify the purity of the desired product using a linear gradient (5% B → 95% B over 27 min). Only peptide fractions that were purified to homogeneity (>95%) were used for the biological assays. TOF-MS was used to validate the presence of synthesized peptides. The observed mass-to-charge (m/z) ratio of the peptide was compared to the expected m/z ratio for each peptide (see Tables S-1 – S-4).
Biological Reagents and Strain Information
All standard biological reagents were purchased from Sigma-Aldrich and used according to enclosed instructions. Donor horse serum (defibrinated) was purchased from Sigma-Aldrich and stored at 4 °C until use in bacterial growth conditions.
To examine the ability of the synthesized CSP analogs to modulate the ComD receptors, and thus the QS circuit in S. pneumoniae, beta-galactosidase assays were performed using D39pcomX∷lacZ (group I) and TIGR4pcomX∷lacZ (group II) reporter strains.
Bacterial Growth Conditions
Freezer stocks were created from 1.5 mL aliquots of bacteria (0.2 OD 600nm) in Todd-Hewitt broth supplemented with 0.5% yeast extract (THY) and 0.5 mL glycerol, and stored at -80 °C. For experiments, bacteria from the freezer stocks were streaked onto a THY agar plate containing 5% serum and chloramphenicol at a final concentration of 4 μg/mL. The plate was incubated for 8-9 hours in a CO2 incubator (37 °C with 5% CO2). Fresh colonies (single colony for D39pcomX∷lacZ; multiple colonies for TIGR4pcomX∷lacZ) were transferred to 5 mL THY broth supplemented with chloramphenicol at a final concentration of 4 μg/mL and the culture was incubated in a CO2 incubator overnight (15 hours). Overnight cultures were then diluted (1:50 for D39pcomX∷lacZ; 1:10 for TIGR4pcomX∷lacZ) with THY and the resulting solution was incubated in a CO2 incubator for 3-4 hours, until the bacteria reached early exponential stage (0.30-0.35 for D39pcomX∷lacZ; 0.20-0.25 for TIGR4pcomX∷lacZ) as determined by using a plate reader.
Beta-Galactosidase Assays
Activation assays
The ability of synthetic CSP analogs to activate the expression of comX was determined using reporter strains grown in THY (pH 7.3). An initial activation screening was performed at high concentration (10 μM) for all CSP analogs. 2 μL of 1 mM solution of CSP analogs in dimethyl sulfoxide (DMSO) were added in triplicate to a clear 96-well microtiter plate. 2 μL of 20 μM solution of CSP1 were added in triplicate and served as the positive control for the group I strain (D39pcomX∷lacZ), while 2μL of 100 μM solution of CSP2 were added as the positive control for the group II strain (TIGR4pcomX∷lacZ). These concentrations were chosen to afford full activation of the QS circuit, as determined from the dose-dependent curves created for the native CSPs. 2 μL DMSO were added in triplicate and served as the negative control for both groups. Then, 198 μL bacterial culture were added to each well containing CSP and analogs, the plate was incubated at 37 °C for 30 minutes, and the OD 600nm was measured. In order to measure the beta-galactosidase activity in the pneumococcal culture, the cells were lysed by incubating the culture for 30 minutes at 37 °C with 20 μL 0.1% Triton X-100. In a new plate, 100 μL Z-buffer solution (60.2 mM Na2HPO4, 45.8 mM NaH2PO4, 10 mM KCl, and 1.0 mM MgSO4 in 18 MΩ H2O; pH was adjusted to 7.0 and the buffer was sterilized before use) containing 2-Nitrophenyl-Beta-D-galactopyranoside (ONPG) at a final concentration of 0.4 mg/mL were added, followed by 100 μL lysate, and the plate was incubated for 3 hours at 37 °C. The reaction was stopped by adding 50 μL of 1 M sodium carbonate solution, and the OD 420nm and OD 550nm were measured using a plate reader. The final results were reported as percent activation, which is the ratio between the Miller units of the analog and of the positive control. For calculation of Miller units, please see data analysis below. Analogs that exhibited high activity in the initial screening (see Figures S-1 – S-5 and S-9 – S-13) were further evaluated using a dose-dependent assay in which peptide stock solutions were diluted with DMSO in serial dilutions (either 1:2, 1:3, or 1:5) and assayed as described above. GraphPad Prism 5 was used to calculate the EC50 values, which are the concentration of a drug that gives half-maximal response.
Inhibition assays
Analogs that exhibited low comX activation in the initial screening (see Figures S-1 – S-5 and S-9 – S-13) were evaluated for competitive inhibition. The ability of synthesized CSP analogs to inhibit the expression of comX by outcompeting CSP for the receptor binding site was evaluated using the same assay conditions as described above, except that in the initial inhibition screening, the native CSP was added to every well in a set concentration (50 nM CSP1 for group I; 250 nM CSP2 for group II) that was chosen to afford full activation of the QS circuit, as determined from the dose-dependent curves created for the native CSPs. 2 μL of native CSP (5 μM solution of CSP1 for group I; 25 μM solution of CSP2 for group II) and 2 μL of 1 mM solution of CSP analogs were added to the same well in triplicate in a clear 96-well microtiter plate. 2 μL native CSP (5 μM solution of CSP1 for group I; 25 μM solution of CSP2 for group II) and 2 μL DMSO were added to the same well in triplicate and served as the positive control. 4 μL DMSO were added in triplicate and served as the negative control. Then, 196 μL bacterial culture were added to the wells and the plate was incubated at 37 °C for 30 minutes. The procedure for lysis, incubation with ONPG and all the measurements were as described in the activation assay. Analogs that exhibited significant competitive inhibition in the initial screening (see Figures S-6 – S-8 and S-14 – S-16) were further evaluated using a dose-dependent assay where peptide stock solutions were diluted with DMSO in serial dilutions (either 1:2, 1:3, or 1:5) and assayed as described above. GraphPad Prism 5 was used to calculate the IC50 values, which are the concentration of an inhibitor where the response (or binding) is reduced by half).
Analysis of Activation/Inhibition Data
Miller units were calculated using the following formula:
Abs420 is the absorbance of o-nitrophenol (ONP). Abs550 is the scatter from cell debris, which, when multiplied by 1.75 approximates the scatter observed at 420 nm. t is the duration of incubation with ONPG in minutes, v is volume of lysate in milliliters, and Abs600 reflects cell density.
