

Abstract
Background
Pulmonary arterial hypertension (PAH) is a severe and progressive disease, a hallmark of which is pulmonary vascular remodeling. Nicotinamide phosphoribosyltransferase (NAMPT), is a cytozyme which regulates intracellular NAD levels and cellular redox state, regulates histone deacetylases, promotes cell proliferation and inhibits apoptosis. We hypothesized that NAMPT promotes pulmonary vascular remodeling, and that inhibition of NAMPT could attenuate pulmonary hypertension.
Methods
Plasma and mRNA and protein levels of NAMPT were measured in the lungs and isolated pulmonary artery endothelial cells (PAECs) from PAH patients, as well as in lungs of rodent models of pulmonary hypertension (PH). Nampt+/− mice were exposed 10% hypoxia and room air for 4 weeks and the preventive and therapeutic effects of NAMPT inhibition were tested in the monocrotaline and Sugen-hypoxia models of PH. The effects on NAMPT activity on proliferation, migration, apoptosis and calcium signaling were tested in human pulmonary artery smooth muscle cell (hPASMC).
Results
Plasma and mRNA and protein levels of NAMPT were increased in the lungs and isolated pulmonary artery endothelial cells (PAECs) from PAH patients, as well as in lungs of rodent models of pulmonary hypertension (PH). Nampt+/− mice were protected from hypoxia-mediated PH. NAMPT activity promoted human pulmonary artery smooth muscle cell (hPASMC) proliferation via a paracrine effect. In addition, recombinant NAMPT stimulated hPASMC proliferation via enhancement of store-operated calcium entry by enhancing expression of Orai2 and STIM2. Finally, inhibition of NAMPT activity attenuated monocrotaline and Sugen hypoxia induced PH in rats.
Conclusions
Our data provide evidence that NAMPT plays a role in pulmonary vascular remodeling and its inhibition could be a potential therapeutic target for PAH.
Keywords: Pulmonary hypertension, NAMPT, pulmonary vascular remodeling
Subject Terms: Pulmonary Biology, Vascular Biology, Smooth Muscle Proliferation and Differentiation
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease caused by functional and structural changes in the pulmonary vasculature, which lead to increased pulmonary vascular resistance and, eventually right ventricular failure and death. PAH is characterized by vasoconstriction, in-situ thrombosis, vascular inflammation and pulmonary artery smooth muscle cell (PASMC) and pulmonary artery endothelial cell (PAEC) proliferation and apoptosis resistance1. PAEC dysfunction and injury are believed to play a critical role in the pathogenesis of the disease and can trigger PASMC proliferation and migration through activation growth factors or disruption of cell survival mediators2, 3. Although important progress has been made in recent years, the precise cellular and molecular basis of pulmonary vascular remodeling in PAH continues to evolve and the discovery of therapeutic targets that attenuate vascular remodeling is still ongoing.
Nicotinamide phosphoribosyltransferase (NAMPT), also called visfatin and pre-B cell colony-enhancing factor (PBEF), was first characterized as a cytokine acting on early B-lineage precursor cells, and is a pleiotropic protein with extracellular proinflammatory cytokine-like activity and intracellular enzymatic activity as a phosphoribosyltransferase4–10. It has been demonstrated as the rate-limiting component of the mammalian NAD biosynthesis pathway from nicotinamide11. NAMPT activity has been linked to several processes involving cellular adaptation to stress responses. These include resistance to senescence, apoptosis and increase in cell proliferation and regulation of cellular redox state6–8, 12, 13. At the pathological level NAMPT promotes inflammatory responses by increasing inflammatory cell survival and increasing pro-inflammatory cytokine production8, 14–16. NAMPT is also up regulated in many types of malignancies where it promotes cell survival, proliferation and resistance to chemotherapeutic agents7, 17–21. In vascular cells NAMPT promotes endothelial cell survival and angiogenic activity as well as smooth muscle cell survival10, 13, 22–24. We therefore hypothesized that NAMPT may play an important role in PAH pathobiology by promoting pulmonary vascular remodeling during pulmonary hypertension development, and that inhibition of NAMPT could attenuate experimental pulmonary hypertension.
Methods
Human Lung Tissues, Human PAECs and Human PASMCs (hPASMCs)
Approval for the collection of plasma and the use of human lung tissues and cells was granted by Institutional Review Boards of participating centers. Plasma samples were obtained from 103 patients with PAH defined by the 2013 Nice classification25 and 53 controls including patients with hypothyroidism, osteoporosis, nephrolithiasis, diabetes mellitus, osteoarthritis and hypertension. De-identified human explanted peripheral lung tissues and PAECS used in this study were from four control subjects (2 unsuitable organ donors and 2 chronic obstructive pulmonary disease patients without PH) and patients with idiopathic PAH (IPAH, 4 patients diagnosed on the basis of National Institutes of Health PAH Registry criteria) obtained at the Cleveland Clinic Foundation. PAECs were isolated as previously described26. In addition, a primary hPASMC and hPAEC cell lines from Lonza (CC-2581 and CC-2530) were used for cell transfection and proliferation assays. Cells were cultured at 37°C in their complete medium with 10% fetal bovine serum and studied at passages 5 to 8.
Reagents, Pharmacologic Inhibitors and Antibodies
The information for reagents, pharmacologic inhibitors and antibodies is provided in the supplementary Materials and Methods.
Western Blotting
Solubilized protein lysates isolated from lung tissues and cells were used to detect NAMPT, STIM2, ORAI2 and β-actin. Cells were lysed in a modified radioimmunoprecipitation assay (mRIPA) lysis buffer with a protease and phosphatase inhibitor cocktail (Sigma Aldrich, St. Louis, MO), and protein quantification and Western blot analysis were performed according to standard procedures.
RNA Isolation and Real-time PCR
Total RNA from lung tissues or cells was isolated using the RNeasy Plus Mini Kit (Cat.#74134, Qiagen). Two micrograms of purified RNA was attenuate transcribed to single-stranded cDNA using the Taqman RNA attenuate transcription kit (Cat.#N8080234, Applied Biosystems, Inc. [ABI]). Real-time PCR was performed on an ABI 7900HT machine. Specific Taqman quantitative real-time PCR assays were ordered from ABI (specific assay IDs available upon request). The relative mRNA expression levels were normalized to the expression of a housekeeping gene, glucose-6-phosphate dehydrogenase (G6PDH), and determined by calculating the ΔΔCt value, as detailed in the manufacturer’s guidelines.
Mouse PASMC isolation
A mixture of 5 ml of medium 199 (M199) growth medium containing 5 g/l low-melting-point agarose type VII (Sigma, St. Louis, MO), 5 g/l iron beads (diameter <10 μM; Sigma), and antibiotics (penicillin and streptomycin) was slowly injected over a period of 60 seconds through the RV, thereby perfusing the PA. M199 growth medium (1 ml) containing 5 g/l agarose type VII was injected in airways through the trachea. The lungs were plunged in cold PBS to cause the agarose to gel. Because of the rapidly solidifying nature of the agarose and the size of the iron particles, the likelihood of traversing the capillary space is minimized. All the lobes were then isolated and finely minced in a Petri dish. The tissue was further disrupted by passing through a 16-gauge followed by an 18-gauge needle approximately five times. The suspension was then mixed in M199 growth medium containing 80 U/ml type IV collagenase (Sigma) and incubated at 37°C for 90 min. With the use of a magnetic column (Invitrogen), the arteries or arterial tissues containing the iron beads were collected. The supernatant was aspirated and the arteries were washed and suspended in 5 ml M199 containing 20% FBS. Aliquots of the suspension were transferred to T25 culture flasks. Cells from the hypoxic group were incubated at 3% O2, whereas cells from the normoxic control were cultured in air. Smooth muscle cell purity was determined by immunostaining with smooth muscle specific α-actin antibody.
Cell Proliferation Assays
Cell proliferation was determined using either a 5-bromo-2′-deoxyuridine (BrdU) incorporation assay or cell counting. BrdU assays (QIA58, Calbiochem, San Diego, CA) were performed in a 96-well format according to manufacturer’s instructions, using starting cell densities of 4000 cells/well. For cell counting, cells were trypsinized and resuspended after experimental procedures; densities were counted with a TC10™ automated cell counter (Bio-Rad). FBS or PDGF was used as positive controls.
Overexpressing NAMPT in hPASMCs and hPAECs
A human wild-type NAMPT expression sequence was cloned downstream of the ubiquitin promoter in a third generation lentiviral plasmid. Separately, a control plasmid was generated by cloning a GFP expression sequence in the same position. Co-transfection of either control-GFP or NAMPT-expressing constructs with three other packaging plasmids in HEK 293FT cells produced lentivirus capable of expressing either GFP or NAMPT. Viral concentration was determined by qPCR using primers spanning the HIV 5′LTR region. hPASMCs and hPAECs were infected with control or NAMPT virus at an MOI of 5 for experiments performed 72 hours later.
Plasma and cell culture medium NAMPT measurement
Plasma NAMPT levels were measured using Bio-Plex Pro Human Diabetes Visfatin Set (Bio-Rad, cat.#171-B7012M). NAMPT secreted from hPAECs during the culture was measured using a Nampt (Visfatin/PBEF) (human) ELISA Kit (Adipogen, cat.#AG-45A-0006YEK-KI01).
[Ca2+]cyt Measurement
[Ca2+]cyt was measured in PASMC loaded with fura-2 (4μM) in a Nikon digital fluorescent imaging system. Cells were loaded with 4 μM fura-2 acetoxymethyl ester (fura-2/AM) for 60 min at 25°C and [Ca2+]cyt was measured using a ratiometric method at 32°C. Cyclepiazonic acid (CPA, a specific Ca2+-ATPase inhibitor) was used to induce store-operated calcium entry (SOCE).
Transfection of Small Interfering RNA (siRNA) in hPASMCs
Using Lipofectamine™ RNAiMAX Reagent (Invitrogen, cat.# 13778-150), hPASMCs were transfected with NAMPT siRNA (L-000458-00), STIM2 siRNA (L-013166-01-0005), ORAI2 siRNA (L-015012-00) or scrambled siRNA (D-001810-02), which were purchased from Dharmacon (Thermo Fisher Scientific, Lafayette, CO, USA) according to the manufacturer’s protocol.
Animal Models of Pulmonary Hypertension and Hemodynamic Measurements
All experiments were approved by the Ethics and Animal Care Committee of the University of Illinois at Chicago. Three rodent models of PH were used in this study. They included a mouse model of hypoxia-induced PH, a rat model of monocrotaline (MCT)-induced PH and a rat model of Sugen-hypoxia induced severe PH. Nampt−/− mice exhibit embryonic lethality thus Nampt+/− mice were used in this study. Male mice (8-week old Nampt−/− mice in C57BL/6 background and their WT siblings) were exposed to hypoxia (10% O2) in a ventilated chamber for four weeks. Male Sprague-Dawley rats (190–200 g) were used for the MCT and Sugen-hypoxia induced PH studies. In the MCT-induced PH model, MCT was dissolved in 0.5N HCl to 200 mg/ml, neutralized to pH 7.4 with 0.5N NaOH, and then diluted with sterile water to 60 mg/ml. One dose of MCT (60 mg/kg body weight) was subcutaneously injected to rats. Control rats were injected with the equivalent volume of dissolvent solution according to their weights. Food and water were provided ad libitum and the rats are checked once per day. For the prevention experiment, FK866 (2.5 mg/kg) was started in day 2 after the MCT injection and continued for two weeks; for the therapeutic experiment, FK866 (2.5mg/kg every 48 hours) was started day 14 after the MCT injection and continued for two weeks. Our historical data shows that rats in day 14 after MCT injection develop pulmonary hypertension as demonstrated by significantly increased RVSP and RVH (Supplemental Figure S1). Each group included 6 to 8 rats. In the Sugen-hypoxia PH model, one dose of Sugen (20 mg/kg) was given subcutaneously at the first day of hypoxia exposure (10% O2). After three-week chronic hypoxia exposure, the rats were placed back to room air. FK866 (2.5 mg/kg every 48 hours) was then started two weeks after reoxygenation and continued for three weeks. Each group included 10 to 12 rats. Hemodynamic data after two-week reoxygenation indicates that rats develop significant PH at this time point (Supplemental Figure S2). Right ventricular systolic pressure (RVSP) was determined by right heart catheterization using a Millar pressure transducer catheter. A weight ratio of the right ventricle divided by the sum of left ventricle and septum (RV/(LV+S)) was measured to determine the extent of right ventricular hypertrophy (RVH). The RV contractility index was calculated as (dp/dt)max/instantaneous RV pressuremax, as previously described.27 Pulmonary artery remodeling was assessed using Aperio ImageScope software (version 11) after lungs were stained with hematoxylin and eosin (H&E). A minimum of 10 microscopic fields were examined for each slide. Approximately twenty muscular arteries with diameters (D) 50–100 μm or D < 50 μm per lung section were outlined and measured. Vessel remodeling was calculated as ((external vessel area – internal vessel area) / external vessel area), as previously described28. Occlusive pulmonary arteries were counted from H&E stained lung slides from each group.
Statistical Analysis
Statistical analysis of experimental data was performed using GraphPad Prism 5.1 (GraphPad Software, Inc., La Jolla, CA). Results are expressed as mean ± standard error of mean (SEM), except where noted, from at least three experiments. Student t - test and ANOVA were used to compare two and three groups, respectively except for the comparison of NAMPT plasma levels between control and PAH subjects where a Mann-Whitney test was used. A P < 0.05 was considered statistically significant.
Results
NAMPT is Up-regulated in Patients with PAH and in Experimental Rodent PH Models
Plasma NAMPT levels were significantly elevated in patients with PAH (n=103, median [IQR] 3197 [1493–7280]) when compared to controls (n=53, median [IQR] 1686 [990–2608]; p<0.001 by Mann-Whitney test, Figure 1A, Supplemental Table 1). mRNA and protein levels of NAMPT were significantly elevated in the lungs and PAECs from patients with PAH when compared to control subjects (Figures 1B–1F and supplemental Figure S3). Pulmonary artery smooth muscle cells isolated from PAH patients did not express higher levels of NAMPT, compared to controls (Supplemental Figure S4).
Figure 1. Plasma NAMPT levels, mRNA and protein levels of NAMPT in the lungs and PAECs are up regulated in patients with PAH.
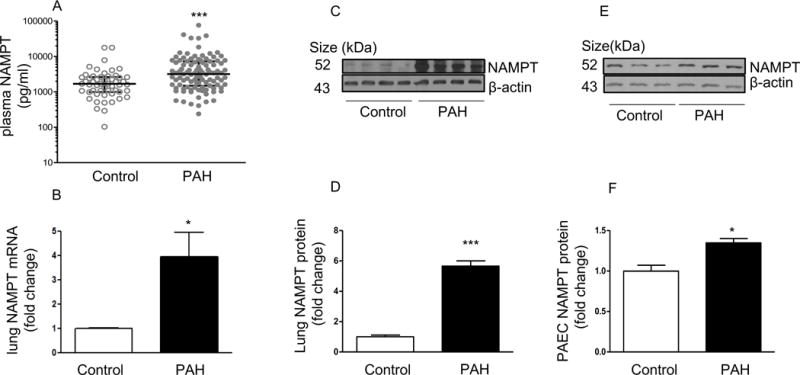
(A) Plasma NAMPT levels are higher in patients with PAH (n = 103) when compared to non-PAH controls (n = 53); (B) NAMPT mRNA levels are upregulated in the lungs of the patients with PAH (n = 4), compared to non-PAH controls (n = 4); (C–D) Representative Western blotting images and β-actin-normalized quantification of protein demonstrate that NAMPT expression is significantly increased in lungs from PAH patients when compared to non-PAH controls; (E–F) Representative Western blotting images and β-actin-normalized quantification of protein demonstrate that NAMPT protein levels were upregulated in PAECs isolated from PAH patients (n = 3) when compared to non-PAH controls (n = 3). Results are expressed as mean ± SEM. *, p < 0.05; ***, p < 0.001 versus control.
A similar pattern was found in different rodent models of PH including a mouse model of hypoxia-mediated pulmonary hypertension (HPH, 10% hypoxia for 4 weeks, Figure 2A), a rat model of monocrotaline-induced PH (2 weeks after administration of monocrotaline [60 mg/kg bw, SQ], Figure 2B) and a rat model of the VEGF receptor blocker sugen plus hypoxia mediated PH (sugen [20 mg/kg bw, SQ] followed by 10% hypoxia for 3 weeks and normoxia for 3 weeks, Figure 2C). In addition, the NAMPT expression profiles in lung tissues from these different rodent models of PH were examined using immunofluorescence staining. As shown in Supplemental Figures S5–6, NAMPT expression in experimental PH appears to increase in the pulmonary vasculature mainly in PAECs and the adventitia, but not in the media, as well as in airway and alveolar epithelial cells. Alveolar macrophages also express NAMPT, especially in the rat model of PH mediated by monocrotaline, as demonstrated by CD68 immunostaining (data not shown).
Figure 2. NAMPT protein levels are significantly increased in lungs from three different rodent models of pulmonary hypertension.
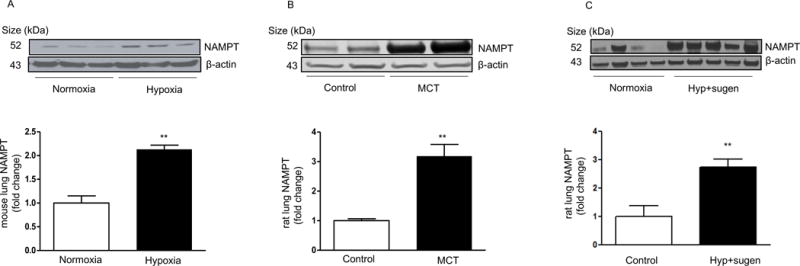
(A) Representative Western blotting images and β-actin-normalized quantification of protein demonstrate that NAMPT expression is significantly increased in mouse lungs after 4-week 10% hypoxia exposure. (B) Representative Western blotting images and β-actin-normalized quantification of protein demonstrate that NAMPT expression is significantly increased in the lungs from a rat model of PH mediated by monocrotaline. (C) Representative Western blotting images and β-actin-normalized quantification of protein demonstrate that NAMPT expression is significantly increased in the lungs from hypoxia plus sugen mediated PH in rats. **,p<0.01 versus controls.