Circular Dichroism (CD) Spectroscopy
CD spectra were recorded with an Aviv Biomedical CD spectrometer (model 202-01). All the measurements were performed with a peptide concentration of 200 μM in PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH was adjusted to 7.4) with 0% or 20% trifluoroethanol (TFE). Measurements were performed at 25 °C with a quartz cuvette (science outlet) with a path length of 0.1 cm. Samples were scanned one time at 3 nm min-1 with a bandwidth of 1 nm and a response time of 20 sec over a wavelength range (195 to 260 nm for CSP1 analogs; 192 to 260 nm for CSP2 analogs). Percent helicity (fH) was calculated for peptides that exhibited significant helical pattern using the following equation:
[θ]222 is the mean residue ellipticity of the sample peptide at 222 nm, [θ∞]222 is the mean residue ellipticity of an ideal peptide with 100% helicity (-44,000 deg cm2 dmol-1),42 n is the number of residues in the potential helical region, and x is an empirical correction for end effects (2.5).42
Results and Discussion
Design and Synthesis of First-Generation CSP Analogs
We started our study by systematic evaluation of each residue of CSP1 and CSP2 by performing full alanine and D-amino acid scans. These 68 peptides were designed to identify key side chains and stereocenters for CSP:ComD interactions. All the CSP analogs were constructed using Fmoc/tBu solid-phase peptide synthesis procedures on 4-Benzyloxybenzyl alcohol (Wang) resin (see Experimental Section). Peptides were cleaved from the solid support, purified to homogeneity by semipreparative RP-HPLC and isolated in acceptable yields (20–30%; see Supporting Information for full characterization details).
S. pneumoniae Reporter Gene Assays
We next tested the ability of the 68 CSP analogs to modulate the activity of the S. pneumoniae ComD1 and ComD2 receptors using β-galactosidase reporter gene assays (see Experimental Section for protocols). Each peptide was tested for its ability to modulate QS in groups I and II S. pneumoniae strains harboring pComX-lacZ reporter plasmids that were incorporated into the bacterial genome (see Experimental Section for strain information).27 In these reporter strains, the comX promoter is fused to the lacZ gene. Thus at high cell densities, which corresponds to high CSP concentrations, the CSP:ComD complex will phosphorylate ComE, which will then bind pcomX and transcribe lacZ (in addition to upregulation of ComX). Hence, ComD modulation can be quantified by measuring β-galactosidase activity. We therefore modified this reporter system to a 96-well microtiter plate format to enable large-scale screening of CSP analogs. We first quantified the ability of the native CSPs to activate their cognate and non-cognate ComD receptors and determined their EC50 values (Table 1). Our activity trends of the native CSPs were comparable to previous reports using different S. pneumoniae reporter strains (less than 4-fold differences).25 We then conducted an initial screening of all the analogs at high peptide concentration (10,000 nM) to identify CSP analogs that are capable of activating the ComD receptors to a level comparable to the native signals (see Figures S-1 – S-4 and S-9 – S-12). Analogs that exhibited high receptor activation, as determined in the initial screening (>75% activation compared to the native signal), were further evaluated to determine their EC50 values, while analogs that failed to activate the receptors (<50% activation compared to the native signal) were evaluated for their ability to competitively inhibit the receptors (see Figures S-6 – S-8 and S-14 – S-16).
Table 1. EC50/IC50 values of the alanine and D-amino acid scan analogs of CSP1 against ComD1 and ComD2 receptorsa.
| Name | EC50/IC50 (nM)b (95% CIc) | Name | EC50/IC50 (nM) (95% CI) | ||
|---|---|---|---|---|---|
|
|
|
||||
| ComD1 | ComD2 | ComD1 | ComD2 | ||
| CSP1 | 10.3 (6.27 - 16.8) | 526 (498 - 556) | CSP2 | 1650 (1190 - 2300) | 50.7 (40.6 - 63.2) |
|
| |||||
| CSP1-E1A | 85.7 (50.8 - 145)* | -- | CSP1-e1 | -- | -- |
| CSP1-M2A | 91.0 (75.4 - 110) | -- | CSP1-m2 | -- | -- |
| CSP1-R3A | --d | -- | CSP1-r3 | -- | -- |
| CSP1-L4A | 205 (179 - 235) | -- | CSP1-l4 | 628 (493 - 799) | -- |
| CSP1-S5A | -- | -- | CSP1-s5 | 10.0 (8.39 - 12.0) | -- |
| CSP1-K6A | 51.0 (37.9 - 68.6) | 24.0 (14.7 - 39.3) | CSP1-k6 | 147 (107 - 200) | 545 (482 - 616) |
| CSP1-F7A | -- | -- | CSP1-f7 | 506 (335 - 765) | 115 (96.2 - 137) |
| CSP1-F8A | >1,000 | -- | CSP1-f8 | 794 (610 - 1030) | >1000 |
| CSP1-R9A | 34.1 (29.9 - 38.7) | 646 (376 - 1110) | CSP1-r9 | -- | 304 (258 - 358) |
| CSP1-D10A | 54.3 (41.5 - 71.1) | 874 (560 - 1360) | CSP1-d10 | 107 (90.9 - 127) | 156 (134 - 181) |
| CSP1-F11A | -- | -- | CSP1-f11 | -- | -- |
| CSP1-I12A | 500 (199 - 1260) | -- | CSP1-i12 | 631 (547 - 728) | >1000 |
| CSP1-L13A | -- | -- | CSP1-l13 | 125 (99.1 - 158) | -- |
| CSP1-Q14A | -- | >1000 | CSP1-q14 | 12.2 (7.74 - 19.2) | -- |
| CSP1-R15A | 16.8 (12.8 - 22.1) | >1000 | CSP1-r15 | 8.37 (7.13 - 9.83) | -- |
| CSP1-K16A | 18.9 (10.0 - 35.6) | >1000 | CSP1-k16 | 8.10 (5.97 - 11.0) | -- |
| CSP1-K17A | 14.2 (6.23 - 32.5) | >1000 | CSP1-k17 | 5.88 (4.23 - 8.16) | >1000 |
See experimental section for detail of reporter strains and methods. See supporting information for plots of agonism or antagonism dose response curves. All assays performed in triplicate.
EC50 or IC50 values determined by testing peptides over a range of concentrations.
95% confidence interval.
EC50 not determined due to the analog's low induction in primary agonism screening assay. See supporting information for detail.
IC50 value.
The reporter gene data revealed several interesting SAR trends for both CSP1 and CSP2 (Tables 1 and 2), as will be discussed below. Furthermore, this analysis also uncovered the first pan-group ComD activator (Table 1) as well as the most potent ComD2 inhibitor known to date (Table 3). Our analysis of the CSP SAR trends is divided to three parts, each discussed in turn below: 1) analysis of the CSP1 scaffold against its cognate receptor, ComD1, 2) analysis of the CSP2 scaffold against its cognate receptor, ComD2, and 3) cross-group interactions between both CSP scaffolds and their non-cognate receptors.