In aggregate, these results suggest that NAMPT may play a role in the development of pulmonary hypertension.
NAMPT Heterozygous (Nampt+/−) Mice are Protected from HPH
To examine the effect of NAMPT on the development of pulmonary hypertension, Nampt+/− mice and their WT siblings in a C57BL6 background were exposed to 10% hypoxia for four weeks. Compared to WT siblings, Nampt+/− mice exhibited significantly lower baseline levels of NAMPT and NAD in lung tissue homogenates (Supplemental Figure S7). After four-week hypoxia exposure, Nampt+/− mice displayed significantly lower right ventricular systolic pressure (RVSP, Figure 3A), decreased RVH (right ventricular hypertrophy, assessed by RV/(LV+S) ratio, Figure 3B), and less severe pulmonary vascular remodeling in response to hypoxia when compared to control mice (Figure 3C–E). Furthermore, PASMCs isolated from Nampt+/− mice were less proliferative compared to WT mice as demonstrated by BrdU incorporation and cell counting (Figure 3F–3G). Similarly, silencing NAMPT via siRNA attenuated complete medium, hypoxia or PDGF-mediated hPASMC proliferation (Supplemental Figure S8).
Figure 3. Nampt+/− mice are protected against hypoxia-mediated pulmonary hypertension and PASMCs isolated from Nampt+/− mice are less proliferative.
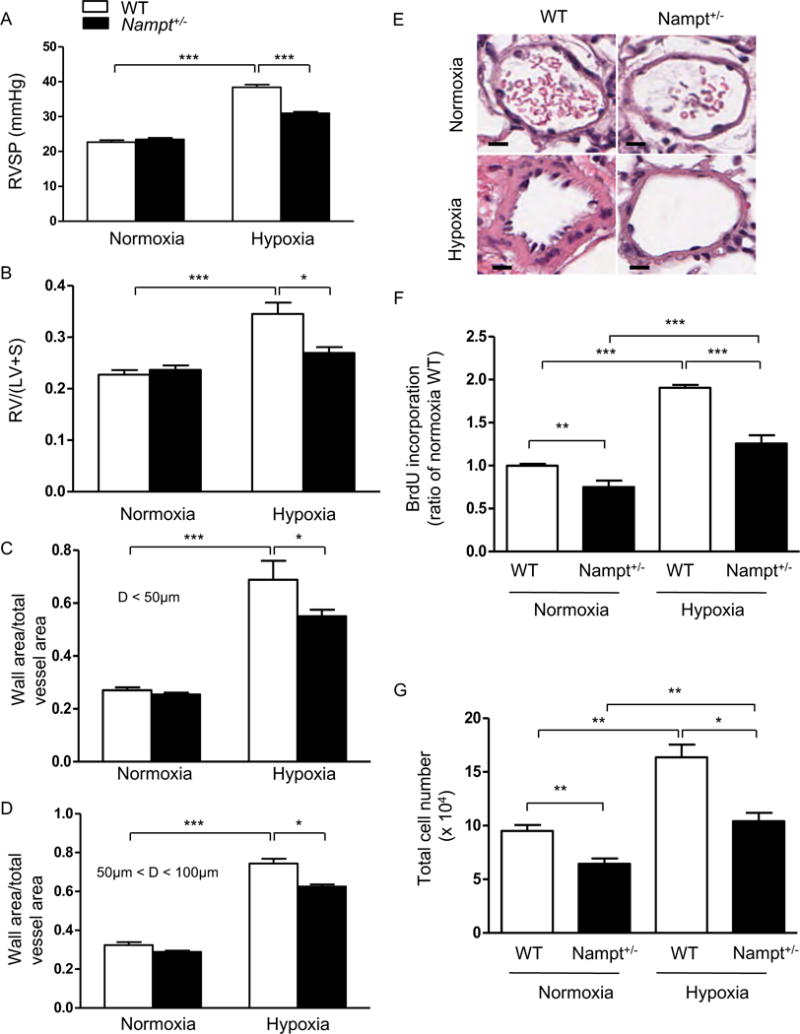
When compared to WT controls, Nampt+/− mice exposed to hypoxia developed (A) less severe elevations in RVSP, (B) less right ventricular hypertrophy (RV/(LV+S)) and lower increases in ratios of wall area to total vessel area of pulmonary arteries less than 50 μm (C) and 50–100 μm in diameter (D). (E) Representative pulmonary artery images in the lung sections of control and Nampt+/− mice exposed to normoxia or hypoxia. BrdU proliferation assays (F) and cell counting data (G) demonstrate that PASMCs isolated from Nampt+/− mice are less proliferative compared to WT mice under either normoxia or hypoxia. Bar size: 20 μm. Results are expressed as mean ± SEM; n = 10 per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus hypoxia WT group.
NAMPT activity promotes PASMC proliferation
In addition to increased expression, PAECs isolated from patients with PAH secreted more NAMPT in culture media than control cells (Figure 4A). Further, conditioned media from PAECs isolated from PAH patients significantly promoted hPASMC proliferation. This effect was significantly attenuated by the NAMPT enzymatic activity inhibitor FK86621 (Figure 4B). Lentivirus-mediated NAMPT overexpression promoted both hPAECs and hPASMC proliferation (Figure 4C–I). When compared to control cells, hPAECs overexpressing NAMPT secreted more NAMPT and conditioned media from these cells stimulated more hPASMC proliferation (Figure 4J). These effects were attenuated by FK866 (Figure 4F, 4I, 4J). Finally, recombinant NAMPT protein (rNAMPT) promoted hPASMC proliferation in a dose-dependent manner (an effect that was attenuated by FK866, Figure 4K) and enhanced hPASMC migration (an effect that was attenuated by FK866, Supplemental Figure S9). Taken together these data suggest that NAMPT promotes hPASMC proliferation and migration in an autocrine and paracrine manner.
Figure 4. NAMPT promotes proliferation of PASMCs or PAECs in a paracrine and autocrine manner.
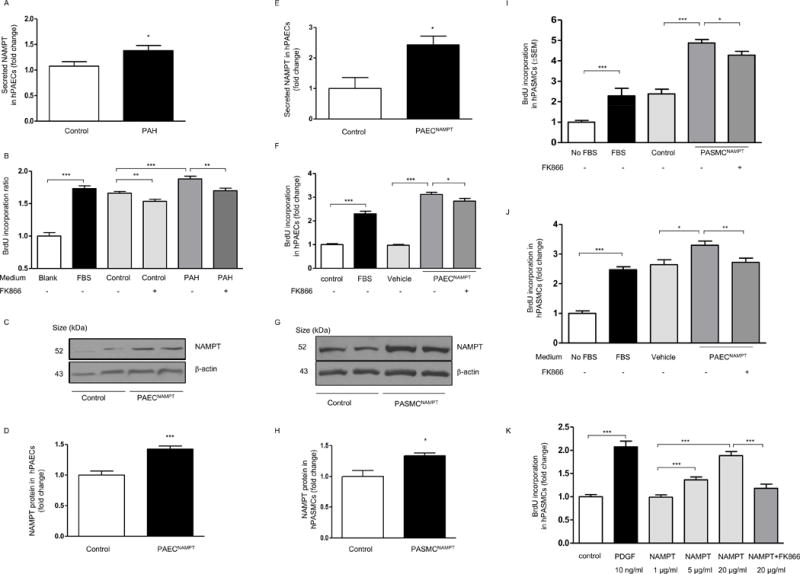
(A) PAECs isolated from PAH patients secret more NAMPT compared to non-PAH controls (n=3 per group); (B) BrdU incorporation assays demonstrate that cell culture medium of PAECs isolated from PAH patients stimulates more PASMC proliferation, and this effect can be attenuated by the specific NAMPT inhibitor, FK866(10 μM) (ANOVA P < 0.05 for the control to PAH comparison). Overexpressing NAMPT in hPAECs increases NAMPT expression and secretion (C–E). NAMPT overexpression promotes cell proliferation and its proliferative effect is attenuated by FK866 (10 μM) (F, ANOVA P < 0.05 for the vehicle to NAMPT and NAMPT+FK-866 comparison). Overexpressing NAMPT in hPASMCs (G–H) promotes cell proliferation and its proliferative effect is attenuated by FK866 (I, ANOVA P < 0.05 for the control to NAMPT and NAMPT+FK-866 comparison). Cell culture medium of PAECs overexpressing NAMPT stimulates more PASMC proliferation, and this effect is attenuated by FK866 (10 μM) (J, ANOVA P < 0.05 for the vehicle to NAMPT and NAMPT+FK-866 comparison). (K) recombinant NAMPT protein stimulates human PASMC proliferation in a dose dependent manner and this effect is attenuated by FK866 (10 μM, ANOVA P < 0.05 for the different NAMPT doses and NAMPT+FK-866 comparison). Results are expressed as mean ± SEM. *, p < 0.05; **, p < 0.01; ***, P < 0.001 versus the pairwise comparison indicated by brackets. FBS or PDGF were used as positive controls for BrdU proliferation assays as stated in the specific figures.
Inhibition of NAMPT activity promotes PASMC apoptosis
Since NAMPT promotes resistance to apoptosis6–8 and resistance to apoptosis is a major contributor to the pathobiology of PAH, we tested whether inhibition of NAMPT activity would promote PASMC apoptosis. As shown in Supplemental Figure S10, treatment with FK866 induced hPASMCs apoptosis. Further, treatment with rNAMPT protected PASMCs from H2O2 induced apoptosis, an effect that was attenuated by concomitant FK866 treatment.
NAMPT Enhances Store-Operated Calcium Entry (SOCE) in PASMCs
NAMPT activity promotes inflammatory responses including the expression of inflammatory cytokines. For instance, NAMPT derived NAD promotes TNF-α synthesis by activated immune cells29. Since inflammatory pathways, particularly the cytokine TNF-α, have been shown to promote SOCE30, 31, we hypothesized that NAMPT-induced PASMC proliferation would be mediated by enhancement of SOCE. To test this hypothesis, we treated PASMCs with rNAMPT (20 μg/ml) for 48 hours. [Ca2+]cyt was measured in PASMCs loaded with fura-2/AM (4 μM) using a fluorescence microscope, and cyclopiazonic acid (CPA) was used to induce SOCE. After 48 hours rNAMPT treatment significantly enhanced CPA-mediated SOCE in PASMCs (Figure 5A).
Figure 5. NAMPT Enhances Store-Operated Calcium Entry (SOCE) in PASMCs.
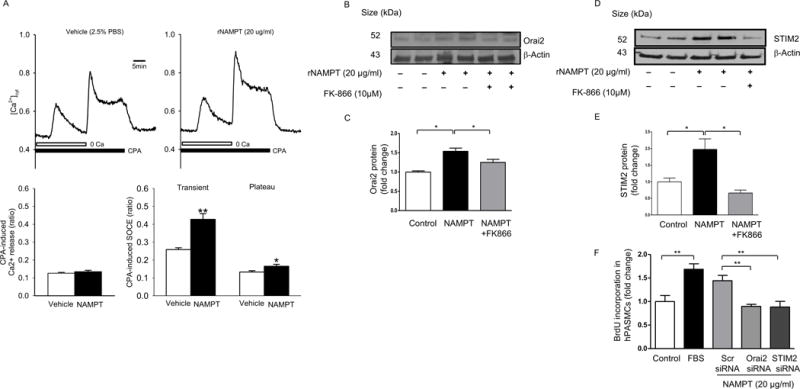
Incubation of hPASMCs with rNAMPT protein (20 μg/ml, 48 hrs) enhances cpA-mediated store operated calcium entry (SOCE) (A), and increases ORAI2/STIM2 expression which is attenuated by FK866 (10 μM) (B–E). Silencing of STIM2 or ORAI2 attenuates rNAMPT-mediated hPASMC proliferation (F). Results are expressed as mean ± SEM, experiments were repeated at least three times. *, p < 0.05; **, p < 0.01.
ORAI calcium release-activated calcium modulator 2 (Orai2) and stromal interaction molecule 2 (STIM2) are up regulated in PASMCs from patients with PAH and contribute to enhanced store-operated calcium entry and to the transition of these cells from a contractile to proliferative phenotype32. Given these findings we tested whether NAMPT activity would enhance SOCE via up regulation of Orai2 and STIM2. NAMPT treatment increased the expression of both Orai2 and STIM2 (Figure 5B–E) but did not change the expression of Orai1 and STIM1 (data not shown). Inhibition of NAMPT via FK866 attenuated NAMPT-mediated STIM2 and ORAI2 upregulation (Figure 5B–E) and silencing of either Orai2 or STIM2 attenuated NAMPT-induced hPASMC proliferation (Figure 5F). This data suggests that SOCE is required for the proliferative effect of NAMPT in PASMCs.
Finally, the effect of NAMPT on the L-type calcium channel mediated signaling was also examined. Blockage of L-type channels with verapamil did not affect rNAMPT mediated hPASMC proliferation (Supplemental Figure S11).
Inhibition of NAMPT by FK866 prevents and attenuates monocrotaline-induced PH in rats
To examine the potential therapeutic role of NAMPT inhibition in PH, we evaluated the effect of FK866 in monocrotaline (MCT)-induced PH model in rats. In PH prevention experiments FK866 (2.5mg/kg every 48 hours IP) or vehicle were given to rats (n = 6–8 animals per group), starting one day after subcutaneous injection of MCT (60 mg/kg bw) and in PH reversal experiments FK866 (2.5mg/kg every 48 hours IP) or vehicle were given to rats (n = 6–8 animals per group), starting fourteen days after MCT injection (60 mg/kg bw). When compared to vehicle-treated rats, treatment with FK866 prevented (Figure 6) and also attenuated (Figure 7) the development of PH. In both experiments, FK866 treatment significantly reduced the PH assessed by the RVSP as well as RVH, RV contractility index and the degree of pulmonary artery muscularization. Furthermore, FK866 treatment decreased PASMCs proliferation, as demonstrated by PCNA and smooth muscle actin double immunostaining of lung tissues (Supplemental Figure S12).
Figure 6. NAMPT inhibition prevents monocrotaline-induced pulmonary hypertension in rats.
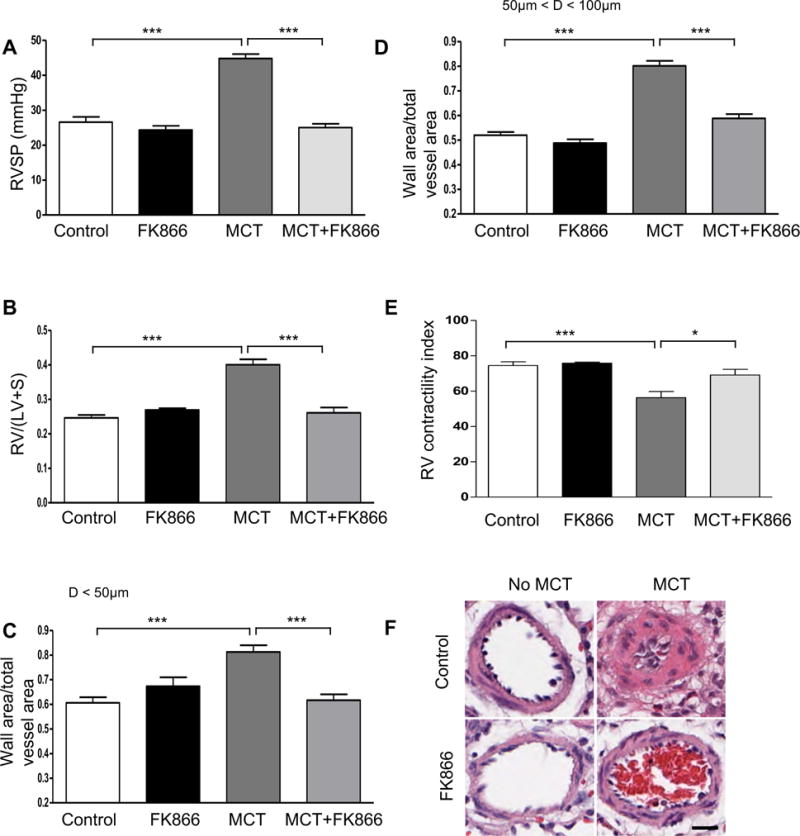
(A) Changes in RVSP. (B) Changes in RV/(LV+S). (C) Changes in ratios of wall area to total vessel area of pulmonary arteries less than 50 μm in diameter in the lung sections of control and FK866 treated (2.5mg/kg every 48 hours IP) groups with or without one dose of monocrotaline (MCT, 60 mg/kg bw, SQ). (D) Changes in ratios of wall area to total vessel area of pulmonary arteries 50–100 μm in diameter in the lung sections of control and FK866 treated groups with or without one dose of MCT (60 mg/kg bw, SQ). (E) Changes of RV contractility index after FK866 treatment. The RV contractility index was calculated as (dp/dt)max/instantaneous RV pressuremax, as demonstrated previously.27 (F) Representative pulmonary artery images in the lung sections of control and FK866 treated (2.5mg/kg every 48 hours IP) groups with or without one dose of MCT (60 mg/kg bw, SQ). Bar size: 20 μm. Results are expressed as mean ± SEM; n = 6–8 per group. *, p < 0.05; ***, p < 0.001.
Figure 7. NAMPT inhibition attenuates monocrotaline-induced pulmonary hypertension in rats.