Table 2. EC50/IC50 values of the alanine and D-amino acid scan analogs of CSP2 against ComD1 and ComD2 receptorsa.
| Name | EC50/IC50 (nM)b (95% CIc) | Name | EC50/IC50 (nM) (95% CI) | ||
|---|---|---|---|---|---|
|
|
|
||||
| ComD1 | ComD2 | ComD1 | ComD2 | ||
| CSP1 | 10.3 (6.27 - 16.8) | 526 (498 - 556) | CSP2 | 1650 (1190 - 2300) | 50.7 (40.6 - 63.2) |
|
| |||||
| CSP2-E1A | >1000* | >1000* | CSP2-e1 | >1000* | >1000* |
| CSP2-M2A | --d | 897 (638 - 1260) | CSP2-m2 | >1000* | >1000* |
| CSP2-R3A | -- | -- | CSP2-r3 | -- | -- |
| CSP2-I4A | -- | 170 (74.3 - 388) | CSP2-i4 | -- | -- |
| CSP2-S5A | -- | 148 (68.4 - 322) | CSP2-s5 | -- | 550 (484 - 625) |
| CSP2-R6A | >1000 | 116 (99.0 - 136) | CSP2-r6 | 830 (624 - 1100) | 182 (158 - 210) |
| CSP2-I7A | >1000 | 250 (212 - 295) | CSP2-i7 | 452 (357 - 573) | 240 (223 - 258) |
| CSP2-I8A | -- | -- | CSP2-i8 | >1000 | 249 (184 - 336) |
| CSP2-L9A | -- | 30.1 (12.2 - 74.6) | CSP2-l9 | >1000* | 111 (103 - 121) |
| CSP2-D10A | >1000* | 319 (274 - 373) | CSP2-d10 | 513 (437 - 602) | 2.86 (1.91 - 4.31) |
| CSP2-F11A | -- | 164 (126 - 212) | CSP2-f11 | -- | 270 (163 - 446) |
| CSP2-L12A | -- | >1,000 | CSP2-l12 | -- | 345 (213 - 559) |
| CSP2-F13A | -- | 505 (388 - 656) | CSP2-f13 | -- | 23.8 (14.7 - 38.6) |
| CSP2-L14A | -- | 175 (107 - 286) | CSP2-l14 | >1000* | 54.2 (52.9 - 55.6) |
| CSP2-R15A | -- | 24.3 (11.2 - 52.7) | CSP2-r15 | >1000* | 41.2 (31.0 - 54.7) |
| CSP2-K16A | -- | 80.5 (34.4 - 188) | CSP2-k16 | >1000* | 38.6 (25.1 - 59.3) |
| CSP2-K17A | -- | -- | CSP2-k17 | -- | 48.2 (44.4 - 52.3) |
See experimental section for detail of reporter strains and methods. See supporting information for plots of agonism or antagonism dose response curves. All assays performed in triplicate.
EC50 or IC50 values determined by testing peptides over a range of concentrations.
95% confidence interval.
EC50 not determined due to the analog's low induction in primary agonism screening assay. See supporting information for detail.
IC50 value.
Table 3. EC50/IC50 values of the second-generation analogs of CSP1 and CSP2 against ComD1 and ComD2 receptorsa.
| Name | EC50 /IC50 (nM)b (95% CIc) | Name | EC50 /IC50 (nM) (95% CI) | ||
|---|---|---|---|---|---|
|
|
|
||||
| ComD1 | ComD2 | ComD1 | ComD2 | ||
| CSP1 | 10.3 (6.27 - 16.8) | 526 (498 - 556) | CSP2 | 1650 (1190 - 2300) | 50.7 (40.6 - 63.2) |
|
| |||||
| CSP1-des-E1 | >1000* | -- | CSP2-des-E1 | >1000* | >1000* |
| CSP1-des-E1M2 | >1000* | -- | CSP2-des-E1M2 | -- | >1000* |
| CSP1-des-E1M2R3 | --d | -- | CSP2-des-E1M2R3 | -- | -- |
| CSP1-des-E1M2R3L4 | -- | -- | CSP2-des-E1M2R3I4 | -- | -- |
| CSP1-des-K17 | 4.86 (3.01 - 7.83) | >1000 | CSP2-des-K17 | -- | 44.5 (30.7 - 64.5) |
| CSP1-des-K16K17 | 8.10 (5.33 - 12.3) | >1000 | CSP2-des-K16K17 | -- | 21.9 (11.5 - 41.6) |
| CSP1-des-R15K16K17 | 139 (125 - 155) | >1000 | CSP2-des-R15K16K17 | -- | 42.3 (21.1 - 84.4) |
| CSP1-des-Q14R15K16K17 | >1000 | >1000 | CSP2-des-L14R15K16K17 | -- | 77.9 (70.6 - 85.9) |
| CSP1-E1AK6A | 104 (55.6 - 195)* | -- | CSP2-E1Ad10 | >1000* | 56.5 (53.5 - 59.6)* |
| CSP1-retro-inverso | -- | -- | CSP2-m2d10 | -- | >1000* |
| CSP2-E1Am2d10 | >1000* | >1000* | |||
| CSP2-retro-inverso | -- | -- | |||
See experimental section for detail of reporter strains and methods. See supporting information for plots of agonism or antagonism dose response curves. All assays performed in triplicate.
EC50 or IC50 values determined by testing peptides over a range of concentrations.
95% confidence interval.
EC50 not determined due to the analog's low induction in primary agonism screening assay. See supporting information for detail.
IC50 value.
SAR of CSP1 against ComD1
The CSP1 scaffold can be separated into three regions: the N-terminus, central region, and the C-terminus. Starting with the N-terminus of the peptide, the alanine and D-amino acid scans revealed that the first three residues have a critical role in receptor binding and activation: alanine or d-amino acid replacements of Arg3 resulted in complete loss of activity, while similar replacements of Met2 significantly reduced the peptide activity (9-fold decrease to complete loss of activity, see Table 1). However, In the case of Glu1, while d-amino acid replacement also eliminated the peptide activity, alanine replacement resulted in the conversion of CSP1 into a potent ComD1 inhibitor (Table 1). Combined, these results suggest that the N-terminus of the peptide binds to a relatively tight binding site that does not accommodate significant side chain orientation changes, as induced by D-amino acid replacements. Furthermore, it appears that Arg3 has a key role in initial receptor recognition, while Glu1 is mainly involved in driving receptor activation, as was suggested in previous studies by Lau and co-workers aimed at identifying CSP1-based QS inhibitors.26, 27
Moving to the central region of the peptide, our analysis revealed that changes of the hydrophobic residues resulted in a dramatic reduction in potency while changes of the hydrophilic residues were more tolerable (compare modifications in Leu4, Phe7, Phe8, Phe11, Ile12, and Leu13 with modifications in Ser5, Lys6, Arg9, and Asp10 in Table 1). This observation implies that signal:receptor recognition is mediated by hydrophobic interactions. Furthermore, for the hydrophilic residues (Ser5, Lys6, Arg9, and Asp10), alanine replacement is generally more tolerable than d-amino acid replacement, suggesting that for these residues the orientation of the side chain, and concomitantly induced local conformation, is more important than the actual side chain residue. The same trend is not apparent for the hydrophobic residues (Leu4, Phe7, Phe8, Phe11, Ile12, and Leu13). For some residues, d-amino acid replacement is more tolerable while for others, alanine replacement is more tolerable. This observation further emphasizes the importance of the hydrophobic side chains and suggests that for this region of the peptide, the receptor binding site may be more promiscuous and thus tolerates more changes in side chain orientation induced by d-amino acid replacements, compared to the N-terminus region. Overall, our analysis of the central region of CSP1 is in agreement with previous work by Johnsborg et al. that showed that the hydrophobic residues in CSP1 are involved in receptor specificity between ComD1 and ComD2.25
With regards to the C-terminus of the peptide, it appears that this region is not involved in either receptor binding or activation: replacement of Arg15, Lys16 or Lys17 with either alanine or the d-amino acid counterparts resulted in analogs with comparable activities to CSP1 (Table 1). Similarly, replacement of Gln14 with its D-amino acid counterpart also resulted in an analog with comparable activity, however replacement of this residue with alanine resulted in significant reduction of activity (Table 1). Overall, our results indicate that at least the last three residues of CSP1 (Arg15, Lys16 and Lys17) are dispensable. It is not clear why these residues were not eliminated over time, but the fact that these three residues are conserved among the two S. pneumoniae CSP signals suggests that they have an important role in the QS circuitry. By examining the entire peptide sequence, it is tempting to speculate that the role of these three positively charged residues is to increase the solubility of the overall relatively hydrophobic peptide sequences. However, further experiments are required to test this hypothesis.