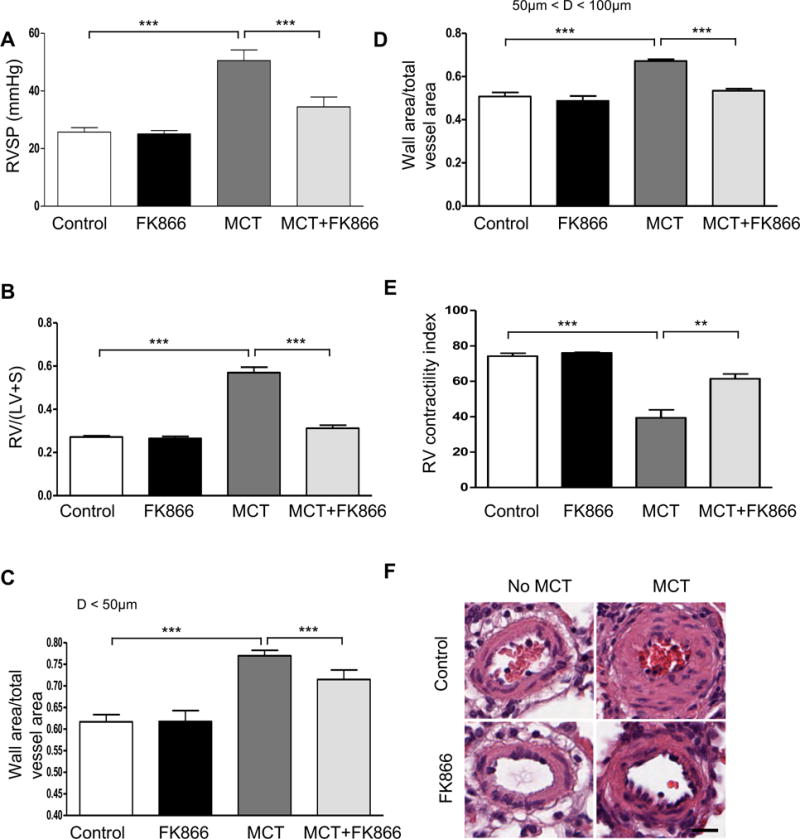
(A) Changes in RVSP. (B) Changes in RV/(LV+S). (C) Changes in ratios of wall area to total vessel area of pulmonary arteries less than 50 μm in diameter in the lung sections of control and FK866 treated (2.5mg/kg every 48 hours IP) groups with or without one dose of monocrotaline (MCT, 60 mg/kg bw, SQ). (D) Changes in ratios of wall area to total vessel area of pulmonary arteries 50–100 μm in diameter in the lung sections of control and FK866 treated (2.5mg/kg every 48 hours IP) groups with or without one dose of MCT (60 mg/kg bw, SQ). (E) Changes of RV contractility index after FK866 treatment. The RV contractility index was calculated as (dp/dt)max/instantaneous RV pressuremax, as demonstrated previously.27 (F) Representative pulmonary artery images in the lung sections of control and FK866 treated (2.5mg/kg every 48 hours IP) groups with or without one dose of MCT (60 mg/kg bw, SQ). Bar size: 20 μm. Results are expressed as mean ± SEM; n = 6–8 per group. **, p < 0.01; ***, p < 0.001.
Inhibition of NAMPT by FK866 attenuates Sugen-hypoxia mediated PH in rats
The rat model of PH mediated by Sugen and hypoxia induces severe pulmonary vascular remodeling characterized by obliterative pulmonary vascular lesions with neointima formation in distal pulmonary arteries. We assessed if NAMPT inhibition by FK866 (2.5 mg/kg every 48 for three weeks) after rats (n= 8–12 per treatment group) were given sugen (20 mg/kg, SQ, once), exposed to chronic hypoxia for 3 weeks, and then kept in normoxia for another two weeks. When compared to controls, FK866 treatment attenuated the development of PH assessed by changes in RVSP, RVH and RV contractility index as well as the development of medial hypertrophy and neointima formation (Figure 8). FK866 treatment also decreased cell proliferation in obliterative lesions (Supplemental Figure S13). Taken together with the results from the MCT studies, these data suggest that NAMPT inhibition is a novel therapeutic target in PAH.
Figure 8. NAMPT inhibition attenuates hypoxia plus sugen-mediated pulmonary hypertension in rats.
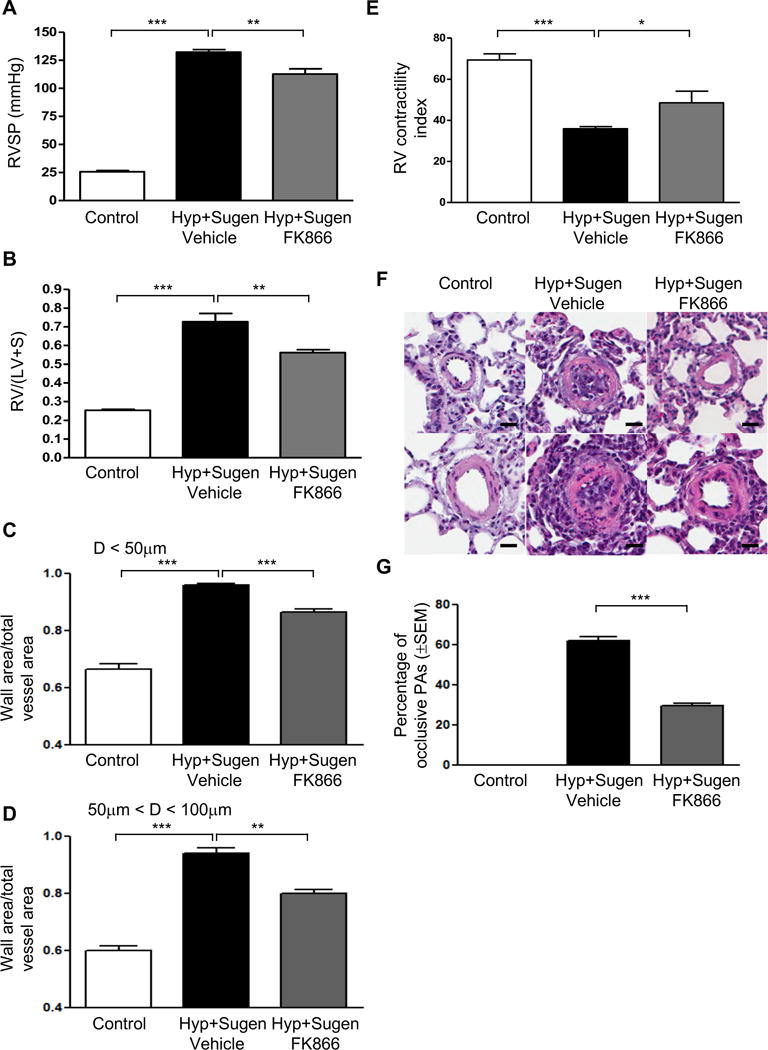
(A) Changes in RVSP. (B) Changes in RV/(LV+S). (C) Changes in ratios of wall area to total vessel area of pulmonary arteries less than 50 μm in diameter in the lung sections of control and FK866 treated (2.5mg/kg every 48 hours IP) groups; (D) Changes in ratios of wall area to total vessel area of pulmonary arteries 50–100 μm in diameter in the lung sections of control and FK866 treated (2.5mg/kg every 48 hours IP) groups; (E) Changes of RV contractility index after FK866 treatment. The RV contractility index was calculated as (dp/dt)max/instantaneous RV pressuremax, as demonstrated previously.27 (F) Representative pulmonary artery images in the lung sections of control and FK866 treated (2.5mg/kg every 48 hours IP) groups; (G) Percentage of occlusive PAs significantly decreases with FK866 therapy (2.5mg/kg every 48 hours IP). Bar size: 20 μm. Results are expressed as mean ± SEM; n = 8–12 per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001
Discussion
Here we show that NAMPT is upregulated in the plasma, lungs and PAECs isolated from patients with PAH, and in different rodent models of PH. PAECs from patients with PAH are more proliferative and secrete more NAMPT which promotes PASMC proliferation in a paracrine fashion via calcium signaling. We also show that NAMPT contributes to pulmonary vascular remodeling in vivo and that pharmacologic inhibition of NAMPT activity attenuates experimental PH. NAMPT is a multifunctional protein with extracellular proinflammatory cytokine-like activity as well as intracellular enzymatic activity as a phosphoribosyltransferase, which regulates NAD levels4. As such, NAMPT regulates the activity of NAD-dependent enzymes, such as sirtuins and poly(ADP-ribose) polymerases, and functions as a master regulator of cellular metabolism, mitochondrial biogenesis and adaptive and maladaptive responses to cellular stress10.
There are several potential mechanisms for the upregulation of NAMPT in patients with IPAH and in experimental models of PH. As major mediators contributing to PAH pathobiology, hypoxia, upregulation of growth factors and inflammatory pathways can also influence NAMPT expression. The NAMPT promoter has hypoxia-inducible factor responsive elements that affect the transcriptional regulation of NAMPT33, 34. The NAMPT promoter also contains binding sites for NF-1, AP-1, and NF-κB and tumor necrosis factor α (TNFα), IL-1β and IL-6 increase NAMPT expression9. Epigenetic regulation of NAMPT expression via micro RNAs has been demonstrated in PAECs35, colorectal cancer cell lines36, hepatocytes37 and TZM-bl HeLa cell lines38. It is possible that similar effects could occur in the context of PAH. Further studies in this area are warranted. NAMPT levels are also increased and correlate with disease severity in metabolic syndrome and diabetes39–41 as well as coronary artery disease42. This suggests that NAMPT should be studied as a prognostic biomarker in PAH.
Uncontrolled PASMC proliferation and resistance to apoptosis are major contributors to the pathobiology of PAH. Our results demonstrate that NAMPT promotes PASMC proliferation in a dose-dependent manner, suggesting that it plays a significant role in the pulmonary vascular remodeling associated with PAH. Ca2+ signaling in PASMCs promotes vascular remodeling through its stimulatory effect on PASMC proliferation and migration. Previous studies have shown that PASMC from idiopathic PAH patients exhibit enhanced SOCE when compared to control PASMCs and that SOCE contributes to PASMC proliferation32, 43–47. Specifically, STIM2 and Orai2 both contribute to SOCE and are upregulated in PASMCs from patients with idiopathic PAH and from animals with experimental pulmonary hypertension in comparison to controls ultimately contributing to the transition of these cells from a contractile to proliferative phenotype32, 47. We show that NAMPT enhances SOCE in PASMCs by increasing the expression of STIM2 and Orai2 and that inhibition of NAMPT activity decreases the expression of these two proteins. In line with these observations, Ye and colleagues have shown that NAMPT knockdown blunted thrombin-mediated increases in Ca2+ entry in human PAEC48. We speculate that NAMPT activity could enhance SOCE by upregulating inflammatory pathways as shown in human aortic smooth muscle cells49. Inflammatory cytokines such as TNF-α have been known to increase agonist-induced [Ca2+]i response and contractility in airway smooth muscle cells31, 50–52 as well as to increase SOCE in these cells30, 31, 53. Furthermore, LPS-induced activation of both the NF-κB and p38 MAPK signaling pathways induce STIM1 expression in PAECs54. Interestingly, by increasing NAD levels, NAMPT promotes TNF synthesis via regulation of SIRT6 activity29, 55. The extracellular cytokine-like activity of NAMPT could also potentially activate NF-κB signaling and further enhance SOCE56, 57.
The paracrine effects of NAMPT have been demonstrated in other studies. Pilai and colleagues58 showed that cardiac-specific overexpressing NAMPT transgenic mice spontaneously developed cardiac hypertrophy and that treatment of cardiomyocytes with recombinant NAMPT induced cardiomyocyte hypertrophy. Yoon and colleagues59 demonstrated that deacetylation of lysine 53 in intracellular NAMPT by SIRT1 predisposes the protein to secretion by adipocytes and increases secreted extracellular NAMPT activity. Adipose tissue-specific NAMPT knockout and knockin mice showed reciprocal changes in circulating NAMPT levels which affected hypothalamic NAD+ and SIRT1 signaling as well as physical activity. Finally, administration of a NAMPT-neutralizing antibody suppressed NAMPT enzymatic activity by 90% for recombinant NAMPT in vitro and by 40% for extracellular NAMPT in plasma and also decreased hypothalamic NAD+ production in vivo.
It is clear that given its pleiotropic effects, NAMPT can exert other effects that promote pulmonary vascular remodeling beyond inducing PASMC proliferation in a paracrine manner. For instance, our results and the work of others suggest that NAMPT activity directly promotes endothelial and smooth muscle cell proliferation12, 13. Our data from Nampt+/− mice and from NAMPT loss of function studies in hPASMCs also suggest that normal NAMPT levels are required to promote cell proliferation both in physiologic and pathologic conditions. By regulating cellular NAD levels and the activity of NAD dependent enzymes such as sirtuins and ADP-ribosyltransferases, NAMPT plays an important role in the regulation of programmed cell death pathways. In models of acute inflammation NAMPT activity prevents the apoptosis of neutrophils60. NAMPT also protects cells from apoptosis by inhibiting the activity of p5361–63. SIRT3 has also been found to prevent loss of mitochondrial membrane potential and cell death in response to hypoxia and staurosporine treatment64. Current and future studies by our group are seeking to elucidate the molecular mechanisms associated with increased proliferation and cell survival of PAECs and PASMCs in the context of PAH. Finally, T and B lymphocytes and macrophages are present in increased numbers in and around the pulmonary arteries of patients with PAH and these cells could be another source of NAMPT within the PAH lung microenvironment.65
Our results show that NAMPT inhibition with FK866 has therapeutic effects in pre-clinical models of PH by attenuating pulmonary vascular remodeling characterized by medial hypertrophy and neointima formation. FK866 specifically inhibits NAMPT activity likely by functioning as a substrate (NAM) mimetic which leads to a decrease is NAD synthesis.21 NAMPT inhibitors have been shown to have therapeutic effects in preclinical studies by inhibiting cell proliferation and promoting apoptosis in cancer models21, inhibiting T-cell proliferation and survival in autoimmune encephalomyelitis55, inhibiting inflammatory cytokine production in collagen-induced arthritis66 and inducing neutrophil apoptosis in LPS-induced acute lung injury60. NAMPT inhibitors have also been studied in phase I-II human clinical trials in patients with cancer67–70. In these studies, the main dose limiting toxicities included hematologic abnormalities (thrombocytopenia, leukopenia and anemia) as well as several gastrointestinal symptoms such as nausea, vomiting, anorexia and abdominal pain.67–70
Our hemodynamic studies are limited by an indirect assessment of PA pressures via measurement of RVSP and lack of a direct assessment of cardiac output or left ventricular function. Further animal and human studies are needed to completely characterize the effects of NAMPT inhibition on the pulmonary and systemic vasculatures as well as its effects on right and left ventricular function. The results of our pre-clinical studies also highlight the relative limitations of the existing experimental PH models. NAMPT inhibition had a much more pronounced therapeutic effect in the monocrotaline than in the Sugen-hypoxia PH model. This is consistent with the notion that the Sugen-hypoxia PH model more closely mimics the severe pulmonary vascular remodeling and pulmonary hypertension associated with human PAH, suggesting that this model is likely more suitable to test the translational potential of novel therapeutic strategies for the disease.
In summary, we have demonstrated that NAMPT promotes pulmonary vascular remodeling and contributes to PAH pathobiology. Most importantly, our results suggest that NAMPT is a novel potential therapeutic target for PAH that should be explored in future preclinical and clinical studies.
Supplementary Material
Clinical Perspective.
What is new?
Nicotinamide phosphoribosyltransferase (NAMPT), a pleiotropic protein with extracellular proinflammatory cytokine-like activity and intracellular enzymatic activity as a phosphoribosyltransferase, is upregulated in human and experimental pulmonary hypertension and promotes pulmonary vascular remodeling in vivo and in vitro.
What are the clinical implications?
Inhibition of NAMPT enzymatic activity is a potential novel therapeutic strategy for pulmonary arterial hypertension.
Acknowledgments
The authors thank Dr. Serpil C. Erzurum and the Lung Biology Tissue and Cell Core of the Department of Pathobiology at the Cleveland Clinic for generously providing human PAH and control PAEC lines used for these studies. We also thank Dr. Guofei Zhou, Dr. Jingbo Dai, Dr. Tianji Chen, Jesse Gerringer and Megh Patel for technical support.
Sources of Funding
This work was supported in part by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (K23HL098454, R01HL111656, R01HL127342 and 1R01HL133951 to R.F.M., R01 HL094394 to J.G.N.G., R01HL066012 and R01HL115014 to J.X.J.Y., F30HL128034 to J.R.S., P01 HL103453, R01 HL115008, and R37 HL060917 to S.C.), University of Illinois at Chicago Area of Excellence Award (to R.F.M.), American Heart Association Pre-doctoral Fellowship (15PRE2190004) (to J.R.S.), and Pulmonary Hypertension Association Proof-of-Concept Research Grant (to J.C.).
Footnotes
Conflict of Interest Statement
The authors have declared that no conflict of interest exists.
Author Contributions
JC and RFM designed the research studies. JC, JRS, SS, SZ, DVJ, TA, KMS, AY, SS, VR, AS, HG, JT and HT performed the experiments. JC, JRS and RFM analyzed the data. SMC, SQY, SC, RD, PH, JXJY and JGNG provided specimens and/or reagents. JXJY and JGNG provided input on the design of portions of the research studies. JC and RFM wrote the manuscript.