SAR of CSP2 against ComD2
Similarly to CSP1, the CSP2 scaffold can also be separated into three regions: the N-terminus, central region, and the C-terminus. Starting with the N-terminus, a similar trend was observed for the first three conserved residues (Glu1, Met2 and Arg3); alanine and D-amino acid replacements of Arg3 resulted in complete loss of activity, while modifications to Glu1 resulted in analogs with weak inhibitory activity (Table 2). In the case of Met2, unlike the CSP1 scaffold, d-amino acid replacement also resulted in a weak inhibitor (Table 2). Thus, in the case of CSP2, it seems that the main role of both Glu1 and Met2 is to drive receptor activation.
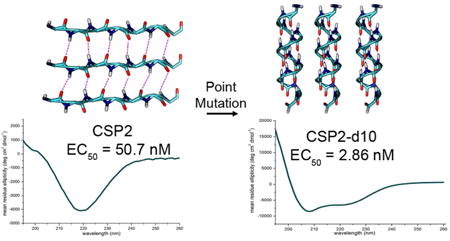
In regards to the central region of CSP2, it appears that this segment is more tolerable to modifications compared to CSP1, as most modifications (alanine or d-amino acid replacements) result in only a minor to moderate change in bioactivity (<11-fold difference, with a few modifications leading to enhanced activity), with the exception of three modification (I8A, L12A and i4). Comparison of the central region sequences of both CSP signals reveal that CSP2 is more hydrophobic than CSP1, having only three hydrophilic residues (Ser5, Agr6 and Asp10). Since hydrophobic interactions are assumed to drive receptor:signal recognition and initial binding, it is possible that the more hydrophobic central region of CSP2 can better accommodate single modifications, and thus compensate for the loss of one hydrophobic side chain contact with the receptor. By far, the most interesting result identified when evaluating this region is the replacement of Asp10 with its d-enantiomer. This modification resulted in >17-fold increased potency compared to the native CSP2 signal, the most significant increase in activity observed in both specificity groups. At this point, the cause of such a significant enhancement in activity is not clear. To address this question, and gain further SAR insights regarding both the CSP1 and CSP2 scaffolds, we conducted structural analysis using circular dichroism (CD) spectroscopy, as will be discussed below.
Evaluation of the C-terminus region of CSP2 revealed a similar trend as that of CSP1. That is, with the exception of alanine replacement at the Lys17 position, replacements of all four residues (Leu14, Arg15, Lys16 or Lys17) resulted in analogs with comparable activities to CSP2 (<3-fold reduction in activity). These results provide additional support that the C-terminus residues (at least Arg15, Lys16 and Lys17) are dispensable.
Cross-group Modulation
A previous study by Johnsborg et al. revealed that the CSP signals have high specificity to their cognate receptors and thus are only capable of activating the cross-group receptors at high concentrations.25 The authors further showed that the specificity of the CSPs towards their cognate ComD receptors can be reversed by replacing the hydrophobic residues at positions 4, 7 and 8 with those of the other signal. As the increased affinity to the non-cognate receptor was always coupled with decreased affinity to the cognate receptor, the authors concluded that the hydrophobic residues in these three positions are essential for receptor specificity. In this study we aimed to identify several CSP-based QS modulators with unique activity profiles. One desired activity profile is a pan-group QS activator. We therefore evaluated all the CSP1 and CSP2 analogs for their cross-group activity. Overall, most of the analogs exhibited similar activity trend as the two native CSPs. That is, most analogs were more active as modulators of their cognate receptor, and in most cases, were relatively inactive as modulators of the non-cognate receptor (Tables 1 and 2). When comparing the CSP1 and CSP2 analogs, a clear trend was apparent in which CSP1 and its analogs were better activators of the ComD2 receptor than CSP2 and its analogs against ComD1. This trend suggests that CSP1 is a better scaffold for the design of pan-group QS activators. Interestingly, with regards to cross-group inhibition, the trend is reversed: while no CSP1 analogs were found to inhibit the ComD2 receptor, several CSP2 analogs were found to inhibit the ComD1 receptor, although only at high concentrations (all the inhibitors exhibited IC50 values that are >1,000 nM, Table 2). However, the most interesting result was the alanine replacement of Lys6 in CSP1 to afford CSP1-K6A (Table 1). This analog exhibited only slightly reduced activity compared to CSP1 against ComD1 (∼5-fold reduction), but showed a significant increase in potency against the ComD2 receptor (∼22-fold increase). Overall, this analog has similar potencies against both ComD receptors (EC50 values of 51.0 and 24.0 nM against ComD1 and ComD2 respectively, Table 1) and is thus the first pan-group QS activator identified to date. This result also suggests that the sixth position of CSP1 is the most critical for receptor specificity, rather than the hydrophobic residues at positions 4, 7 and 8.