References
- 1.Malenfant S, Neyron AS, Paulin R, Potus F, Meloche J, Provencher S, Bonnet S. Signal transduction in the development of pulmonary arterial hypertension. Pulm Circ. 2013;3:278–293. doi: 10.4103/2045-8932.114752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 3.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 4.Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol. 2007;23:164–170. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- 5.Sommer G, Garten A, Petzold S, Beck-Sickinger AG, Bluher M, Stumvoll M, Fasshauer M. Visfatin/PBEF/Nampt: structure, regulation and potential function of a novel adipokine. Clin Sci (Lond) 2008;115:13–23. doi: 10.1042/CS20070226. [DOI] [PubMed] [Google Scholar]
- 6.Imai S. Nicotinamide phosphoribosyltransferase (Nampt): a link between NAD biology, metabolism, and diseases. Curr Pharm Des. 2009;15:20–28. doi: 10.2174/138161209787185814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galli M, Van Gool F, Rongvaux A, Andris F, Leo O. The nicotinamide phosphoribosyltransferase: a molecular link between metabolism, inflammation, and cancer. Cancer Res. 2010;70:8–11. doi: 10.1158/0008-5472.CAN-09-2465. [DOI] [PubMed] [Google Scholar]
- 8.Dahl TB, Holm S, Aukrust P, Halvorsen B. Visfatin/NAMPT: a multifaceted molecule with diverse roles in physiology and pathophysiology. Annu Rev Nutr. 2012;32:229–243. doi: 10.1146/annurev-nutr-071811-150746. [DOI] [PubMed] [Google Scholar]
- 9.Sun Z, Lei H, Zhang Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor Rev. 2013;24:433–442. doi: 10.1016/j.cytogfr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11:535–546. doi: 10.1038/nrendo.2015.117. [DOI] [PubMed] [Google Scholar]
- 11.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 12.Borradaile NM, Pickering JG. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell. 2009;8:100–112. doi: 10.1111/j.1474-9726.2009.00453.x. [DOI] [PubMed] [Google Scholar]
- 13.van der Veer E, Ho C, O’Neil C, Barbosa N, Scott R, Cregan SP, Pickering JG. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem. 2007;282:10841–10845. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 14.Moschen AR, Gerner RR, Tilg H. Pre-B cell colony enhancing factor/NAMPT/visfatin in inflammation and obesity-related disorders. Curr Pharm Des. 2010;16:1913–1920. doi: 10.2174/138161210791208947. [DOI] [PubMed] [Google Scholar]
- 15.Grahnert A, Grahnert A, Klein C, Schilling E, Wehrhahn J, Hauschildt S. Review: NAD +: a modulator of immune functions. Innate Immun. 2011;17:212–233. doi: 10.1177/1753425910361989. [DOI] [PubMed] [Google Scholar]
- 16.Zhang LQ, Heruth DP, Ye SQ. Nicotinamide Phosphoribosyltransferase in Human Diseases. J Bioanal Biomed. 2011;3:13–25. doi: 10.4172/1948-593X.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bi TQ, Che XM. Nampt/PBEF/visfatin and cancer. Cancer Biol Ther. 2010;10:119–125. doi: 10.4161/cbt.10.2.12581. [DOI] [PubMed] [Google Scholar]
- 18.Jieyu H, Chao T, Mengjun L, Shalong W, Xiaomei G, Jianfeng L, Zhihong L. Nampt/Visfatin/PBEF: a functionally multi-faceted protein with a pivotal role in malignant tumors. Curr Pharm Des. 2012;18:6123–6132. doi: 10.2174/138161212803582531. [DOI] [PubMed] [Google Scholar]
- 19.Montecucco F, Cea M, Bauer I, Soncini D, Caffa I, Lasiglie D, Nahimana A, Uccelli A, Bruzzone S, Nencioni A. Nicotinamide phosphoribosyltransferase (NAMPT) inhibitors as therapeutics: rationales, controversies, clinical experience. Curr Drug Targets. 2013;14:637–643. doi: 10.2174/1389450111314060003. [DOI] [PubMed] [Google Scholar]
- 20.Shackelford RE, Mayhall K, Maxwell NM, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase in malignancy: a review. Genes Cancer. 2013;4:447–456. doi: 10.1177/1947601913507576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sampath D, Zabka TS, Misner DL, O’Brien T, Dragovich PS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Ther. 2015;151:16–31. doi: 10.1016/j.pharmthera.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Montecucco F, Cea M, Cagnetta A, Damonte P, Nahimana A, Ballestrero A, Del Rio A, Bruzzone S, Nencioni A. Nicotinamide phosphoribosyltransferase as a target in inflammation- related disorders. Curr Top Med Chem. 2013;13:2930–2938. doi: 10.2174/15680266113136660208. [DOI] [PubMed] [Google Scholar]
- 23.Romacho T, Sanchez-Ferrer CF, Peiro C. Visfatin/Nampt: an adipokine with cardiovascular impact. Mediators Inflamm. 2013;2013:946427. doi: 10.1155/2013/946427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang P, Li WL, Liu JM, Miao CY. NAMPT and NAMPT-controlled NAD metabolism in vascular repair. J Cardiovasc Pharmacol. 2015;67:474–481. doi: 10.1097/FJC.0000000000000332. [DOI] [PubMed] [Google Scholar]
- 25.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 26.Comhair SA, Xu W, Mavrakis L, Aldred MA, Asosingh K, Erzurum SC. Human primary lung endothelial cells in culture. Am J Respir Cell Mol Biol. 2012;46:723–730. doi: 10.1165/rcmb.2011-0416TE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeineh NS, Bachman TN, El-Haddad H, Champion HC. Effects of acute intravenous iloprost on right ventricular hemodynamics in rats with chronic pulmonary hypertension. Pulm Circ. 2014;4:612–618. doi: 10.1086/677358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Tang H, Sysol JR, Moreno-Vinasco L, Shioura KM, Chen T, Gorshkova I, Wang L, Huang LS, Usatyuk PV, Sammani S, Zhou G, Raj JU, Garcia JG, Berdyshev E, Yuan JX, Natarajan V, Machado RF. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;190:1032–1043. doi: 10.1164/rccm.201401-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Gool F, Galli M, Gueydan C, Kruys V, Prevot PP, Bedalov A, Mostoslavsky R, Alt FW, De Smedt T, Leo O. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009;15:206–210. doi: 10.1038/nm.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia L, Delmotte P, Aravamudan B, Pabelick CM, Prakash YS, Sieck GC. Effects of the inflammatory cytokines TNF-alpha and IL-13 on stromal interaction molecule-1 aggregation in human airway smooth muscle intracellular Ca(2+) regulation. Am J Respir Cell Mol Biol. 2013;49:601–608. doi: 10.1165/rcmb.2013-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sathish V, Abcejo AJ, Thompson MA, Sieck GC, Prakash YS, Pabelick CM. Caveolin-1 regulation of store-operated Ca(2+) influx in human airway smooth muscle. Eur Respir J. 2012;40:470–478. doi: 10.1183/09031936.00090511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D, Yuan JX. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol. 2015;308:C581–593. doi: 10.1152/ajpcell.00202.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bae SK, Kim SR, Kim JG, Kim JY, Koo TH, Jang HO, Yun I, Yoo MA, Bae MK. Hypoxic induction of human visfatin gene is directly mediated by hypoxia-inducible factor-1. FEBS Lett. 2006;580:4105–4113. doi: 10.1016/j.febslet.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 34.Segawa K, Fukuhara A, Hosogai N, Morita K, Okuno Y, Tanaka M, Nakagawa Y, Kihara S, Funahashi T, Komuro R, Matsuda M, Shimomura I. Visfatin in adipocytes is upregulated by hypoxia through HIF1alpha-dependent mechanism. Biochem Biophys Res Commun. 2006;349:875–882. doi: 10.1016/j.bbrc.2006.07.083. [DOI] [PubMed] [Google Scholar]
- 35.Adyshev DM, Elangovan VR, Moldobaeva N, Mapes B, Sun X, Garcia JG. Mechanical stress induces pre-B-cell colony-enhancing factor/NAMPT expression via epigenetic regulation by miR-374a and miR-568 in human lung endothelium. Am J Respir Cell Mol Biol. 2014;50:409–418. doi: 10.1165/rcmb.2013-0292OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C, Tong J, Huang G. Nicotinamide phosphoribosyl transferase (Nampt) is a target of microRNA-26b in colorectal cancer cells. PLoS One. 2013;8:e69963. doi: 10.1371/journal.pone.0069963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, Kang Y, Li X, Kemper B, Kemper JK. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell. 2013;12:1062–1072. doi: 10.1111/acel.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen XY, Zhang HS, Wu TC, Sang WW, Ruan Z. Down-regulation of NAMPT expression by miR-182 is involved in Tat-induced HIV-1 long terminal repeat (LTR) transactivation. Int J Biochem Cell Biol. 2013;45:292–298. doi: 10.1016/j.biocel.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Takebayashi K, Suetsugu M, Wakabayashi S, Aso Y, Inukai T. Association between plasma visfatin and vascular endothelial function in patients with type 2 diabetes mellitus. Metabolism. 2007;56:451–458. doi: 10.1016/j.metabol.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 40.Kadoglou NP, Sailer N, Moumtzouoglou A, Kapelouzou A, Tsanikidis H, Vitta I, Karkos C, Karayannacos PE, Gerasimidis T, Liapis CD. Visfatin (nampt) and ghrelin as novel markers of carotid atherosclerosis in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2010;118:75–80. doi: 10.1055/s-0029-1237360. [DOI] [PubMed] [Google Scholar]