Structural Analysis using CD
Our SAR analysis of CSP1 and CSP2 revealed several interesting activity trends, however this analysis was lacking key information as to how different modifications affect the overall peptide structure. To gain additional structural information and correlate it to bioactivity we set out to assess the overall structures of the two native CSPs and their analogs using CD spectroscopy. CD enables the evaluation of the overall structural motifs of peptides and proteins. More importantly, this analysis allows for the assessment of a large number of analogs and the identification of structural motifs required for activity. Previous analysis of CSP1 revealed that this peptide adopted an α-helix conformation in membrane mimicking conditions. This observation led to the hypothesis that CSP1 adopts a helix conformation upon binding to its transmembrane receptor, ComD1.25 Unfortunately, no additional data to support this hypothesis was provided, nor any structural data of CSP2. We therefore assessed all the CSP analogs in both aqueous (PBS buffer, pH 7.4) and membrane mimicking conditions (20% TFE in PBS, pH 7.4). Starting with CSP1, this peptide was unfolded in aqueous solution (Figure S-17) but adopted a helix conformation in membrane mimicking conditions (Figure 2 and S-18). We quantified the percent helicity of CSP1 using the mean residue ellipticity at 222 nm as previously reported,42 and found that 20% of the CSP1 sequence is helical (Table S-5). Further evaluation of the alanine and d-amino acid analogs of CSP1 revealed a very strong correlation between bioactivity and helicity. That is, analogs that exhibited low helicity also showed significant reduction in activity (see Figures 2, S-18, S-20, and Table S-5). Interestingly, with the exception of CSP1-L4A, all the alanine replacements in CSP1 yielded analogs with comparable helicity to CSP1, suggesting that reduced activity in any of these analogs is due to the loss of a key receptor-ligand interaction as opposed to conformational change. Contrary to the alanine scan, d-amino acid replacements showed more variation with regards to helicity of the different analogs. Specifically, analogs bearing modifications in central region residues (positions 6 to 13) showed significantly lower helicity compared to CSP1 (Figure 2, S-20 and Table S-5). This observation is in agreement with the 3D structure obtained for CSP1 using NMR techniques, where the central region (positions 6 to 12) was found to be helical.25 By combining the biological results with the CD analysis, we concluded that the reduced activity, as a result of D-amino acid replacements in the central regions of CSP1, is caused by a conformational change of the peptide, presumably due to steric hindrance between the reversibly oriented D-amino acid and the residues surrounding it.
Figure 2. CD spectra of select CSP1 analogs in membrane mimicking conditions.
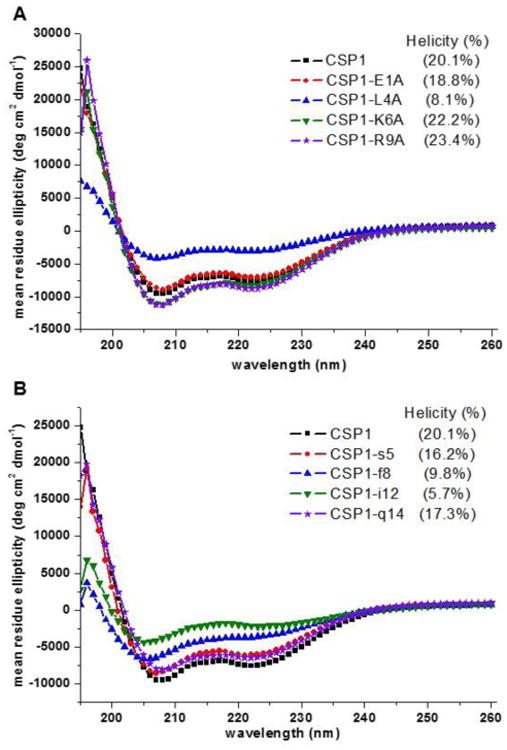
A) CD spectra of select alanine-scanning analogs. B) CD spectra of select d-amino acid scanning analogs. There is a clear correlation between a helical conformation and biological activity for the CSP1 analogs, as can be concluded from comparing the CD spectra, percent helicity, and the biological data in Table 1.
Moving to CSP2, due to the similarity in sequence to CSP1 and identical function, we assumed this peptide should have a similar mechanism of action and thus should adopt a helix conformation in membrane mimicking conditions. Unexpectedly, the CD analysis revealed that CSP2 adopted a β-sheet conformation in both aqueous and membrane mimicking conditions (Figure 3, S-23 and S-24). For relatively short peptide sequences such as CSP2 (17 residues), this result implies that the peptide interacts with other peptides to form β-sheet aggregates. The more hydrophobic nature of CSP2 compared to CSP1, along with our relative difficulties handling and purifying this peptide due to the viscous solutions that it forms, further support this hypothesis and suggest that CSP2 aggregates in solution when it reaches a high concentration. Analysis of the alanine replacement analogs revealed that, with a few exceptions, all the CSP2 analogs adopt a β-sheet conformation in both aqueous and membrane mimicking conditions, suggesting that this conformation may have biological relevance (Figure 3, S-23 and S-24). The d-amino acid replacement analogs exhibited more heterogeneous spectra, although the β-sheet conformation was still the most dominant feature observed (Figure 3, S-25 and S-26). Perhaps the most interesting result of this analysis was the CD spectrum of CSP2-d10 in membrane mimicking conditions. This analog, which was the most potent ComD2 activator (EC50 value of 2.86 nM against ComD2), exhibited an α-helix conformation in these conditions (Figure 3). As β-sheets are stabilized by inter-chain interactions while α-helices are stabilized by intra-chain interactions, we hypothesize that the bioactive conformation of CSP2 is that of an α-helix. However, as this peptide tends to aggregate as a β-sheet, the effective concentration of the monomeric active form is relatively low. Thus, shifting the equilibrium towards the bioactive conformation should increase the potency of the peptide. We further hypothesize that the D-amino acid replacement of Asp10 causes a critical steric hindrance that disrupts the inter-chain forces that stabilize the β-sheet, resulting in the equilibrium shifting towards the monomeric α-helix active form. Thus, CSP2-d10 exerts its increased potency through a conformational change and the stabilization of the bioactive conformation. Additional structural analysis is needed to further validate this hypothesis and gain better insight regarding the CSP2 mechanism of action.
Figure 3. CD spectra of select CSP2 analogs in membrane mimicking conditions.
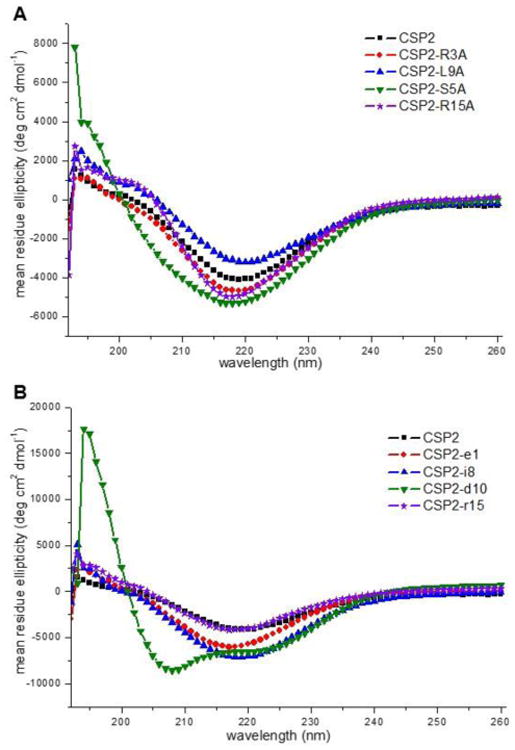
A) CD spectra of select alanine-scanning analogs. B) CD spectra of select d-amino acid scanning analogs. CSP2 and most of its analogs adopt a β-sheet conformation. However, CSP2-d10, the most active CSP2 analog identified, adopts an α-helix conformation.
Second-generation CSP Analogs
Our initial analysis of CSP1 and CSP2 revealed several interesting activity trends. To further characterize these trends and identify CSP-based QS modulators with unique activity profiles (i.e. potent group II inhibitors, pan-group QS inhibitors), we designed a second-generation library of CSP analogs. First, our analysis revealed that the N-terminal residues may be involved primarily in receptor activation, while the C-terminal residues may be dispensable. To further test the role of these residues, we synthesized a library of sequentially truncated CSP1 and CSP2 analogs. This library consisted of sequential truncation of either the four N-terminal residues, or the four C-terminal residues of both native signals. Second, our analysis revealed that replacement of Asp10 with its enantiomer in CSP2 resulted in increased potency, while replacement of Glu1 with alanine and Met2 with its enantiomer produced weak ComD2 inhibitors. We therefore hypothesized that combining these mutations together would result in potent ComD2 inhibitors. Thus, we synthesized a set of three double or triple mutated CSP2 analogs (CSP2-E1Ad10, CSP2-m2d10, and CSP2-E1Am2d10). Third, we found that replacement of Lys6 with alanine in CSP1 converted this peptide to a pan-group activator, while replacement of Glu1 with alanine converted CSP1 to a potent ComD1 inhibitor. Thus, we hypothesized that combining these mutations would yield a potent pan-group inhibitor. Lastly, our analysis revealed a strong correlation between α-helicity and bioactivity in both groups. To gain further insights regarding key interactions between the peptides and the ComD receptors, we synthesized the two retro-inverso analogs of CSP1 and CSP2. In these analogs, the entire peptide sequence is constructed using d-amino acids in the reversed order of that of the native CSP. The retro-inverso modification produces topologically equivalent peptidomimetics where only the backbone of the peptide is affected, switching amide nitrogen and carbonyl, while the side chain orientation remains the same.43-45 Thus, this approach can be used to dissect the role of the peptide backbone in bioactivity.
Starting with the truncated CSP analogs, a similar trend was observed for the N-terminal residues in both groups: the removal of the first two residues (Glu1 and Met2) resulted in weak QS inhibitors (IC50 >1,000 nM against the cognate receptor), with CSP2-des-E1 exhibiting weak inhibitory activity against both ComD receptors (Table 3). Removal of additional residues from the N-terminus (Arg3 and Leu/Ile4 in CSP1 and CSP2 respectively) resulted in complete loss of activity (Table 3). With regards to the C-terminal residues, a slightly different trend was observed for CSP1 and CSP2: in CSP1 removal of the last two C-terminal residues (Lys17 and Lys16) resulted in analogs with comparable activities to CSP1, whereas removal of additional residues (Arg15 or Gln14) led to significant reduction in potency (Table 3). Contrary, in CSP2 all four C-terminal residues (Lys17, Lys16, Arg15 and Leu14) were found to be dispensable and removal of these residues resulted in analogs with comparable activities to CSP2 (Table 3). Overall, this analysis confirmed that the first two residues at the N-terminus of both CSP signals are required for receptor activation, while the C-terminal residues have no major impact on activity. CD analysis of the truncated analogs further confirmed the importance of α-helicity to bioactivity of CSP1, as all the active analogs retained relatively high helicity while most of the inactive analogs exhibited lower helicity (Figures S-21 and S-22, and Table S-6). With regards to the truncated CSP2 analogs, most modifications resulted in some conformational change (Figures S-27 and S-28): At the N-terminus, removal of more than two residues resulted in a conformational change from a β-sheet structure to an α-helix, while all changes at the C-terminus led to analogs with very weak β-sheet characteristics (Figure S-28).
As expected, biological evaluation of the three double and triple mutation CSP2 analogs, CSP2-E1Ad10, CSP2-m2d10, and CSP2-E1Am2d10, revealed three ComD2 inhibitors. Surprisingly, two of them were also ComD1 inhibitors (CSP2-E1Ad10 and CSP2-E1Am2d10, See Table 3). However, most importantly, one of these analogs, CSP2-E1Ad10, was found to be a potent ComD2 inhibitor (IC50 = 56.5 nM), while the other two analogs were only weak inhibitors (IC50 > 1,000 nM, Table 3). Interestingly, the CD spectra of these three analogs was that of an α-helix, supporting our hypothesis regarding the role of the d10 substitution in shifting the conformational equilibrium towards a monomeric α-helix (Figure 4A). Furthermore, these results also support our hypothesis that an α-helix is the bioactive conformation of CSP2.
Figure 4. CD spectra of select second-generation CSP analogs in membrane mimicking conditions.
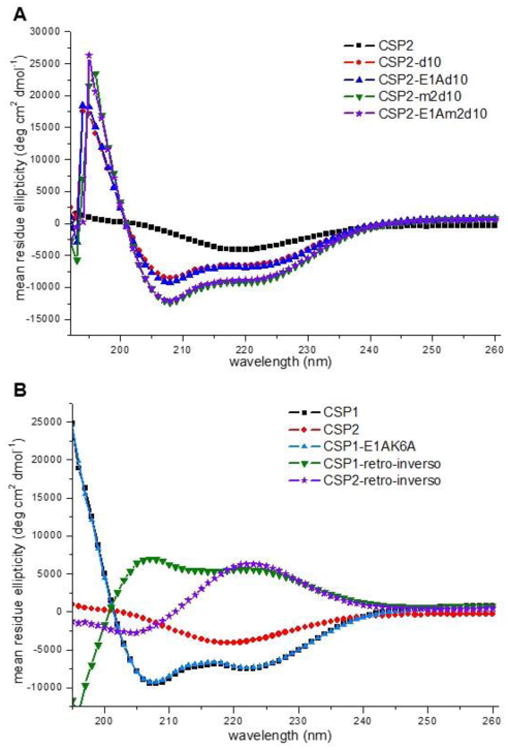
A) CD spectra of double and triple mutation CSP2 analogs. All CSP2 analogs bearing the d10 mutation adopt an α-helix conformation. B) CD spectra of CSP1-E1AK6A and the two retro-inverso peptides. CSP1-E1AK6A adopts an α-helix conformation, while the two retro-inverso peptides adopt a reversed pattern compared to their parent signals.
As for the double mutant CSP1 analog, CSP1-E1AK6A, as expected this analog exhibited potent inhibitory activity against the ComD1 receptor (IC50 = 104 nM, Table 3). Moreover, this analog was also found to adopt an α-helix conformation (Figure 4B). Unfortunately, contrary to our expectation, this analog was found to be inactive against the ComD2 receptor (Table 3). Moving to the retro-inverso analogs, both analogs exhibited reversed conformational pattern compared to their parent CSP signal (reversed α-helix for CSP1 retro-inverso and reversed β-sheet for CSP2 retro-inverso, Figure 4B), suggesting that the side chain residues adopt topologically equivalent orientation. However, when evaluating these analogs for their ability to modulate QS, both analogs were found to be relatively inactive against both ComD receptors (Table 3). This result implies that the signal backbone is directly involved in receptor binding, presumably through extensive hydrogen bonding, and that the reversed backbone amides in the retro-inverso analogs are no longer capable of effectively forming these hydrogen bonds, resulting in relatively inactive analogs.