- 41.Zhong M, Tan HW, Gong HP, Wang SF, Zhang Y, Zhang W. Increased serum visfatin in patients with metabolic syndrome and carotid atherosclerosis. Clin Endocrinol (Oxf) 2008;69:878–884. doi: 10.1111/j.1365-2265.2008.03248.x. [DOI] [PubMed] [Google Scholar]
- 42.Liu SW, Qiao SB, Yuan JS, Liu DQ. Association of plasma visfatin levels with inflammation, atherosclerosis and acute coronary syndromes (ACS) in humans. Clin Endocrinol (Oxf) 2009;71:202–207. doi: 10.1111/j.1365-2265.2008.03453.x. [DOI] [PubMed] [Google Scholar]
- 43.Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L144–155. doi: 10.1152/ajplung.00412.2001. [DOI] [PubMed] [Google Scholar]
- 44.Brevnova EE, Platoshyn O, Zhang S, Yuan JX. Overexpression of human KCNA5 increases IK V and enhances apoptosis. Am J Physiol Cell Physiol. 2004;287:C715–722. doi: 10.1152/ajpcell.00050.2004. [DOI] [PubMed] [Google Scholar]
- 45.Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation. 2009;119:2313–2322. doi: 10.1161/CIRCULATIONAHA.108.782458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang S, Dong H, Rubin LJ, Yuan JX. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292:C2297–2305. doi: 10.1152/ajpcell.00383.2006. [DOI] [PubMed] [Google Scholar]
- 47.Song MY, Makino A, Yuan JX. STIM2 Contributes to Enhanced Store-operated Ca Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension. Pulm Circ. 2011;1:84–94. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye SQ, Zhang LQ, Adyshev D, Usatyuk PV, Garcia AN, Lavoie TL, Verin AD, Natarajan V, Garcia JG. Pre-B-cell-colony-enhancing factor is critically involved in thrombin-induced lung endothelial cell barrier dysregulation. Microvasc Res. 2005;70:142–151. doi: 10.1016/j.mvr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 49.Romacho T, Azcutia V, Vazquez-Bella M, Matesanz N, Cercas E, Nevado J, Carraro R, Rodriguez-Manas L, Sanchez-Ferrer CF, Peiro C. Extracellular PBEF/NAMPT/visfatin activates pro-inflammatory signalling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia. 2009;52:2455–2463. doi: 10.1007/s00125-009-1509-2. [DOI] [PubMed] [Google Scholar]
- 50.Sathish V, Delmotte PF, Thompson MA, Pabelick CM, Sieck GC, Prakash YS. Sodium-calcium exchange in intracellular calcium handling of human airway smooth muscle. PLoS One. 2011;6:e23662. doi: 10.1371/journal.pone.0023662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sathish V, Thompson MA, Bailey JP, Pabelick CM, Prakash YS, Sieck GC. Effect of proinflammatory cytokines on regulation of sarcoplasmic reticulum Ca2+ reuptake in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2009;297:L26–34. doi: 10.1152/ajplung.00026.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White TA, Xue A, Chini EN, Thompson M, Sieck GC, Wylam ME. Role of transient receptor potential C3 in TNF-alpha-enhanced calcium influx in human airway myocytes. Am J Respir Cell Mol Biol. 2006;35:243–251. doi: 10.1165/rcmb.2006-0003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sieck GC, White TA, Thompson MA, Pabelick CM, Wylam ME, Prakash YS. Regulation of store-operated Ca2+ entry by CD38 in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2008;294:L378–385. doi: 10.1152/ajplung.00394.2007. [DOI] [PubMed] [Google Scholar]
- 54.DebRoy A, Vogel SM, Soni D, Sundivakkam PC, Malik AB, Tiruppathi C. Cooperative signaling via transcription factors NF-kappaB and AP1/c-Fos mediates endothelial cell STIM1 expression and hyperpermeability in response to endotoxin. J Biol Chem. 2014;289:24188–24201. doi: 10.1074/jbc.M114.570051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bruzzone S, Fruscione F, Morando S, Ferrando T, Poggi A, Garuti A, D’Urso A, Selmo M, Benvenuto F, Cea M, Zoppoli G, Moran E, Soncini D, Ballestrero A, Sordat B, Patrone F, Mostoslavsky R, Uccelli A, Nencioni A. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS One. 2009;4:e7897. doi: 10.1371/journal.pone.0007897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oita RC, Ferdinando D, Wilson S, Bunce C, Mazzatti DJ. Visfatin induces oxidative stress in differentiated C2C12 myotubes in an Akt- and MAPK-independent, NFkB-dependent manner. Pflugers Arch. 2010;459:619–630. doi: 10.1007/s00424-009-0752-1. [DOI] [PubMed] [Google Scholar]
- 57.Camp SM, Ceco E, Evenoski CL, Danilov SM, Zhou T, Chiang ET, Moreno-Vinasco L, Mapes B, Zhao J, Gursoy G, Brown ME, Adyshev DM, Siddiqui SS, Quijada H, Sammani S, Letsiou E, Saadat L, Yousef M, Wang T, Liang J, Garcia JG. Unique Toll-Like Receptor 4 Activation by NAMPT/PBEF Induces NFkappaB Signaling and Inflammatory Lung Injury. Sci Rep. 2015;5:13135. doi: 10.1038/srep13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pillai VB, Sundaresan NR, Kim G, Samant S, Moreno-Vinasco L, Garcia JG, Gupta MP. Nampt secreted from cardiomyocytes promotes development of cardiac hypertrophy and adverse ventricular remodeling. Am J Physiol Heart Circ Physiol. 2013;304:H415–426. doi: 10.1152/ajpheart.00468.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, Nakagawa T, Yoshino J, Imai S. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD+ and Function in Mice. Cell Metab. 2015;21:706–717. doi: 10.1016/j.cmet.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moreno-Vinasco L, Quijada H, Sammani S, Siegler J, Letsiou E, Deaton R, Saadat L, Zaidi RS, Messana J, Gann PH, Machado RF, Ma W, Camp SM, Wang T, Garcia JG. Nicotinamide phosphoribosyltransferase inhibitor is a novel therapeutic candidate in murine models of inflammatory lung injury. Am J Respir Cell Mol Biol. 2014;51:223–228. doi: 10.1165/rcmb.2012-0519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gonfloni S, Iannizzotto V, Maiani E, Bellusci G, Ciccone S, Diederich M. P53 and Sirt1: routes of metabolism and genome stability. Biochem Pharmacol. 2014;92:149–156. doi: 10.1016/j.bcp.2014.08.034. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Shen L, Sun LQ. The regulation of radiosensitivity by p53 and its acetylation. Cancer Lett. 2015;363:108–118. doi: 10.1016/j.canlet.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 63.van Leeuwen IM, Higgins M, Campbell J, McCarthy AR, Sachweh MC, Navarro AM, Lain S. Modulation of p53 C-terminal acetylation by mdm2, p14ARF, and cytoplasmic SirT2. Mol Cancer Ther. 2013;12:471–480. doi: 10.1158/1535-7163.MCT-12-0904. [DOI] [PubMed] [Google Scholar]
- 64.Pellegrini L, Pucci B, Villanova L, Marino ML, Marfe G, Sansone L, Vernucci E, Bellizzi D, Reali V, Fini M, Russo MA, Tafani M. SIRT3 protects from hypoxia and staurosporine-mediated cell death by maintaining mitochondrial membrane potential and intracellular pH. Cell Death Differ. 2012;19:1815–1825. doi: 10.1038/cdd.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cohen-Kaminsky S, Hautefort A, Price L, Humbert M, Perros F. Inflammation in pulmonary hypertension: what we know and what we could logically and safely target first. Drug Discov Today. 2014;19:1251–1256. doi: 10.1016/j.drudis.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 66.Busso N, Karababa M, Nobile M, Rolaz A, Van Gool F, Galli M, Leo O, So A, De Smedt T. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS One. 2008;3:e2267. doi: 10.1371/journal.pone.0002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hovstadius P, Larsson R, Jonsson E, Skov T, Kissmeyer AM, Krasilnikoff K, Bergh J, Karlsson MO, Lonnebo A, Ahlgren J. A Phase I study of CHS 828 in patients with solid tumor malignancy. Clin Cancer Res. 2002;8:2843–2850. [PubMed] [Google Scholar]
- 68.Ravaud A, Cerny T, Terret C, Wanders J, Bui BN, Hess D, Droz JP, Fumoleau P, Twelves C. Phase I study and pharmacokinetic of CHS-828, a guanidino-containing compound, administered orally as a single dose every 3 weeks in solid tumours: an ECSG/EORTC study. Eur J Cancer. 2005;41:702–707. doi: 10.1016/j.ejca.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 69.Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs. 2008;26:45–51. doi: 10.1007/s10637-007-9083-2. [DOI] [PubMed] [Google Scholar]
- 70.von Heideman A, Berglund A, Larsson R, Nygren P. Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharmacol. 2010;65:1165–1172. doi: 10.1007/s00280-009-1125-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.