Summary and Conclusions
S. pneumoniae uses the CSP-mediated QS circuit to acquire genetic information from the environment, including antibiotic resistance genes, and regulate phenotypes associated with virulence and infectivity. Synthetic signal analogs that modulate QS in S. pneumoniae could thus be applied as chemical probes to study the role of QS in pneumococcal infections. In the current study, we report the design, synthesis, and biological and structural evaluation of a series of 90 non-native CSP analogs, comprised of 44 peptides based on the CSP1 scaffold and 46 peptides based on the CSP2 scaffold. Our work revealed the first pan-group ComD activator, a potent ComD2 inhibitor, and overall represents the most in-depth analysis of the SARs defining the activities of CSP1 and CSP2 against the two ComD receptors.
There are several important outcomes of this study. First, the SAR analyses of CSP1 and CSP2 revealed a series of interesting activity trends for ComD receptor modulation in both QS specificity groups. Our results both validate and expand on previous observations regarding the role of the N-terminus region of the CSP signals in ComD activation and the importance of the hydrophobic residues at the central region of the peptides for ComD recognition.25-27 Moreover, we demonstrated that the C-terminal region of CSP1 and CSP2 is dispensable and can be removed without affecting the signal activity. Second, to gain additional structural insights regarding the activity trends of the CSP analogs, we conducted a comprehensive structural analysis of all the CSP analogs using CD spectroscopy. This analysis revealed a strong correlation between α-helicity and bioactivity for the CSP1 scaffold. Moreover, although CSP2, and most of its analogs, were found to adopt a β-sheet conformation, our analysis suggests that CSP2 adopts an α-helix conformation upon binding ComD2, as the most potent CSP2 analogs identified exhibited α-helix conformation.
Lastly, this study uncovered several synthetic CSP analogs with unique activity trends: our analysis of CSP1 revealed the first pan-group ComD activator, CSP1-K6A, that exhibits similar potencies against the two ComD receptors, while our analysis of CSP2 led us to the development of the most potent ComD2 inhibitor, CSP2-E1Ad10. Importantly, the latter also exhibited weak inhibitory activity against ComD1 and can thus be used as a template for the design of potent pangroup ComD inhibitors. Such studies are ongoing in our laboratory, along with additional in-depth structural and biochemical studies aimed at characterizing the mechanism of action of lead CSP analogs, as well as identifying new avenues to improve their potency and stability, and will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by the Nevada INBRE through a grant from the National Institute of General Medical Sciences (GM103440) from the National Institutes of Health (NIH). S. pneumoniae D39pcomX∷lacZ and TIGR4pcomX∷lacZ reporter strains were generous gifts from G. W. Lau (University of Illinois at Urbana-Champaign).
Footnotes
Supporting Information: Full details of peptide synthesis and characterization, initial screening results, dose response curves for CSP analogs, and CD spectra of all the CSP analogs. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rutherford ST, Bassler BL. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med. 2012;2:a012427. doi: 10.1101/cshperspect.a012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Bodman SB, Willey JM, Diggle SP. Cell-cell communication in bacteria: united we stand. J Bacteriol. 2008;190:4377–4391. doi: 10.1128/JB.00486-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams P, Winzer K, Chan WC, Camara M. Look who's talking: communication and quorum sensing in the bacterial world. Philos Trans R Soc Lond B Biol Sci. 2007;362:1119–1134. doi: 10.1098/rstb.2007.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsek MR, Greenberg EP. Sociomicrobiology: the connections between quorum sensing and biofilms. Trends Microbiol. 2005;13:27–33. doi: 10.1016/j.tim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Palmer AG, Senechal AC, Mukherjee A, Ane JM, Blackwell HE. Plant responses to bacterial N-acyl L-homoserine lactones are dependent on enzymatic degradation to L-homoserine. ACS Chem Biol. 2014;9:1834–1845. doi: 10.1021/cb500191a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang B, Muir TW. Regulation of Virulence in Staphylococcus aureus: Molecular Mechanisms and Remaining Puzzles. Cell Chem Biol. 2016;23:214–224. doi: 10.1016/j.chembiol.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon CP, Williams P, Chan WC. Attenuating Staphylococcus aureus virulence gene regulation: a medicinal chemistry perspective. J Med Chem. 2013;56:1389–1404. doi: 10.1021/jm3014635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Welsh MA, Blackwell HE. Chemical probes of quorum sensing: from compound development to biological discovery. FEMS Microbiol Rev. 2016;40:774–794. doi: 10.1093/femsre/fuw009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shepherd NE, Harrison RS, Fairlie DP. Targeting quorum sensing and competence stimulation for antimicrobial chemotherapy. Curr Drug Targets. 2012;13:1348–1359. doi: 10.2174/138945012803530233. [DOI] [PubMed] [Google Scholar]
- 10.Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 11.Galloway WR, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Quorum sensing in Gram-negative bacteria: small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chem Rev. 2011;111:28–67. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]
- 12.Novick RP, Geisinger E. Quorum sensing in staphylococci. Annu Rev Genet. 2008;42:541–564. doi: 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- 13.Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. Peptide Signaling in the Staphylococci. Chem Rev. 2011;111:117–151. doi: 10.1021/cr100370n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, Blackwell HE. Highly Potent Inhibitors of Quorum Sensing in Staphylococcus aureus Revealed Through a Systematic Synthetic Study of the Group-III Autoinducing Peptide. J Am Chem Soc. 2013;135:7869–7882. doi: 10.1021/ja3112115. [DOI] [PubMed] [Google Scholar]
- 15.Broderick AH, Stacy DM, Tal-Gan Y, Kratochvil MJ, Blackwell HE, Lynn DM. Surface Coatings that Promote Rapid Release of Peptide-Based AgrC Inhibitors for Attenuation of Quorum Sensing in Staphylococcus aureus. Adv Healthcare Mater. 2014;3:97–105. doi: 10.1002/adhm.201300119. [DOI] [PubMed] [Google Scholar]
- 16.Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP, Muir TW. Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence. Proc Natl Acad Sci U S A. 1999;96:1218–1223. doi: 10.1073/pnas.96.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang T, Tal-Gan Y, Paharik AE, Horswill AR, Blackwell HE. Structure-Function Analyses of a Staphylococcus epidermidis Autoinducing Peptide Reveals Motifs Critical for AgrC-type Receptor Modulation. ACS Chem Biol. 2016;11:1982–1991. doi: 10.1021/acschembio.6b00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Buttner H, Dunman PM, Rohde H, Cech NB, Fey PD, Horswill AR. Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. J Bacteriol. 2014;196:3482–3493. doi: 10.1128/JB.01882-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin X, Singh KV, Weinstock GM, Murray BE. Characterization of fsr, a regulator controlling expression of gelatinase and serine protease in Enterococcus faecalis OG1RF. J Bacteriol. 2001;183:3372–3382. doi: 10.1128/JB.183.11.3372-3382.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishiguchi K, Nagata K, Tanokura M, Sonomoto K, Nakayama J. Structure-activity relationship of gelatinase biosynthesis-activating pheromone of Enterococcus faecalis. J Bacteriol. 2009;191:641–650. doi: 10.1128/JB.01029-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakayama J, Yokohata R, Sato M, Suzuki T, Matsufuji T, Nishiguchi K, Kawai T, Yamanaka Y, Nagata K, Tanokura M, Sonomoto K. Development of a peptide antagonist against fsr quorum sensing of Enterococcus faecalis. ACS Chem Biol. 2013;8:804–811. doi: 10.1021/cb300717f. [DOI] [PubMed] [Google Scholar]
- 22.Vidal JE, Ma M, Saputo J, Garcia J, Uzal FA, McClane BA. Evidence that the Agr-like quorum sensing system regulates the toxin production, cytotoxicity and pathogenicity of Clostridium perfringens type C isolate CN3685. Mol Microbiol. 2012;83:179–194. doi: 10.1111/j.1365-2958.2011.07925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma M, Li J, McClane BA. Structure-function analysis of peptide signaling in the Clostridium perfringens Agr-like quorum sensing system. J Bacteriol. 2015;197:1807–1818. doi: 10.1128/JB.02614-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Håvarstein LS, Coomaraswamy G, Morrison DA. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc Natl Acad Sci U S A. 1995;92:11140–11144. doi: 10.1073/pnas.92.24.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnsborg O, Kristiansen PE, Blomqvist T, Havarstein LS. A hydrophobic patch in the competence-stimulating Peptide, a pneumococcal competence pheromone, is essential for specificity and biological activity. J Bacteriol. 2006;188:1744–1749. doi: 10.1128/JB.188.5.1744-1749.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duan C, Zhu L, Xu Y, Lau GW. Saturated alanine scanning mutagenesis of the pneumococcus competence stimulating peptide identifies analogs that inhibit genetic transformation. PLoS One. 2012;7:e44710. doi: 10.1371/journal.pone.0044710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu L, Lau GW. Inhibition of competence development, horizontal gene transfer and virulence in Streptococcus pneumoniae by a modified competence stimulating peptide. PLoS Pathog. 2011;7:e1002241. doi: 10.1371/journal.ppat.1002241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehr S, Wood N. Streptococcus pneumoniae--a review of carriage, infection, serotype replacement and vaccination. Paediatr Respir Rev. 2012;13:258–264. doi: 10.1016/j.prrv.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell PK, Lipsitch M, Hanage WP. Carriage burden, multiple colonization and antibiotic pressure promote emergence of resistant vaccine escape pneumococci. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140342. doi: 10.1098/rstb.2014.0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang SS, Johnson KM, Ray GT, Wroe P, Lieu TA, Moore MR, Zell ER, Linder JA, Grijalva CG, Metlay JP, Finkelstein JA. Healthcare utilization and cost of pneumococcal disease in the United States. Vaccine. 2011;29:3398–3412. doi: 10.1016/j.vaccine.2011.02.088. [DOI] [PubMed] [Google Scholar]
- 31.Hamborsky J, Kroger A, Wolfe S, editors. Pneumococcal Disease. 13th. Centers for Disease Control and Prevention, Epidemiology and Prevention of Vaccine-Preventable Diseases; Public Health Foundation; Washington D.C: 2015. pp. 279–294. [Google Scholar]
- 32.Pestova EV, Havarstein LS, Morrison DA. Regulation of competence for genetic transformation in Streptococcus pneumoniae by an auto-induced peptide pheromone and a two-component regulatory system. Mol Microbiol. 1996;21:853–862. doi: 10.1046/j.1365-2958.1996.501417.x. [DOI] [PubMed] [Google Scholar]
- 33.Hui FM, Morrison DA. Genetic transformation in Streptococcus pneumoniae: nucleotide sequence analysis shows comA, a gene required for competence induction, to be a member of the bacterial ATP-dependent transport protein family. J Bacteriol. 1991;173:372–381. doi: 10.1128/jb.173.1.372-381.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cvitkovitch DG, Li YH, Ellen RP. Quorum sensing and biofilm formation in Streptococcal infections. J Clin Invest. 2003;112:1626–1632. doi: 10.1172/JCI20430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pozzi G, Masala L, Iannelli F, Manganelli R, Havarstein LS, Piccoli L, Simon D, Morrison DA. Competence for genetic transformation in encapsulated strains of Streptococcus pneumoniae: two allelic variants of the peptide pheromone. J Bacteriol. 1996;178:6087–6090. doi: 10.1128/jb.178.20.6087-6090.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lau GW, Haataja S, Lonetto M, Kensit SE, Marra A, Bryant AP, McDevitt D, Morrison DA, Holden DW. A functional genomic analysis of type 3 Streptococcus pneumoniae virulence. Mol Microbiol. 2001;40:555–571. doi: 10.1046/j.1365-2958.2001.02335.x. [DOI] [PubMed] [Google Scholar]
- 37.Hava DL, Camilli A. Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol Microbiol. 2002;45:1389–1406. [PMC free article] [PubMed] [Google Scholar]
- 38.Oggioni MR, Trappetti C, Kadioglu A, Cassone M, Iannelli F, Ricci S, Andrew PW, Pozzi G. Switch from planktonic to sessile life: a major event in pneumococcal pathogenesis. Mol Microbiol. 2006;61:1196–1210. doi: 10.1111/j.1365-2958.2006.05310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu L, Lin J, Kuang Z, Vidal JE, Lau GW. Deletion analysis of Streptococcus pneumoniae late competence genes distinguishes virulence determinants that are dependent or independent of competence induction. Mol Microbiol. 2015;97:151–165. doi: 10.1111/mmi.13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oggioni MR, Iannelli F, Ricci S, Chiavolini D, Parigi R, Trappetti C, Claverys JP, Pozzi G. Antibacterial activity of a competence-stimulating peptide in experimental sepsis caused by Streptococcus pneumoniae. Antimicrob Agents Chemother. 2004;48:4725–4732. doi: 10.1128/AAC.48.12.4725-4732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis. Vol. 222. Oxford University Press; Oxford: 2000. [Google Scholar]
- 42.Luo P, Baldwin RL. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 43.Chorev M, Goodman M. A dozen years of retro-inverso peptidomimetics. Acc Chem Res. 1993;26:266–273. [Google Scholar]
- 44.Guichard G, Benkirane N, Zeder-Lutz G, van Regenmortel MH, Briand JP, Muller S. Antigenic mimicry of natural L-peptides with retro-inverso-peptidomimetics. Proc Natl Acad Sci U S A. 1994;91:9765–9769. doi: 10.1073/pnas.91.21.9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cardo-Vila M, Giordano RJ, Sidman RL, Bronk LF, Fan Z, Mendelsohn J, Arap W, Pasqualini R. From combinatorial peptide selection to drug prototype (II): targeting the epidermal growth factor receptor pathway. Proc Natl Acad Sci U S A. 2010;107:5118–5123. doi: 10.1073/pnas.0915146107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.